
Abstract
Purpose
To provide an overview on positron emission tomography (PET) imaging of tau pathology in Alzheimer’s disease (AD) and other neurodegenerative disorders.
Results
Different classes of tau tracers such as flortaucipir, THK5317, and PBB3 have been developed and utilized in previous clinical studies. In AD, the topographical distribution of tracer binding follows the known distribution of neurofibrillary tangles and is closely associated with neurodegeneration as well as the clinical phenotype of dementia. Significant retention of tracers has also been observed in the frequent site of the 4-repeat (4R) tau isoform deposits in non-AD tauopathies, such as in progressive supranuclear palsy. However, in vitro binding studies indicate that most tau tracers are less sensitive to straight tau filaments, in contrast to their high binding affinity to paired helical filaments of tau (PHF-tau). The first-generation of tau tracers shows off-target binding in the basal ganglia, midbrain, thalamus, choroid plexus, and venous sinus. Off-target binding of THK5351 to monoamine oxidase B (MAO-B) has been observed in disease-associated brain regions linked to neurodegeneration and is associated with astrogliosis in areas of misfolded protein accumulation. The second generation of tau tracers, such as [18F]MK-6240, is highly selective to PHF-tau with little off-target binding and have enabled the reliable assessment of PHF-tau burden in aging and AD.
Conclusions
Tau PET tracers have enabled in vivo quantification of PHF-tau burden in human brains. Tau PET can help in understanding the underlying cause of dementia symptoms, and in patient selection for clinical trials of anti-dementia therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Misfolded protein accumulation is a common pathogenic mechanism in several neurodegenerative disorders, such as Alzheimer’s disease (AD) and Parkinson’s disease. The neuropathology of AD is characterized by the deposition of senile plaques (SPs) and neurofibrillary tangles (NFTs), which are misfolded aggregates of amyloid-β (Aβ) and hyperphosphorylated tau proteins, respectively [1]. Definitive diagnosis of AD can be made by postmortem autopsy confirmation of these protein lesions in the brain. Aβ is a 39–43 amino acid peptide derived from the proteolytic cleavage of the amyloid precursor protein (APP) by β-secretase (BACE1) and γ-secretase. This protein has been regarded as an initiator of the neurodegenerative process in AD [2]. Pathogenic mutations in the APP gene or presenilin genes cause the overproduction of Aβ and early-onset familial AD. In sporadic AD, impaired clearance of Aβ is considered to be the main cause for the accumulation of Aβ in the brain [3]. The amyloid cascade hypothesis has been widely accepted for explaining the etiology and pathogenesis of AD [4]. The deposition of Aβ is the initial pathological event in AD leading to the formation of SPs and then to NFTs, neuronal loss, and ultimately cognitive decline. Amyloid PET studies have demonstrated that neocortical accumulation of Aβ precedes neurodegenerative changes and cognitive decline in AD [5].
Tau protein, another key player in the pathogenesis of AD, is a microtubule-associated protein (MAP), which is abundantly expressed in the central nervous system (CNS) [6]. The accumulation of hyperphosphorylated tau protein in neurons is considered to be crucial in the neurodegenerative process of AD. Abnormal hyperphosphorylation of tau causes protein self-aggregation and the formation of paired helical filaments (PHF). Abnormal tau aggregation occurs not only in the AD brain, but also in a family of protein-misfolding diseases called tauopathies, which include frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), Pick’s disease, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and chronic traumatic encephalopathy [7]. There are six molecular isoforms of tau generated by alternative pre-mRNA splicing of a single gene transcript and classified according to their tubulin-binding domains as 3-repeat (3R) and 4-repeat (4R) tau proteins. Equal amounts of 3R and 4R tau isoforms exist in the normal brain. In PHF-tau, there is also an equal balance of 3R and 4R isoforms. The changes in the 3R and 4R tau ratio can cause abnormal tau accumulation and lead to neurodegeneration as in tauopathies [8]. For example, the 3R tau isoform is predominantly accumulated in Pick’s disease, whereas the 4R tau is more abundant in PSP, CBD, and argyrophilic grain disease [8]. In 4R tauopathies, tau aggregates are also observed in glial cells and form disease-specific astroglial lesions, such as tufted astrocytes in PSP and astrocytic plaques in CBD. Frontotemporal lobar degeneration linked to microtubule-associated protein tau (MAPT) mutations involves 3R tau or 4R tau, or a combination of both isoforms. The spatial distribution of tau deposits is different for each tauopathy and is strongly associated with the clinical phenotype of these diseases.
Characteristics of tau PET images
Table 1 summarizes the comparison between amyloid and tau PET images in AD. In contrast to diffuse and widespread distribution of amyloid tracers in the neocortex, retention of tau tracers is mainly observed in the inferior temporal and parietal cortices of AD patients. Neocortical PHF-tau formation is considered to be a downstream event after Aβ deposition in AD [9, 10]. Therefore, tau protein deposits are not frequent in the neocortex, even if widespread amyloid-β deposits exist during asymptomatic stages of AD [2]. Normal aging is also associated with accumulation of PHF-tau in the medial temporal lobe. The formation of NFTs in the absence of Aβ deposition is neuropathologically classified as primary age-related tauopathy (PART) [11]. Significant tau tracer retention in the temporal lobe has been reported even in cognitively normal older people [12]. Amyloid PET studies have shown little association between amyloid burden and the clinical severity of dementia [13]. In contrast, tau PET studies have shown a close relationship between tau pathology and the clinical severity of dementia [5]. During aging and in the very early stages of AD, tau tracer retention is usually confined to the medial temporal cortex. After dementia symptoms appear, neocortical tau pathology progresses rapidly following the onset of dementia and adhering to a stereotypical spatial pattern as the clinical symptoms progress [1]. Thus, tau imaging may allow the accurate staging of AD [14] and could prove useful in predicting the prognosis of dementia. Recent PET studies have shown that the topographical distribution of tau tracer retention overlaps with that of other neurodegenerative markers such as fluorodeoxyglucose (FDG)-PET and structural magnetic resonance imaging (MRI). PET imaging enables the longitudinal assessment of tau pathology progression and the understanding of the natural history of tau pathology progression.
The development of disease-modifying therapies (DMTs) is ongoing as an initiative to decrease the number of patients with AD. Considering that the accumulation of Aβ starts before the clinical onset of AD, it is ideal to begin using DMTs before the clinical symptoms appear. Amyloid PET has been widely used for patient selection and the evaluation of treatment efficacy in clinical trials of anti-amyloid drugs. However, Aβ-targeted therapies have been unsuccessful to date. Tau plays a key part in the formation of NFTs and might, therefore, represent an important therapeutic target in AD [5, 15]. Based on previous studies, anti-tau immunotherapies and tau aggregation inhibitors appear to be promising therapeutic strategies in AD [16]. Tau PET has the potential to facilitate the monitoring of treatment efficacy of new drugs and the identification of suitable patients who could enroll in the clinical trials evaluating these drugs. Progressive tau accumulation has also been observed in many neurodegenerative diseases and chronic traumatic encephalopathies [16]. Recent PET studies have shown the potential utility of tau imaging for the differential diagnosis of non-AD tauopathies [5]. Although it is uncertain whether currently available tau tracers can be used as biomarkers of non-AD tau deposits, early and accurate diagnosis of these diseases will facilitate preventive clinical trials in a broad range of tauopathies.
First generation of tau tracers
Tau tracers are expected to bind tau protein fibrils at low nanomolar concentrations and should be highly selective for tau over Aβ, given that the concentration of Aβ is substantially higher than that of tau in the AD neocortex [17]. In vitro autoradiography of human brain sections has been used for the assessment of binding selectivity of radiotracers at low nanomolar concentrations [18]. High blood–brain barrier (BBB) permeability is also essential for brain PET tracers. Therefore, low molecular weight compounds have been used as tau PET tracers [18, 19]. Radiotracers should show sufficient amount of brain uptake after administration and be cleared rapidly from normal brain tissue. For quantitative analysis of PET images, it is also ideal that the radiolabeled metabolites do not enter the brain and/or bind to specific targets. The 18F-labeled tracer may be more suitable for high-throughput screening of tau burden than the 11C-labeled tracer. As shown in Fig. 1, several different classes of tau tracers have been developed and utilized in human studies. In contrast with the development of receptor ligands, there were no pre-existing small molecules that selectively bind to PHF-tau with high affinity. Furthermore, there are multiple binding sites on PHF-tau. This makes it difficult to conduct a blocking study using gold-standard ligand and limits the proof that the proposed radiotracers bind specifically to tau pathology in the human brain. Therefore, the binding specificity to tau protein deposits must be carefully validated in the development of new tau tracers.

Chemical structures of tau PET tracers
Quinoline derivatives
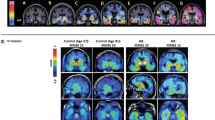
Quinoline derivatives, BF-158, BF-170, and BF-242 (THK-523), were first identified as potential candidates for tau tracers through the screening of β-sheet-binding compounds [20]. Notably, the [18F]THK523 showed higher affinity for tau fibrils than for Aβ fibrils [21]. Autoradiographic studies have demonstrated the accumulation of this tracer in AD brain regions containing a high density of PHF-tau. Small animal PET studies have also demonstrated significant tracer retention in brains of tau transgenic mice [22]. However, clinical PET studies have failed to visualize tau deposits in the gray matter of AD patients’ brains [23], possibly due to the insufficient binding affinity of this tracer to native tau conformation. [18F]THK5105 and [18F]THK5117 were additionally developed to improve the binding affinity to PHF-tau. These tracers exhibited binding affinity and selectivity to PHF-tau over Aβ in AD brain tissues [24]. In proof-of-concept PET studies, these radiotracers clearly differentiated the brains of patients with AD from those of healthy controls [25,26,27]. Furthermore, the amount of tracer retention in the neocortex was significantly correlated with the clinical severity of dementia [25]. Longitudinal PET studies have also demonstrated a significant correlation in changes of tracer binding with cognitive decline in AD patients [28]. The following clinical studies have been performed using the (S)-enantiomer of THK5117, because the (S)-enantiomer of quinoline derivatives showed better pharmacokinetics than the (R)-enantiomer [29, 30]. High [18F]THK5317 ((S)-[18F]THK5117) retention has been observed in cases of mild cognitive impairment (MCI) and AD dementia [31] and has been found to be negatively associated with cognitive performance [32]. One of the drawbacks of THK5117 and THK5317 is a non-negligible white matter binding, which is possibly due to the binding of the radiotracers to the β-sheet structures of the myelin basic protein. THK5351 was then developed to reduce its binding to the white matter tissue [33]. In human study, THK5351 exhibited faster white matter clearance and higher specific binding to AD tau-associated regions of interest (ROI) than THK5317 [34]. The spatial pattern of THK5351 binding in the neocortex was similar to that of THK5317 and was also found to be associated with the clinical phenotype of dementia [35]. Moreover, THK5351 retention was found to be correlated with extra-hippocampal subregional atrophy rather than hippocampal subfields, suggesting different underlying mechanisms of atrophy in early AD [36]. As shown in Fig. 2, significant THK5351 retention was also observed in the midbrain and the globus pallidus of PSP patients [37,38,39], which was found to be highly correlated to the clinical severity of PSP [39]. In patients with corticobasal syndrome (CBS), asymmetric THK5351 retention was observed in the frontal, parietal, and globus pallidus, contralaterally to the side associated with greater cortical dysfunction and parkinsonism [40]. In recent studies, doubt was cast on the selectivity of THK5351 to tau, because the tracer uptake was high in the basal ganglia, the thalamus, and the brainstem [41]. Furthermore, significant tracer retention has been observed in the brain of patients with semantic variant primary progressive aphasia (Fig. 3), which is infrequently associated with tau protein accumulation [42, 43]. These findings may be explained by off-target binding of THK5351, which will be discussed below.

[18F]THK5351 PET images in patients with Alzheimer’s disease (AD), corticobasal syndrome (CBS), and progressive supranuclear palsy (PSP). In AD patients, THK5351 retention was high in the basal ganglia and the thalamus, reflecting the off-target binding to monoamine oxidase B (MAO-B). CBS patients showed prominent tracer retention in the basal ganglia, the precentral gyrus, and the midbrain. THK5351 retention in the midbrain was also elevated in PSP

Figure courtesy of Dr. Ryota Kobayashi, Yamagata University
[18F]THK5351 PET images in patient with semantic variant primary progressive aphasia. Significant tracer retention was observed in the left anterior temporal pole.
PBB3
PBB3 was designed to detect a broad range of tau pathologies in the human brain [44]. In an autoradiography of the hippocampus of AD patients, specific tracer binding was observed in the CA1 and subiculum areas, where high density of fibrillar tau aggregates exist [44]. In an in vitro binding analysis, PBB3 were found to bind preferentially to dystrophic neurites as well as NFTs in AD brains than flortaucipir [45]. In clinical PET studies, the binding of [11C]PBB3 was detected in the medial temporal region of AD patients. Tracer retention in the medial temporal region, the precuneus, and the frontal cortex was well correlated with cognitive decline. [11C]PBB3 binding was also associated with gray matter atrophy [46]. As is the case with THK5351, [11C]PBB3 retention was also observed in the putamen, the midbrain, the globus pallidus, and the substantia nigra of patients with PSP [47]. Laterality of tracer signals in the basal ganglia was also observed in patient with CBS [44], suggesting the binding of this tracer to 4R tau lesions. Recent PET studies have suggested different binding targets between PBB3 and THK5351 [48] with no significant correlation in the load and regional distribution between PBB3 and THK5351. There are several technical issues on the use of [11C]PBB3, including lower dynamic range, metabolic instability, and off-target binding in the basal ganglia, the longitudinal sinus and the choroid plexus [5]. The 18F-labeled derivatives ([18F]AM-PBB3 and [18F]PM-PBB3) have been developed and clinically tested to improve these drawbacks of [11C]PBB3.
Flortaucipir ([18F]AV-1451)
Benzimidazole pyrimidine derivatives were identified as candidates for tau PET tracers. [18F] flortaucipir, also known as [18F]AV-1451 (T807), and [18F]T808 exhibit high binding affinities to the PHF-tau, and high binding selectivity for tau over Aβ in AD brains [49,50,51]. Autoradiography studies have demonstrated that flortaucipir binding colocalizes with tau deposits, but not with Aβ, α-synuclein, or TDP-43 protein deposits [49, 52]. Additionally, postmortem analysis revealed that flortaucipir binding correlates with postmortem NFT Braak staging in AD brains [53]. Clinical PET studies have shown preferential tracer retention in the inferior temporal and the posterior parietal cortices of AD patients (Fig. 4) [12, 54]. In amnestic AD cases, the spatial patterns of tracer distribution follow the known distribution of NFTs in AD [55]. Furthermore, the amount and area of tracer retention significantly correlate with the clinical severity of dementia. Compelling test–retest reproducibility for flortaucipir was observed across the neocortical and the mesial temporal lobe structures in healthy controls and AD patients [56].

Figure courtesy of Dr. Victor Villemagne, University of Melbourne
[18F]Flortaucipir PET images in patient with Alzheimer’s disease (AD) and healthy control (HC). Tracer retention was markedly elevated in the neocortex of AD patient
The relationship between flortaucipir binding and cognition was observed even in cognitively sound older people [12]. Interestingly, neocortical flortaucipir retention was found in some of the preclinical AD cases, but was rarely found in amyloid-negative cases [57, 58]. These observations suggest that the neocortical Aβ deposition may trigger the spread of tau pathology from the medial temporal area to the broad neocortical areas. Age-related increases in flortaucipir retention was observed in the medial temporal lobe of amyloid-negative, normal subjects, suggesting the existence of age-related tau deposition in this population, which is neuropathologically classified as PART [11]. However, whether flortaucipir retention in this area truly reflects age-related tau pathology in cognitively normal subjects is yet to be confirmed.
The diversity of flortaucipir distribution has been observed in amyloid-positive MCI and AD [58, 59], which is associated with the age of onset. Early-onset AD cases exhibited greater flortaucipir uptake than late-onset AD cases in the prefrontal and the premotor, as well as in the inferior parietal cortex [60]. There is also a diversity of flortaucipir distribution in the neocortex of atypical AD cases [61, 62]. Patients with logopenic variant primary progressive aphasia demonstrated asymmetric, left greater than right, hemisphere tracer retention. In a case with posterior cortical atrophy, preferential flortaucipir retention was observed in the posterior cortical regions, which showed a compelling association with glucose hypometabolism and the clinical phenotype observed [63]. Notably, flortaucipir retention was correlated with regional gray matter atrophy in this population [62]. Increased neocortical flortaucipir retention has also been observed in some cases of dementia with Lewy bodies (DLB) [64, 65]. However, the amount of flortaucipir binding in DLB patients was relatively lower than that of AD patients [65]. Intriguingly, 4 out of 17 of the Lewy body disease cases with low amyloid burden had elevated flortaucipir binding in the inferior temporal cortex, suggesting that tau pathology in DLB may extend to the neocortex without amyloid burden [64]. Many researchers have examined the relationship between flortaucipir PET and cerebrospinal fluid (CSF) biomarkers. To this end, flortaucipir binding in the para-hippocampal gyrus and neocortex was shown to correlate with CSF-tau levels [66, 67]. However, flortaucipir retention was more strongly correlated with neurodegeneration and cognitive decline than with CSF-tau levels. Cross-sectional analysis suggested that these two biomarkers showed different dynamics at different stages of AD [68].
Furthermore, clinical studies of flortaucipir have been extended to a broad range of tauopathies. In PSP patients, significant flortaucipir retention was observed in the globus pallidus, the putamen, the subthalamic nucleus, and in the midbrain of PSP patients [69,70,71,72,73]. The globus pallidus is one of the brain regions where PSP patients can be distinguished from control and PD patients’ brains. The spatial pattern of tracer binding may be associated with the clinical phenotype of PSP although the clinical severity of PSP did not significantly correlate with flortaucipir binding in any region [72]. These clinical findings suggest a binding ability of flortaucipir to the 4R tau lesion in PSP. However, postmortem analysis of PSP cases failed to show detectable flortaucipir binding to 4R tau deposits and also showed a lack of significant correlation between in vivo flortaucipir retention and the accumulation of straight tau filaments [73]. Several studies have shown a poor association of flortaucipir binding with non-AD tauopathies that have preferential accumulation of either 3R or 4R tau [52, 73, 74] although some studies have shown moderate tracer binding in brain tissues from patients with Pick’s disease and FTDP-17 [75]. Technical problems may underlie the discrepancies between the in vivo PET and the in vitro autoradiography results. In the autoradiogram of brain tissues, alcohol is frequently used for washing the slides after incubation. The treatment with alcohol could remove the specific tracer binding on tau deposits and cause the underestimation of specific binding in the autoradiogram. Significant PET findings have also been observed in patients with CBS. CBS patients exhibited distinct patterns of flortaucipir retention in the motor cortex, the corticospinal tract, and the basal ganglia contralateral to the affected body side [76, 77]. In an autopsy-confirmed case with corticobasal degeneration (CBD), regional flortaucipir binding showed an excellent correlation with 4R tau burden, suggesting higher sensitivity of flortaucipir to 4R tau deposits in CBD, compared with other non-AD tauopathies [78]. In another autopsy-confirmed CBD case, flortaucipir retention was highly related to tau-positive threads, when compared to tangle pathology in CBD [79].
Current challenges in tau imaging
Recent studies have exhibited high radiotracer retention in the area where tau pathology is not frequently observed in AD and other tauopathies (Table 2). In flortaucipir PET studies, off-target binding has been observed in the midbrain, the meninges, the choroid plexus, and the striatum. In the analysis of autopsy brain samples, flortaucipir was found to bind to vessels, iron-associated regions, the substantia nigra, calcifications in the choroid plexus, and to leptomeningeal melanin [74]. High radiotracer signals in the substantia nigra and in the meninges are considered to be associated with neuromelanin and melanocyte binding. Therefore, this tracer could be also used to measure the density of pigmented dopaminergic neurons in the substantia nigra [80]. Significant reduction of flortaucipir binding was demonstrated in the substantia nigra of PD patients [81]. Off-target binding in extra-brain regions has been also observed in tau PET tracers. High tracer signal in the eye might be associated with tracer binding to melanin in retinal pigment epithelial cell [52]. Furthermore, high density of flortaucipir binding was observed in brain hemorrhage lesions, which may reflect the binding of this tracer to hemosiderin. Another frequent site of off-target binding is the choroid plexus which is closely located to the hippocampus. It should then be noted that high tracer signals in the choroid plexus can spill over into the hippocampus and cause a misinterpretation of tau PET results. Nonetheless, it is still not very well understood why the tracers accumulate in the choroid plexus. The choroid plexus produces the CSF and is known as the major route of blood–cerebrospinal fluid barrier exchange [82]. It may be possible that radiotracers and/or their metabolites are trapped in the epithelial cells of the choroid plexus during the transport from the blood into the CSF. Another possibility is that tau deposits in epithelial cells contribute to PET signals in the choroid plexus [83]. Striatum is also a frequent site of off-target binding of tau tracers. High amount of [18F]flortaucipir, [18F]THK5351, and [11C]PBB3 binding has been observed in the basal ganglia of aged normal subjects. The amount of these tracers was significantly elevated in patients with PSP and CBS. However, there is a debate as to whether the tracer retentions reflect the specific binding of PET tracers to tau deposits or not. Age-related increases of flortaucipir binding have been observed in the basal ganglia of healthy controls. This binding may be associated with the accumulation of iron in this area [74, 84]. There is also a possibility that neuroinflammatory changes or other concomitant proteinopathies are associated with significant tracer retention in PSP-related brain regions [59]. Monoamine oxidase (MAO) is one of identified off-targets of tau PET tracers. MAO is present in the outer mitochondrial membrane and is involved in the degradation of monoamines such as dopamine and serotonin. This enzyme is classified into two isoforms, MAO-A and MAO-B. The concentration of MAO-B in the brain is ten times higher than that of MAO-A. Flortaucipir is reported to show high binding affinity to MAO-A [85]. Another study also showed high affinity binding of flortaucipir to MAO-B in vitro [86]. However, MAO-B blocking studies in humans demonstrated that flortaucipir did not significantly bind to MAO-B in vivo [87, 88]. Therefore, it is unlikely that MAO-B is the off-target substrate of flortaucipir in the basal ganglia. In clinical studies of patients with semantic variant of primary progressive aphasia, flortaucipir binding was detected in regions that are likely to contain TDP-43 [89]. Considering low binding affinity of flortaucipir to TDP-43 in vitro [52], significant flortaucipir signals may be caused by unidentified molecules that are associated with non-specific neurodegenerative processes. THK5351 showed greater off-target binding in the midbrain, the thalamus, and the basal ganglia with greater cortical uptake in FTD than flortaucipir [5, 90]. Off-target binding of THK5351 can be attributed to MAO-B [41, 91]. Our initial investigation using fixed brain tissues failed to detect the binding of this tracer to MAO-B in the basal ganglia [33], possibly due to the fact that fixation of brain tissues with formalin and paraformaldehyde can denature the native structures of enzymes. In vitro autoradiography of [18F]THK5351 using fresh-frozen sections demonstrated a reduction of [18F]THK-5351 binding after treatment with MAO-B inhibitors [41, 91]. Furthermore, prominent reduction of PET signal has been observed in the brain of patients with AD and PSP, after single oral administration of 10 mg selegiline, an irreversible MAO-B inhibitor [92]. For this reason, THK5351 should not be used as a biomarker of tau. Compared with THK5351, THK5117 and THK5317 showed lower binding affinity to MAO-B in a competitive binding assay using 3H-Deprenyl [93]. High density of focal THK5351 binding has also been reported in patients with semantic variant primary progressive aphasia [45] and cerebral infarction [94], suggesting the binding of this tracer to MAO-B-positive astrogliosis [41, 91]. There are several molecular targets for in vivo imaging of neuroinflammation. For instance, many translocator protein (TSPO) tracers have been developed and used for the imaging of activated microglia. Considering the high dynamic range of THK5351 signals in various neurodegenerative diseases, MAO-B would be a promising target for the imaging of neuroinflammation in the brain.
Second-generation tau tracers
After the development of the first-generation tracers, several pharmaceutical companies have been trying to improve the binding selectivity and pharmacokinetics of tau PET tracers. [18F]RO6958948 (RO-948), [11C]RO6931643 (RO-643), and [11C]RO6924963 (RO-963) were identified as high-affinity competitors at the 3H-T808 binding site on native tau aggregates in AD brain tissue [95]. These tracers showed high affinity for NFTs and excellent selectivity against Aβ plaques in AD brain tissues. Among them, [18F]RO-948 showed appropriate pharmacokinetic and metabolic properties in mice and non-human primates [95, 96]. Preclinical binding analysis also has suggested lower binding affinity of these compounds to MAO-A and MAO-B than the binding affinity of flortaucipir and THK5351. Importantly, the results from a first-in-human PET study were consistent with preclinical data [97]. [18F]RO-948 showed a better signal-to-background ratio than [11C]RO-643 and [11C]RO-963 in AD patients [98]. [18F]GTP1, a deuterated analogue of [18F]T808, was designed to improve the stability of radiotracer to defluorination. A clinical PET study of [18F]GTP1 successfully prevented tracer accumulation in the skull and clearly differentiated AD patients from healthy control subjects [98]. As mentioned above, 18F-labeled PBB derivatives, [18F]AM-PBB3 and [18F]PM-PBB3, have been developed by the National Institute of Radiological Sciences in Japan. These 18F-labeled PBB3 s showed a greater signal-to-background ratio and less off-target signals in the basal ganglia than [11C]PBB3. [18F]MK-6240, a novel pyridine isoquinoline amine derivative developed by Merck, showed a high sensitivity and specificity for PHF-tau binding [99]. In autoradiographic studies, MK-6240 showed high affinity to tangle-rich AD brain homogenates and large amounts of displaceable binding in the gray matter of AD brain sections [85]. Absence of off-target binding of MK-6240 to MAO-A and MAO-B was confirmed in preclinical studies as well [85]. Recent clinical studies have demonstrated that spatial patterns of MK-6240 binding were consistent with neuropathological staging of NFTs [100]. Unlike flortaucipir and THK5351, off-target binding of MK-6240 was not observed in the basal ganglia and choroid plexus although mild tracer retention was observed in the substantia nigra and meninges [100]. A novel tracer [18F]PI-2620 has also shown a lack of off-target binding in the basal ganglia [101]. Intriguingly, a preclinical analysis has suggested a high-affinity binding of PI-2620 to PSP and Pick’s disease brains [102]. To confirm these initial observations, several clinical studies are ongoing in non-AD patients.
Conclusions and future prospects
Recent progress in the development of tau PET tracers has enabled the non-invasive monitoring of PHF-tau accumulation in aging and AD brains. This technology will further contribute to our understanding of the natural course of tau pathology progression and to the development of DMTs in AD. Nevertheless, the existing tau tracers have not been completely validated by autopsy studies as of yet, in contrast to amyloid PET tracers that have already been validated in clinical trials. To establish tau PET imaging as a biomarker of PHF-tau, imaging–autopsy correlation studies are necessary to confirm whether the amount and topographical distribution of the tracer retention truly reflect the formation of PHF-tau in the brain. It is also important to establish methods for accurate quantification of tracer binding, given that tau PET is used for therapeutic monitoring of PHF-tau density. There is as yet limited information about the speed and direction of tau pathology progression during the course of aging and AD. Current ongoing longitudinal studies may help clarify the natural history of PHF-tau formation.
The currently available tau tracers are less sensitive to detect non-AD tau lesions. Therefore, efforts should be focused on the development of PET tracers for selective imaging of 3R or 4R tauopathies. The same strategy could be applied to radiotracer development of other misfolded proteins such as α-synuclein and TDP-43. Neuroinflammation, which is characterized by the activation of glial cells, such as microglia and astrocytes, is considered to play a key role in the neurodegenerative process of various protein-misfolding diseases. Activated microglia stimulates the release of cytokines and chemokines and may cause synaptic dysfunction and neuronal loss. Reactive astrogliosis is also known to be strongly involved in AD progression and other neurodegenerative diseases. Sensitive PET tracers for activated microglia and reactive astrocytosis are indeed necessary to fully understand the neurodegenerative processes after misfolded protein accumulation. These tracers could be applied as surrogate markers of therapeutic efficacy of DMTs.
References
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Morris JC, Price JL (2001) Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci 17:101–118
Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E et al (2015) Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 11:457–470. https://doi.org/10.1038/nrneurol.2015.119
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185
Villemagne VL, Dore V, Burnham SC, Masters CL, Rowe CC (2018) Imaging tau and amyloid-beta proteinopathies in Alzheimer disease and other conditions. Nat Rev Neurol 14:225–236. https://doi.org/10.1038/nrneurol.2018.9
Goedert M, Crowther RA, Garner CC (1991) Molecular characterization of microtubule-associated proteins tau and MAP2. Trends Neurosci 14:193–199
Villemagne VL, Fodero-Tavoletti MT, Masters CL, Rowe CC (2015) Tau imaging: early progress and future directions. Lancet Neurol 14:114–124. https://doi.org/10.1016/S1474-4422(14)70252-2
Harada R, Okamura N, Furumoto S, Tago T, Yanai K, Arai H et al (2016) Characteristics of tau and its ligands in PET imaging. Biomolecules 6:7. https://doi.org/10.3390/biom6010007
Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS et al (2013) Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12:207–216. https://doi.org/10.1016/S1474-4422(12)70291-0
Perrin RJ, Fagan AM, Holtzman DM (2009) Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature 461:916–922. https://doi.org/10.1038/nature08538
Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I et al (2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128:755–766. https://doi.org/10.1007/s00401-014-1349-0
Scholl M, Lockhart SN, Schonhaut DR, O’Neil JP, Janabi M, Ossenkoppele R et al (2016) PET imaging of tau deposition in the aging human brain. Neuron 89:971–982. https://doi.org/10.1016/j.neuron.2016.01.02813
Rabinovici GD, Jagust WJ (2009) Amyloid imaging in aging and dementia: testing the amyloid hypothesis in vivo. Behav Neurol 21:117–128. https://doi.org/10.3233/BEN-2009-0232
Schwarz AJ, Shcherbinin S, Slieker LJ, Risacher SL, Charil A, Irizarry MC et al (2018) Topographic staging of tau positron emission tomography images. Alzheimers Dement (Amst) 10:221–231. https://doi.org/10.1016/j.dadm.2018.01.006
Giacobini E, Gold G (2013) Alzheimer disease therapy–moving from amyloid-beta to tau. Nat Rev Neurol 9:677–686. https://doi.org/10.1038/nrneurol.2013.223
DeKosky ST, Blennow K, Ikonomovic MD, Gandy S (2013) Acute and chronic traumatic encephalopathies: pathogenesis and biomarkers. Nat Rev Neurol 9:192–200. https://doi.org/10.1038/nrneurol.2013.36
Villemagne VL, Furumoto S, Fodero-Tavoletti MT, Harada R, Mulligan RS, Kudo Y et al (2012) The challenges of tau imaging. Future Neurol 7:409–421. https://doi.org/10.2217/fnl.12.34
Furumoto S, Tago T, Harada R, Kudo Y, Okamura N (2017) 18F-Labeled 2-arylquinoline derivatives for tau imaging: chemical, radiochemical, biological and clinical features. Curr Alzheimer Res 14:178–185
Okamura N, Harada R, Furumoto S, Arai H, Yanai K, Kudo Y (2014) Tau PET imaging in Alzheimer’s disease. Curr Neurol Neurosci Rep 14:500. https://doi.org/10.1007/s11910-014-0500-6
Okamura N, Suemoto T, Furumoto S, Suzuki M, Shimadzu H, Akatsu H et al (2005) Quinoline and benzimidazole derivatives: candidate probes for in vivo imaging of tau pathology in Alzheimer’s disease. J Neurosci 25:10857–10862. https://doi.org/10.1523/JNEUROSCI.1738-05.2005
Harada R, Okamura N, Furumoto S, Tago T, Maruyama M, Higuchi M et al (2013) Comparison of the binding characteristics of [18F]THK-523 and other amyloid imaging tracers to Alzheimer’s disease pathology. Eur J Nucl Med Mol Imaging 40:125–132. https://doi.org/10.1007/s00259-012-2261-2
Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA et al (2011) 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain 134:1089–1100. https://doi.org/10.1093/brain/awr038
Villemagne VL, Furumoto S, Fodero-Tavoletti MT, Mulligan RS, Hodges J, Harada R et al (2014) In vivo evaluation of a novel tau imaging tracer for Alzheimer’s disease. Eur J Nucl Med Mol Imaging 41:816–826. https://doi.org/10.1007/s00259-013-2681-7
Okamura N, Furumoto S, Harada R, Tago T, Yoshikawa T, Fodero-Tavoletti M et al (2013) Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J Nucl Med 54:1420–1427. https://doi.org/10.2967/jnumed.112.117341
Okamura N, Furumoto S, Fodero-Tavoletti MT, Mulligan RS, Harada R, Yates P et al (2014) Non-invasive assessment of Alzheimer’s disease neurofibrillary pathology using 18F-THK5105 PET. Brain 137:1762–1771. https://doi.org/10.1093/brain/awu064
Li Y, Tsui W, Rusinek H, Butler T, Mosconi L, Pirraglia E et al (2015) Cortical laminar binding of PET amyloid and tau tracers in Alzheimer disease. J Nucl Med 56:270–273. https://doi.org/10.2967/jnumed.114.149229
Harada R, Okamura N, Furumoto S, Furukawa K, Ishiki A, Tomita N et al (2015) [18F]THK-5117 PET for assessing neurofibrillary pathology in Alzheimer’s disease. Eur J Nucl Med Mol Imaging 42:1052–1061. https://doi.org/10.1007/s00259-015-3035-4
Ishiki A, Okamura N, Furukawa K, Furumoto S, Harada R, Tomita N et al (2015) Longitudinal assessment of tau pathology in patients with Alzheimer’s disease using [18F]THK-5117 positron emission tomography. PLoS One 10:e0140311. https://doi.org/10.1371/journal.pone.0140311
Tago T, Furumoto S, Okamura N, Harada R, Adachi H, Ishikawa Y et al (2016) Preclinical evaluation of [18F]THK-5105 enantiomers: effects of chirality on its effectiveness as a tau imaging radiotracer. Mol Imaging Biol 18:258–266. https://doi.org/10.1007/s11307-015-0879-8
Tago T, Furumoto S, Okamura N, Harada R, Adachi H, Ishikawa Y et al (2016) Structure-activity relationship of 2-Arylquinolines as PET imaging tracers for tau pathology in Alzheimer disease. J Nucl Med 57:608–614. https://doi.org/10.2967/jnumed.115.166652
Chiotis K, Saint-Aubert L, Savitcheva I, Jelic V, Andersen P, Jonasson M et al (2016) Imaging in vivo tau pathology in Alzheimer’s disease with THK5317 PET in a multimodal paradigm. Eur J Nucl Med Mol Imaging 43:1686–1699. https://doi.org/10.1007/s00259-016-3363-z
Saint-Aubert L, Almkvist O, Chiotis K, Almeida R, Wall A, Nordberg A (2016) Regional tau deposition measured by [18F]THK5317 positron emission tomography is associated to cognition via glucose metabolism in Alzheimer’s disease. Alzheimer’s Res Ther 8:38. https://doi.org/10.1186/s13195-016-0204-z
Harada R, Okamura N, Furumoto S, Furukawa K, Ishiki A, Tomita N et al (2016) 18F-THK5351: a novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer’s disease. J Nucl Med 57:208–214. https://doi.org/10.2967/jnumed.115.164848
Betthauser TJ, Lao PJ, Murali D, Barnhart TE, Furumoto S, Okamura N et al (2017) In vivo comparison of tau radioligands 18F-THK-5351 and 18F-THK-5317. J Nucl Med 58:996–1002. https://doi.org/10.2967/jnumed.116.182980
Kang JM, Lee SY, Seo S, Jeong HJ, Woo SH, Lee H et al (2017) Tau positron emission tomography using [18F]THK5351 and cerebral glucose hypometabolism in Alzheimer’s disease. Neurobiol Aging 59:210–219. https://doi.org/10.1016/j.neurobiolaging.2017.08.008
Sone D, Imabayashi E, Maikusa N, Okamura N, Furumoto S, Kudo Y et al (2017) Regional tau deposition and subregion atrophy of medial temporal structures in early Alzheimer’s disease: a combined positron emission tomography/magnetic resonance imaging study. Alzheimers Dement (Amst) 9:35–40. https://doi.org/10.1016/j.dadm.2017.07.001
Ishiki A, Harada R, Okamura N, Tomita N, Rowe CC, Villemagne VL et al (2017) Tau imaging with [18F]THK-5351 in progressive supranuclear palsy. Eur J Neurol 24:130–136. https://doi.org/10.1111/ene.13164
Shimizu S, Imabayashi E, Takenoshita N, Okamura N, Furumoto S, Kudo Y et al (2018) Case of progressive supranuclear palsy detected by tau imaging with [18F]THK-5351 before the appearance of characteristic clinical features. Geriatr Gerontol Int 18:501–502. https://doi.org/10.1111/ggi.13229
Brendel M, Schonecker S, Hoglinger G, Lindner S, Havla J, Blautzik J et al (2017) [18F]-THK5351 PET correlates with topology and symptom severity in progressive supranuclear palsy. Front Aging Neurosci 9:440. https://doi.org/10.3389/fnagi.2017.00440
Kikuchi A, Okamura N, Hasegawa T, Harada R, Watanuki S, Funaki Y et al (2016) In vivo visualization of tau deposits in corticobasal syndrome by 18F-THK5351 PET. Neurology 87:2309–2316. https://doi.org/10.1212/WNL.0000000000003375
Harada R, Ishiki A, Kai H, Sato N, Furukawa K, Furumoto S et al (2018) Correlations of 18F-THK5351 PET with postmortem burden of tau and astrogliosis in Alzheimer disease. J Nucl Med 59:671–674. https://doi.org/10.2967/jnumed.117.197426
Lee H, Seo S, Lee SY, Jeong HJ, Woo SH, Lee KM et al (2018) [18F]-THK5351 PET imaging in patients with semantic variant primary progressive aphasia. Alzheimer Dis Assoc Disord 32:62–69. https://doi.org/10.1097/WAD.0000000000000216
Takaya M, Ishii K, Hosokawa C, Saigoh K, Shirakawa O (2018) Tau accumulation in two patients with frontotemporal lobe degeneration showing different types of aphasia using 18F-THK-5351 positron emission tomography: a case report. Int Psychogeriatr 30:641–646. https://doi.org/10.1017/S1041610217002277
Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J et al (2013) Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 79:1094–1108. https://doi.org/10.1016/j.neuron.2013.07.037
Ono M, Sahara N, Kumata K, Ji B, Ni R, Koga S et al (2017) Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain 140:764–780. https://doi.org/10.1093/brain/aww339
Shimada H, Kitamura S, Shinotoh H, Endo H, Niwa F, Hirano S et al (2017) Association between Abeta and tau accumulations and their influence on clinical features in aging and Alzheimer’s disease spectrum brains: a [11C]PBB3-PET study. Alzheimers Dement (Amst) 6:11–20. https://doi.org/10.1016/j.dadm.2016.12.009
Perez-Soriano A, Arena JE, Dinelle K, Miao Q, McKenzie J, Neilson N et al (2017) PBB3 imaging in Parkinsonian disorders: evidence for binding to tau and other proteins. Mov Disord 32:1016–1024. https://doi.org/10.1002/mds.27029
Chiotis K, Stenkrona P, Almkvist O, Stepanov V, Ferreira D, Arakawa R et al (2018) Dual tracer tau PET imaging reveals different molecular targets for 11C-THK5351 and 11C-PBB3 in the Alzheimer brain. Eur J Nucl Med Mol Imaging. https://doi.org/10.1007/s00259-018-4012-5
Xia CF, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D et al (2013) [18F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement 9:666–676. https://doi.org/10.1016/j.jalz.2012.11.008
Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY et al (2013) Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis 34:457–468. https://doi.org/10.3233/JAD-122059
Chien DT, Szardenings AK, Bahri S, Walsh JC, Mu FR, Xia CF et al (2014) Early clinical PET imaging results with the novel PHF-tau radioligand [F18]-T808. J Alzheimers Dis 38:171–184. https://doi.org/10.3233/JAD-130098
Marquie M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG et al (2015) Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol 78:787–800. https://doi.org/10.1002/ana.24517
Marquie M, Siao Tick Chong M, Anton-Fernandez A, Verwer EE, Saez-Calveras N, Meltzer AC et al (2017) [F-18]-AV-1451 binding correlates with postmortem neurofibrillary tangle Braak staging. Acta Neuropathol 134:619–628. https://doi.org/10.1007/s00401-017-1740-8
Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, Rentz D et al (2016) Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 79:110–119. https://doi.org/10.1002/ana.24546
Schwarz AJ, Yu P, Miller BB, Shcherbinin S, Dickson J, Navitsky M et al (2016) Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 139:1539–1550. https://doi.org/10.1093/brain/aww023
Devous MD Sr, Joshi AD, Navitsky M, Southekal S, Pontecorvo MJ, Shen H et al (2017) Test-retest reproducibility for the tau PET imaging agent Flortaucipir F 18. J Nucl Med. https://doi.org/10.2967/jnumed.117.200691
Sperling R, Mormino E, Johnson K (2014) The evolution of preclinical Alzheimer’s disease: implications for prevention trials. Neuron 84:608–622. https://doi.org/10.1016/j.neuron.2014.10.038
Pontecorvo MJ, Devous MD Sr, Navitsky M, Lu M, Salloway S, Schaerf FW et al (2017) Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 140:748–763. https://doi.org/10.1093/brain/aww334
Okamura N, Yanai K (2017) Brain imaging: applications of tau PET imaging. Nat Rev Neurol 13:197–198. https://doi.org/10.1038/nrneurol.2017.38
Scholl M, Ossenkoppele R, Strandberg O, Palmqvist S, Fs Swedish Bio, Jogi J et al (2017) Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer’s disease. Brain 140:2286–2294. https://doi.org/10.1093/brain/awx171
Ossenkoppele R, Schonhaut DR, Scholl M, Lockhart SN, Ayakta N, Baker SL et al (2016) Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 139:1551–1567. https://doi.org/10.1093/brain/aww027
Nasrallah IM, Chen YJ, Hsieh MK, Phillips JS, Ternes K, Stockbower GE et al (2018) 18F-Flortaucipir PET/MRI correlations in nonamnestic and amnestic variants of Alzheimer disease. J Nucl Med 59:299–306. https://doi.org/10.2967/jnumed.117.194282
Ossenkoppele R, Schonhaut DR, Baker SL, O’Neil JP, Janabi M, Ghosh PM et al (2015) Tau, amyloid, and hypometabolism in a patient with posterior cortical atrophy. Ann Neurol 77:338–342. https://doi.org/10.1002/ana.24321
Gomperts SN, Locascio JJ, Makaretz SJ, Schultz A, Caso C, Vasdev N et al (2016) Tau positron emission tomographic imaging in the Lewy body diseases. JAMA Neurol 73:1334–1341. https://doi.org/10.1001/jamaneurol.2016.3338
Kantarci K, Lowe VJ, Boeve BF, Senjem ML, Tosakulwong N, Lesnick TG et al (2017) AV-1451 tau and beta-amyloid positron emission tomography imaging in dementia with Lewy bodies. Ann Neurol 81:58–67. https://doi.org/10.1002/ana.24825
Gordon BA, Friedrichsen K, Brier M, Blazey T, Su Y, Christensen J et al (2016) The relationship between cerebrospinal fluid markers of Alzheimer pathology and positron emission tomography tau imaging. Brain 139:2249–2260. https://doi.org/10.1093/brain/aww139
Chhatwal JP, Schultz AP, Marshall GA, Boot B, Gomez-Isla T, Dumurgier J et al (2016) Temporal T807 binding correlates with CSF tau and phospho-tau in normal elderly. Neurology 87:920–926. https://doi.org/10.1212/WNL.0000000000003050
Mattsson N, Scholl M, Strandberg O, Smith R, Palmqvist S, Insel PS et al (2017) 18F-AV-1451 and CSF T-tau and P-tau as biomarkers in Alzheimer’s disease. EMBO Mol Med. 9:1212–1223. https://doi.org/10.15252/emmm.201707809
Hammes J, Bischof GN, Giehl K, Faber J, Drzezga A, Klockgether T et al (2017) Elevated in vivo [18F]-AV-1451 uptake in a patient with progressive supranuclear palsy. Mov Disord 32:170–171. https://doi.org/10.1002/mds.26727
Smith R, Schain M, Nilsson C, Strandberg O, Olsson T, Hagerstrom D et al (2017) Increased basal ganglia binding of 18F-AV-1451 in patients with progressive supranuclear palsy. Mov Disord 32:108–114. https://doi.org/10.1002/mds.26813
Cho H, Choi JY, Hwang MS, Lee SH, Ryu YH, Lee MS et al (2017) Subcortical 18F-AV-1451 binding patterns in progressive supranuclear palsy. Mov Disord 32:134–140. https://doi.org/10.1002/mds.26844
Schonhaut DR, McMillan CT, Spina S, Dickerson BC, Siderowf A, Devous MD Sr et al (2017) 18F-flortaucipir tau positron emission tomography distinguishes established progressive supranuclear palsy from controls and Parkinson disease: a multicenter study. Ann Neurol 82:622–634. https://doi.org/10.1002/ana.25060
Marquie M, Normandin MD, Meltzer AC, Siao Tick Chong M, Andrea NV, Anton-Fernandez A et al (2017) Pathological correlations of [F-18]-AV-1451 imaging in non-alzheimer tauopathies. Ann Neurol 81:117–128. https://doi.org/10.1002/ana.24844
Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE et al (2016) An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun 4:58. https://doi.org/10.1186/s40478-016-0315-6
Sander K, Lashley T, Gami P, Gendron T, Lythgoe MF, Rohrer JD et al (2016) Characterization of tau positron emission tomography tracer [18F]AV-1451 binding to postmortem tissue in Alzheimer’s disease, primary tauopathies, and other dementias. Alzheimers Dement 12:1116–1124. https://doi.org/10.1016/j.jalz.2016.01.003
Smith R, Scholl M, Widner H, van Westen D, Svenningsson P, Hagerstrom D et al (2017) In vivo retention of 18F-AV-1451 in corticobasal syndrome. Neurology 89:845–853. https://doi.org/10.1212/WNL.0000000000004264
Cho H, Baek MS, Choi JY, Lee SH, Kim JS, Ryu YH et al (2017) 18F-AV-1451 binds to motor-related subcortical gray and white matter in corticobasal syndrome. Neurology 89:1170–1178. https://doi.org/10.1212/WNL.0000000000004364
Josephs KA, Whitwell JL, Tacik P, Duffy JR, Senjem ML, Tosakulwong N et al (2016) [18F]AV-1451 tau-PET uptake does correlate with quantitatively measured 4R-tau burden in autopsy-confirmed corticobasal degeneration. Acta Neuropathol 132:931–933. https://doi.org/10.1007/s00401-016-1618-1
McMillan CT, Irwin DJ, Nasrallah I, Phillips JS, Spindler M, Rascovsky K et al (2016) Multimodal evaluation demonstrates in vivo 18F-AV-1451 uptake in autopsy-confirmed corticobasal degeneration. Acta Neuropathol 132:935–937. https://doi.org/10.1007/s00401-016-1640-3
Hansen AK, Knudsen K, Lillethorup TP, Landau AM, Parbo P, Fedorova T et al (2016) In vivo imaging of neuromelanin in Parkinson’s disease using 18F-AV-1451 PET. Brain 139:2039–2049. https://doi.org/10.1093/brain/aww098
Coakeley S, Cho SS, Koshimori Y, Rusjan P, Ghadery C, Kim J et al (2018) [18F]AV-1451 binding to neuromelanin in the substantia nigra in PD and PSP. Brain Struct Funct 223:589–595. https://doi.org/10.1007/s00429-017-1507-y
Laterra J, Keep R, Betz LA, Goldstein GW (1999) Blood-cerebrospinal fluid barrier. Basic neurochemistry: molecular, cellular and medical aspects, 6th edn. Lippincott-Raven, Philadelphia
Ikonomovic MD, Abrahamson EE, Price JC, Mathis CA, Klunk WE (2016) [F-18]AV-1451 positron emission tomography retention in choroid plexus: more than “off-target” binding. Ann Neurol 80:307–308. https://doi.org/10.1002/ana.24706
Choi JY, Cho H, Ahn SJ, Lee JH, Ryu YH, Lee MS et al (2018) Off-Target 18F-AV-1451 binding in the basal ganglia correlates with age-related iron accumulation. J Nucl Med 59:117–120. https://doi.org/10.2967/jnumed.117.195248
Hostetler ED, Walji AM, Zeng Z, Miller P, Bennacef I, Salinas C et al (2016) Preclinical characterization of 18F-MK-6240, a promising PET tracer for in vivo quantification of human neurofibrillary tangles. J Nucl Med 57:1599–1606. https://doi.org/10.2967/jnumed.115.171678
Vermeiren C, Motte P, Viot D, Mairet-Coello G, Courade JP, Citron M et al (2018) The tau positron-emission tomography tracer AV-1451 binds with similar affinities to tau fibrils and monoamine oxidases. Mov Disord 33:273–281. https://doi.org/10.1002/mds.27271
Hansen AK, Brooks DJ, Borghammer P (2018) MAO-B inhibitors do not block in vivo flortaucipir ([18F]-AV-1451) binding. Mol Imaging Biol 20:356–360. https://doi.org/10.1007/s11307-017-1143-1
Smith R, Scholl M, Londos E, Ohlsson T, Hansson O (2018) 18F-AV-1451 in Parkinson’s disease with and without dementia and in dementia with Lewy bodies. Sci Rep 8:4717. https://doi.org/10.1038/s41598-018-23041-x
Bevan-Jones WR, Cope TE, Jones PS, Passamonti L, Hong YT, Fryer TD et al (2017) [18F]AV-1451 binding in vivo mirrors the expected distribution of TDP-43 pathology in the semantic variant of primary progressive aphasia. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2017-316402
Jang YK, Lyoo CH, Park S, Oh SJ, Cho H, Oh M et al (2018) Head to head comparison of [18F] AV-1451 and [18F] THK5351 for tau imaging in Alzheimer’s disease and frontotemporal dementia. Eur J Nucl Med Mol Imaging 45:432–442. https://doi.org/10.1007/s00259-017-3876-0
Ishiki A, Harada R, Kai H, Sato N, Totsune T, Tomita N et al (2018) Neuroimaging-pathological correlations of [18F]THK5351 PET in progressive supranuclear palsy. Acta Neuropathol Commun 6:53. https://doi.org/10.1186/s40478-018-0556-7
Ng KP, Pascoal TA, Mathotaarachchi S, Therriault J, Kang MS, Shin M et al (2017) Monoamine oxidase B inhibitor, selegiline, reduces 18F-THK5351 uptake in the human brain. Alzheimer’s Res Ther 9:25. https://doi.org/10.1186/s13195-017-0253-y
Lemoine L, Gillberg PG, Svedberg M, Stepanov V, Jia Z, Huang J et al (2017) Comparative binding properties of the tau PET tracers THK5117, THK5351, PBB3, and T807 in postmortem Alzheimer brains. Alzheimer’s Res Ther 9:96. https://doi.org/10.1186/s13195-017-0325-z
Ishibashi K, Kameyama M, Tago T, Toyohara J, Ishii K (2017) Potential use of 18F-THK5351 PET to identify Wallerian degeneration of the pyramidal tract caused by cerebral infarction. Clin Nucl Med 42:e523–e524. https://doi.org/10.1097/RLU.0000000000001868
Honer M, Gobbi L, Knust H, Kuwabara H, Muri D, Koerner M et al (2018) Preclinical evaluation of 18F-RO6958948, 11C-RO6931643, and 11C-RO6924963 as novel PET radiotracers for imaging tau aggregates in Alzheimer disease. J Nucl Med 59:675–681. https://doi.org/10.2967/jnumed.117.196741
Gobbi LC, Knust H, Korner M, Honer M, Czech C, Belli S et al (2017) Identification of three novel radiotracers for imaging aggregated tau in Alzheimer’s disease with positron emission tomography. J Med Chem 60:7350–7370. https://doi.org/10.1021/acs.jmedchem.7b00632
Wong DF, Comley R, Kuwabara H, Rosenberg PB, Resnick SM, Ostrowitzki S et al (2018) First in-human PET study of 3 novel tau radiopharmaceuticals: [11C]RO6924963, [11C]RO6931643, and [18F]RO6958948. J Nucl Med. https://doi.org/10.2967/jnumed.118.209916
Bohorquez S, Barret O, Tamagnan G, Alagille D, Marik J, Ayalon G et al (2016) Evaluation of tau burden in a cross-sectional cohort of Alzheimer’s disease subjects using [18F]GTP1 (Genentech Tau Probe 1). Alzheimer’s Dementia. https://doi.org/10.1016/j.jalz.2016.07.096
Walji AM, Hostetler ED, Selnick H, Zeng Z, Miller P, Bennacef I et al (2016) Discovery of 6-(Fluoro-18F)-3-(1H-pyrrolo[2,3-c]pyridin-1-yl)isoquinolin-5-amine ([18F]-MK-6240): a positron emission tomography (PET) imaging agent for quantification of neurofibrillary tangles (NFTs). J Med Chem 59:4778–4789. https://doi.org/10.1021/acs.jmedchem.6b00166
Betthauser TJ, Cody KA, Zammit MD, Murali D, Converse AK, Barnhart TE et al (2018) In vivo characterization and quantification of neurofibrillary tau PET radioligand [18F]MK-6240 in humans from Alzheimer’s disease dementia to young controls. J Nucl Med. https://doi.org/10.2967/jnumed.118.209650
Barret O, Seibyl J, Stephans A, Madonia J, Alagille D, Mueller A et al (2017) First in human characterization of PI-2620, a next generation PET tracer for assessing tau in AD and other tauopathies. AD/PD 2017 Poster Presentation. https://www.acimmune.com/en/ad-pd-2017/. Accessed 28 June 2018
Stephans A (2017) Characterization of novel PET tracer for the assessment of tau pathology in Alzheimer’s disease and other tauopathies. AD/PD 2017 Oral Presentation. https://www.acimmune.com/en/ad-pd-2017/. Accessed 28 June 2018
Acknowledgements
This work was partly supported by grants to study PET imaging from Clino Ltd. and grants from a Grant-in-Aid for Scientific Research (B) (18H02771) and a Grant-in-Aid for Scientific Research on Innovative Areas (26117003) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Author information
Authors and Affiliations
Contributions
NO: literature search and review, and manuscript writing. RH: literature search, manuscript writing and editing. AI: manuscript editing. AK: manuscript writing and editing. TN: manuscript editing. YK: manuscript editing
Corresponding author
Ethics declarations
Conflict of interest
Nobuyuki Okamura and Yukitsuka Kudo have received research support from Clino Ltd. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008.
Informed consent
Informed consent was obtained from all patients for being included in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Okamura, N., Harada, R., Ishiki, A. et al. The development and validation of tau PET tracers: current status and future directions. Clin Transl Imaging 6, 305–316 (2018). https://doi.org/10.1007/s40336-018-0290-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40336-018-0290-y