
Abstract
Background
A fixed-dose combination of daclatasvir (DCV; hepatitis C virus NS5A inhibitor), asunaprevir (ASV; non-structural protein 3 inhibitor), and beclabuvir (BCV; non-structural protein 5B inhibitor) is approved in Japan for hepatitis C virus genotype 1.
Objective
The objective of this study was to assess the combination’s drug–drug interaction potential in vivo using a validated cocktail of eight cytochrome P450 (CYP) and transporter probes.
Methods
We conducted an open-label single-sequence study in healthy adults (n = 20) given single-dose caffeine (CYP1A2 substrate), metoprolol (CYP2D6), flurbiprofen (CYP2C9), montelukast (CYP2C8), omeprazole (CYP2C19), midazolam (CYP3A4), digoxin (P-glycoprotein), and pravastatin (organic anion-transporting polypeptide), alone or with steady-state twice-daily DCV/ASV/BCV 30/200/75 mg (with or without additional BCV 75 mg to adjust for higher exposure in hepatitis C virus infection).
Results
Daclatasvir/asunaprevir/beclabuvir did not affect CYP1A2, CYP2C8, or CYP2C9; the probe maximum observed concentration and area under the concentration–time curve extrapolated to infinite time geometric mean ratios and 90% confidence intervals were all within the 0.8–1.25 bioequivalence range. Beclabuvir showed moderate dose-dependent CYP2C19 induction; omeprazole maximum observed concentration and area under the concentration–time curve from 0 to the last quantifiable concentration were lower with additional BCV [geometric mean ratio 0.36 (90% confidence interval 0.23–0.55) and 0.34 (0.25–0.46), respectively] than without [0.57 (0.42–0.78), 0.48 (0.39–0.59)]. Weak-to-moderate CYP3A4 induction was observed, plus weak CYP2D6, P-glycoprotein, and organic anion-transporting polypeptide inhibition [maximum observed concentration and area under the concentration–time curve extrapolated to infinite time without additional BCV: midazolam 0.57 (0.50–0.65), 0.53 (0.47–0.60); metoprolol 1.40 (1.20–1.64), 1.71 (1.49–1.97); digoxin 1.23 (1.12–1.35), 1.23 (1.17–1.29); pravastatin 2.01 (1.63–2.47), 1.68 (1.43–1.97)].
Conclusions
No dose adjustments with DCV/ASV/BCV are indicated for CYP1A2, CYP2C8, CYP2C9, or P-glycoprotein substrates. CYP3A4, CYP2D6, and OATP substrates should be co-administered with caution. Co-administration with agents solely metabolized by CYP2C19 is not recommended.
Similar content being viewed by others

The potential of a fixed-dose co-formulation of the hepatitis C antiviral agents daclatasvir, asunaprevir, and beclabuvir (DCV/ASV/BCV) to perpetrate pharmacokinetic interactions with substrates of cytochrome P450 (CYP) and drug transporters was evaluated in healthy volunteers using a novel eight-drug probe cocktail. |
Steady-state DCV/ASV/BCV showed weak-to-moderate induction of CYP3A4 and inhibition of CYP2D6; moderate induction of CYP2C19; minor inhibition of P-glycoprotein; and inhibition of organic anion-transporting polypeptide. No effect was seen on substrates of CYP1A2, CYP2C8, or CYP2C9. |
A priori dose adjustments are not indicated for sensitive substrates of CY1A2, CYP2C8, CYP2C9, or P-glycoprotein administered to patients with hepatitis C virus treated with DCV/ASV/BCV; however, in the absence of direct evidence with individual substrates, CYP3A4, CYP2D6, and organic anion-transporting polypeptide substrates with narrow therapeutic windows should be administered with caution and agents solely metabolized by CYP2C19 should be avoided. |
1 Introduction
The hepatitis C virus (HCV) pandemic continues to have an important impact on global health, with more than 180 million people worldwide estimated to be chronically infected with HCV and at risk for progression to cirrhosis, liver failure, and hepatocellular carcinoma [1]. Over the past decade, the treatment of chronic HCV infection has undergone a major evolution with the development of combination regimens of oral direct-acting antiviral agents (DAAs) that have rapidly superseded the previous standard of care based on pegylated interferon-α and ribavirin. There are now several effective, well-tolerated all-oral treatment options available and both cure rates (i.e., post-treatment sustained virologic response) and treatment tolerability have dramatically improved.
Daclatasvir (DCV), a pan-genotypic inhibitor of the multifunctional HCV non-structural protein 5A (NS5A) [2], and asunaprevir (ASV), an inhibitor of HCV non-structural protein 3 protease active against genotype (GT)-1, GT-4, GT-5, and GT-6 [3], have been evaluated for their drug–drug interactions both alone and in combination [4,5,6]. Both are substrates of cytochrome P450 (CYP) 3A4; DCV is a very weak CYP3A4 inducer in vivo with minimal effects on midazolam or other CYP3A4 substrates, while ASV is a weak inducer of CYP3A4 and a moderate inhibitor of CYP2D6. Both are substrates and weak inhibitors of P-glycoprotein (P-gp) and organic anion-transporting polypeptide (OATP). Neither DCV nor ASV has any clinically meaningful interactions with beclabuvir (BCV) [7], a non-nucleoside inhibitor of the HCV non-structural protein 5B (NS5B) RNA-dependent RNA polymerase that is active against GT-1, -3, -4, and -5, and with variable activity against GT-6 [8, 9]. A fixed-dose co-formulation of DCV, ASV, and BCV (DCV/ASV/BCV 30/200/75 mg) has recently been approved for use in Japan as a single-tablet regimen (Ximency Bristol-Myers Squibb, Tokyo, Japan) administered twice daily for the treatment of HCV GT-1 following evaluation in four phase III studies [10,11,12,13].
Multi-agent therapeutic regimens are at significant risk of pharmacokinetic (PK) interactions with concomitant medications, and patients treated for HCV infection are commonly receiving a variety of other therapies for co-infections such as human immunodeficiency virus [14, 15] or co-morbidities of liver disease or older age [16]. The impact of any such interactions is potentially exacerbated for fixed-dose co-formulations that do not allow adjustment of component dosing.
Empirical studies of specific drug–drug interactions may be informed by an initial assessment of the potential for a given agent or combination to affect the activity of major metabolic or transport pathways. Sequential testing of individual mechanisms is a laborious process, but “cocktails” of several readily assayed, non-interacting substrates for individual metabolic enzymes or transporters allow simultaneous in-vivo assessment of multiple pathways [17, 18]. The metabolic cocktail approach is well established for the assessment of CYP effects, but until recently there were no clinically evaluated cocktails covering both CYP and major transporter pathways such as P-gp and OATP. A novel eight-probe cocktail has now been developed [19] that includes substrate probes for six CYP enzymes—CYP1A2 (caffeine), CYP2D6 (metoprolol), CYP2C8 (montelukast), CYP2C19 (flurbiprofen), CYP2C19 (omeprazole), and CYP3A4 (midazolam)—plus probes for P-gp (digoxin) and OATP (pravastatin). These eight common clinical agents have well-characterized and acceptable safety profiles, and have shown acceptable pre-absorption compatibility in vitro [19]. Here, we report the use of this eight-agent cocktail to assess in vivo the drug–drug interaction potential of steady-state DCV, ASV, and BCV, administered as a fixed-dose co-formulation to healthy volunteers without HCV infection.
2 Methods
2.1 Study Drugs and Probes
DCV, ASV, and BCV were administered orally twice daily as a fixed-dose single tablet comprising DCV 30 mg, ASV 200 mg, and BCV 75 mg. Previous clinical data have identified an approximately two-fold increase in BCV area under the concentration–time curve (AUC) and maximum plasma concentration in HCV-infected patients over that in uninfected subjects; hence, an additional tablet of BCV 75 mg was also administered with each dose for part of the study to normalize BCV exposure for the concentrations observed in clinical practice.
The probe cocktail was administered orally as separate doses of caffeine 200 mg, metoprolol 50 mg, montelukast 10 mg, flurbiprofen 50 mg, omeprazole 40 mg, midazolam 5 mg, digoxin 0.25 mg, and pravastatin 40 mg. All probes were administered as a single tablet except for omeprazole (one delayed-release capsule) and midazolam (2.5 mL of oral syrup).
Plasma exposure to ASV in a tablet formulation is elevated by food [6] and it is recommended that the DCV/ASV/BCV combination be administered with food. All study treatment doses were therefore administered with a standard meal (≈500 calories, of which ≈30% was from fat) following an overnight (10-h) fast.
2.2 Study Population and Design
This was a single-center, open-label, single-sequence five-period study open to healthy volunteers between the ages of 18 and 45 years, with a body mass index between 18 and 32 kg/m2. Women of child-bearing potential were ineligible. Health status was determined by medical history, physical examination, vital sign measurements, 12-lead electrocardiogram (ECG) measurements, and clinical laboratory test results.
Subjects were admitted to the clinical site the day before dosing (day −1) and remained there until discharge on day 36. Treatments were administered sequentially per the schema shown in Fig. 1. The probe cocktail was administered as a single dose on day 1 (Treatment A). The DCV/ASV/BCV tablet was administered twice daily for 15 days both with and without an additional twice-daily tablet of BCV 75 mg: without additional BCV from days 6 to 15 (Treatment B) then continuing without additional BCV from day 16 through day 20 with a single dose of probe cocktail on day 16 (Treatment C); with additional BCV (DCV/ASV/BCV + BCV) from days 21 to 30 (Treatment D) then continuing with additional BCV from day 31 through day 35 with a single dose of probe cocktail on day 31 (Treatment E).

Study design. ASV asunaprevir, BCV beclabuvir, BID twice daily, D study discharge, DAA direct-acting antiviral (DCV, ASV, BCV, BMS-794712), DCV daclatasvir, PK pharmacokinetic, S study start
2.3 Sample Collection and Pharmacokinetic Analyses
Serial blood samples for PK analysis of cocktail probes and their major metabolites were collected pre-dose on days 1, 16, and 31, and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 48, 72, 96, and 120 h post-dose. Serial samples for determination of plasma concentrations of DCV, ASV, and BCV were collected pre-dose on days 16 and 31, and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, and 12 h post-dose. The major N-desmethyl metabolite of BCV, BMS-794712, was also assayed owing to its equipotent activity to the parent compound and potential to contribute to drug interactions. Pre-dose samples were also obtained on days 14, 15, 30, and 31 to determine steady-state trough concentrations of DCV, ASV, BCV, and BMS-794712.
All analytes were assayed by validated high-performance liquid chromatography/tandem mass spectrometry procedures during the period of known analyte stability. Analysis of DCV, ASV, BCV, and BMS-794712 was performed by Tandem Labs (West Trenton, NJ, USA) as previously described [20]. Analysis of cocktail probes and relevant metabolites (1′-hydroxymidazolam, α-hydroxymetoprolol, 5-hydroxyomeprazole, 36-hydroxymontelukast, pravastatin lactone) was performed by PPD® Laboratories (Richmond, VA, USA). For DCV, ASV, BCV, and BMS-794712, inter-assay coefficients of variation were < 8% and mean deviations from nominal concentrations within 7%. For probe substrates and metabolites, inter-assay coefficients of variation were < 11% and mean deviations from nominal concentrations within 8%. Assay performance data for all analytes are shown in Table S1 of the Electronic Supplementary Material (ESM).
Plasma concentration vs. time data were used to derive both single- and multiple-dose PK parameters using non-compartmental methods. Single-dose parameters for cocktail probes and metabolites included maximum observed concentration (C max), time to C max, AUC from 0 to the last quantifiable concentration (AUC0−T), AUC extrapolated to infinite time (AUCinf), and half-life (T ½). Molecular weight-corrected metabolite-to-parent ratios for C max, AUC0–T, and AUCinf were also determined. Multiple-dose PK parameters for DCV, ASV, BCV, and BMS-794712 included C max, time to C max, concentration at 12 h post-dose (C 12), AUC for one dosing interval (AUCτ), and pre-dose trough concentration.
2.4 Safety Evaluation
Safety was assessed throughout the study by a medical review of adverse events (AEs) and vital sign measurements, 12-lead ECG measurements, pulse oximetry measurements, physical examination findings, and clinical laboratory test results.
2.5 Statistics
The overall PK population included all subjects who received study treatment and had any available concentration vs. time data. The evaluable PK population comprised subjects with adequate data for the accurate estimation of the relevant parameters. Poor metabolizers for a given CYP enzyme, as predicted by the presence of CYP genetic polymorphisms of known effect on enzyme activity, were excluded from the relevant analysis (two or fewer poor metabolizers) or included as a subgroup analysis (more than two poor metabolizers).
A linear mixed-effect model was used on log-transformed data of the PK parameters of each analyte with treatment as a fixed effect and measurements within each subject as repeated measures. Point estimates and associated 90% confidence intervals (CIs) for the geometric mean ratios (GMRs) for C max, AUC0−T, and AUCinf of probe substrates administered with vs. without DCV/ASV/BCV or DCV/ASV/BCV+BCV were derived from the log-transformed data and back-exponentiated. No adjustments were made for multiplicity. SAS® software, Version 9.2 (SAS Institute, Inc., Cary, NC, USA) was used for statistical analyses.
3 Results
3.1 Subjects
A total of 20 subjects were enrolled and received treatment, and 19 (95.0%) completed the study. Subjects were predominantly male (95.0%), white (50.0%), or black/African American (35.0%), with a mean age of 31 years (range 18–43 years). Subject baseline characteristics are shown in Table 1.
One subject discontinued treatment following an AE on day 11 (see Sect. 3.5) and was included only in the day 1 assessment of probe exposure parameters without concomitant DAA treatment. Two subjects were predicted to be CYP2C19 poor metabolizers on the basis of a homozygous CYP2C19*2 genotype (rs4244285 A/A) [21] and were excluded from the analyses of omeprazole and its metabolites.
3.2 Concentration–Time Profiles of Steady-State DCV, ASV, and BCV With and Without Additional BCV 75 mg
DCV and ASV exposure parameters over the 12-h dosing interval when DCV/ASV/BCV was administered with the probe cocktail were comparable both without (day 16) or with (day 31) an additional dose of BCV 75 mg (Fig. S1 of the ESM), and similar to historic data in healthy subjects and HCV-infected patients [5]. Geometric mean parameters for DCV in subjects given DCV/ASV/BCV, with or without additional BCV, were: C max, 974–975 ng/mL; AUCτ, 6960–7144 h·ng/mL; C 12, 336–356 ng/mL. Geometric mean parameters for ASV with or without additional BCV were: C max, 473–492 ng/mL; AUCτ, 1272–1387 h·ng/mL; C 12, 13.4–15.5 ng/mL. Exposure to BCV and BMS-794712 was approximately dose proportional (Fig. S1 of the ESM), and geometric mean BCV parameters when DCV/ASV/BCV was administered with an additional 75 mg of BCV (C max, 3141 ng/mL; AUCτ, 14,670 h·ng/mL; C 12, 309 ng/mL) were comparable to those observed in clinical studies of DCV/ASV/BCV in HCV-infected patients (data not shown).
3.3 Effect of Daclatasvir/Asunaprevir/Beclabuvir on the Pharmacokinetics of Cytochrome P450 Substrate Probes
Table 2 shows summary statistics for all recorded probe exposure parameters for a single dose of the eight-drug probe cocktail administered with or without steady-state DCV/ASV/BCV or DCV/ASV/BCV + BCV 75 mg. Rapid decline of omeprazole concentrations in the terminal phase resulted in low numbers of subjects with evaluable AUCinf, T ½, and apparent total body clearance; AUC0–T was therefore used as the exposure measure for omeprazole and 5-hydroxyomeprazole in these analyses.
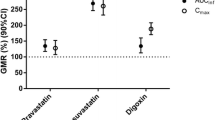
Figure 2 shows statistical comparisons (GMRs and 90% CIs) for C max and AUC for probe administration with vs. without DCV/ASV/BCV ± BCV. There was no meaningful effect of DCV/ASV/BCV on CYP1A2, CYP2C8, or CYP2C9 activity, with or without additional BCV 75 mg. All exposure parameters for caffeine, montelukast, and flurbiprofen were comparable with or without concomitant DAA administration (Table 2), and the GMRs and 90% CIs for C max and AUCinf were all contained within the standard bioequivalence range of 0.80–1.25.

Geometric mean ratios and 90% confidence intervals for single-dose, probe substrate maximum observed concentration (C max) and area under the concentration–time curve extrapolated to infinite time (AUCinf) [area under the concentration–time curve from 0 to the last quantifiable concentration (AUC0–T) for omeprazole] administered with vs. without steady-state twice-daily daclatasvir (DCV), asunaprevir (ASV), and beclabuvir (BCV). Shaded regions denote standard bioequivalence range (0.8–1.25). BID twice daily, CI confidence interval, CYP cytochrome P450, GMR geometric mean ratio, OATP organic anion-transporting polypeptide, P-gp P-glycoprotein
By contrast, weak-to-moderate inductions of CYP2C19 and CYP3A4 were observed on the basis of omeprazole and midazolam exposures, respectively, with evidence for a moderate dose-dependent effect of BCV on CYP2C19. Omeprazole C max decreased by 43% and AUC0–T by 52% with DCV/ASV/BCV, and additional BCV 75 mg further decreased C max (64% reduction) and AUC0–T (66% reduction). The poor CYP2C19 metabolizers excluded from the main omeprazole analysis (subjects S1 and S2) showed variable reductions in omeprazole AUC0–T when administered with DCV/ASV/BCV alone (S1: 17% reduction; S2: 46% reduction) vs. DCV/ASV/BCV plus BCV 75 mg (S1: 38% reduction; S2: 51% reduction). For midazolam, C max and AUCinf decreased by 43 and 47%, respectively, under co-administration with DCV/ASV/BCV. Midazolam parameter reductions were also slightly higher with additional BCV (C max reduced 52% and AUCinf reduced 58%), but to a lesser extent than for omeprazole.
Weak-to-moderate inhibition of CYP2D6, P-gp, and OATP was also observed. For CYP2D6 and P-gp, inhibition was comparable irrespective of BCV dose: metoprolol C max and AUCinf increased by 40 and 71%, respectively, with DCV/ASV/BCV, and by 30 and 60%, respectively, with DCV/ASV/BCV + BCV 75 mg. Similarly, small increases in digoxin exposures with DCV/ASV/BCV were not further increased by DCV/ASV/BCV + BCV (C max elevated by 23% with or without additional BCV, AUCinf elevated by 23% without and 17% with additional BCV). By contrast, larger increases in pravastatin exposure with DCV/ASV/BCV (C max elevated by 101%, AUCinf by 68%) were elevated slightly further by additional BCV (C max 130%, AUCinf 81%).
3.4 Effect of Daclatasvir/Asunaprevir/Beclabuvir on Cytochrome P450 Substrate Probe Metabolite Pharmacokinetics
Changes in the molecular weight-adjusted GMRs for the ratios of CYP probe metabolites to parent compound C max and AUC0–T were generally consistent with the effects of DCV/ASV/BCV ± BCV on the parent compounds (Fig. 3). A small increase (27–29%) in the ratio of C max for 36-hydroxymontelukast to montelukast was observed independent of the BCV dose, which was not reflected in AUC0–T (9% increase), consistent with the minimal effect of the DAA combination inferred for CYP2C8. Relative exposure to α-hydroxymetoprolol was reduced relative to the parent compound, without a BCV dose effect, with the α-hydroxymetoprolol/metoprolol C max and AUC0–T GMR being 40–48% lower with DCV/ASV/BCV ± BCV, consistent with inhibition of CYP2D6. In contrast, 5-hydroxomeprazole and 1′-hydroxymidazolam exposures were both elevated relative to their parent compounds, with evidence of a small BCV dose effect for 5-hydroxyomeprazole and a larger effect for 1′-hydroxymidazolam. The 1′-hydroxymidazolam/midazolam C max and AUC0–T GMR were 57–64% higher with DCV/ASV/BCV without additional BCV, and 71–83% higher with additional BCV, while 5-hydroxyomeprazole/omeprazole C max and AUC0–T were 63–68% higher without additional BCV and 113–116% higher with additional BCV.

Geometric mean ratios and 90% confidence intervals for metabolite-to-parent ratio maximum observed concentration (C max) and area under the concentration–time curve from 0 to the last quantifiable concentration (AUC0–T) for probe administration with vs. without steady-state twice-daily daclatasvir (DCV), asunaprevir (ASV), and beclabuvir (BCV). BID twice daily, CI confidence interval, CYP cytochrome P450, GMR geometric mean ratio, OATP organic anion-transporting polypeptide
The relative exposure of pravastatin lactone was reduced under DAA co-administration, with a BCV dose effect observed: C max and AUC0–T were 48–51% lower with DCV/ASV/BCV, and 66–69% lower with DCV/ASV/BCV + BCV. The data were consistent with inhibition of OATP cellular influx transporters and the associated rise in plasma pravastatin concentrations observed.
3.5 Safety
Daclatasvir/asunaprevir/beclabuvir with or without an additional 75 mg of BCV was generally well tolerated when given twice daily over a period of 30 days, with or without single doses of the eight-probe cocktail. Twelve subjects reported AEs, of which the most common were constipation, nausea, and dizziness (n = 3 each); all events were mild or moderate in intensity and resolved over time. There were no clinically significant changes in laboratory parameters, physical measurements, or vital signs. One subject discontinued from study treatment for a mild AE of hemorrhoidal hemorrhage on day 11, considered to be related to flurbiprofen; this event resolved without treatment after 3 days.
Both metoprolol and digoxin are associated with cardiac PR interval prolongation and sinus bradycardia [22, 23]. Overall, five subjects had a PR interval > 200 ms on treatment with the probe cocktail, DCV/ASV/BCV ± BCV, or both. Of these five, only one had a PR interval > 210 ms at any ECG assessment; this subject had a pre-treatment PR interval of 206 ms at screening and an interval of 201–231 ms at every on-treatment assessment and at discharge on day 36. No PR prolongation was considered clinically significant by the investigator, or reported as an AE. Eight subjects had a change from baseline in uncorrected QT interval > 30 ms, all of which occurred after cocktail administration with or without DCV/ASV/BCV ± BCV and were the result of a reduced heart rate. No subject had a corrected (QTcF) interval that was out of range or a change from baseline > 30 ms. The mean values of ECG parameters at screening, baseline, on-treatment, and at discharge were similar. Overall, 19 subjects demonstrated one or more abnormal ECG interpretations across the observation period from screening to discharge, consisting primarily of sinus bradycardia or first-degree atrioventricular block. No ECG abnormality was reported as an AE.
4 Discussion
This phase I study used a novel eight-drug metabolic cocktail to simultaneously evaluate the drug-interaction potential of a fixed-dose combination of DCV 30 mg, ASV 200 mg, and BCV 75 mg across several CYP450 and transporter pathways. Healthy volunteers received an additional 75 mg of BCV for part of the study period to account for the higher exposure observed in patients with HCV infection relative to uninfected adults. To our knowledge, this is the first use of this cocktail to evaluate drug interaction potentials in vivo. The probe compounds were generally well tolerated under simultaneous single-dose administration, both with and without steady-state DCV/ASV/BCV ± BCV, with only one discontinuation for a mild flurbiprofen-associated AE, which resolved spontaneously, and a number of minor asymptomatic ECG abnormalities consistent with the known safety profiles of metoprolol and digoxin.
No clinically meaningful effect of DCV/ASV/BCV, with or without additional BCV, was noted on exposure to sensitive substrate probes for CYP1A2 (caffeine), CYP2C8 (montelukast), or CYP2C9 (flurbiprofen), inferring a minimal impact of this combination on these metabolic pathways that is consistent with historical data for ASV when administered alone with probes for CYP1A2 (caffeine) or CYP2C9 (losartan) [6].
Weak-to-moderate induction of CYP3A4 was noted using US Food and Drug Administration definitions [24], with reductions in midazolam exposure parameters and corresponding increases in 1′-hydroxymidazolam. For patients receiving 150 mg of BCV (i.e., those receiving DCV/ASV/BCV with the additional BCV tablet), reductions in midazolam C max and AUCinf (52–58%) were comparable to earlier data from the phase I study AI443-006, assessing steady-state twice-daily BCV 150 mg on midazolam exposure in healthy subjects (C max reduced 34%, AUCinf reduced 50%) [25]. Historical data have shown a minimal effect of DCV alone on midazolam [4] and only a very minor effect of ASV (midazolam AUC reduced by ~ 20%) [6]. The current study therefore suggests that neither DCV nor ASV has any meaningful additive or synergistic effect on BCV-associated induction of CYP3A4 in this combination.
Moderate induction of CYP2C19 by DCV/ASV/BCV was also observed, with a 52% reduction in omeprazole AUC0–T for administration without additional BCV, which increased to 66% for subjects receiving additional BCV whose systemic BCV exposure would have been more representative of HCV-infected patients. This level of induction is substantially higher than historic data for 200 mg of twice-daily ASV alone, where a 20% reduction in omeprazole AUCinf was observed [6]. Overall omeprazole plasma exposure includes a minor contribution by the CYP3A4 pathway via metabolism to omeprazole sulfone. However, the magnitude of the reduction in omeprazole parameters compared with those of midazolam and the corresponding increases in 5-hydroxyomeprazole, the primary CYP2C19 metabolite, suggest that DCV/ASV/BCV acts as a stronger inducer of CYP2C19 than of CYP3A4. Simultaneous induction of both CYP3A4 and CYP2C19 may potentially be due to a BCV-mediated effect on the human pregnane X receptor activity known to regulate expression of both CYP3A4 and the CYP2C family [26]. Transactivation of pregnane X receptor was originally noted as an off-target activity of several of the candidate cyclopropyl-fused indolobenzazepine compounds from which the BCV molecule is derived, although BCV itself did not show a significant transactivation potential in vitro (50% effective concentration > 50 µM) [27]. It is a feature of interest here that the degree of induction by DCV/ASV/BCV appeared to differ between CYP3A4 and CYP2C19, while neither CYP2C8 nor CYP2C9 was significantly affected.
Changes in the CYP2D6 substrate metoprolol and its primary metabolite were broadly unaffected by the BCV dose, suggesting a minimal contribution from this component of the combination. Daclatasvir does not influence CYP2D6, but previous data have shown ASV to be a moderate in-vivo inhibitor of CYP2D6 using dextromethorphan as a probe [6]. The 71% increase in metoprolol AUCinf observed with DCV/ASV/BCV in this study would be considered weak inhibition by US Food and Drug Administration guidance [24], but because metoprolol is a less sensitive substrate for CYP2D6 than dextromethorphan [28], the DCV/ASV/BCV combination is best considered a weak-to-moderate CYP2D6 inhibitor.
Both DCV and ASV are weak inhibitors of P-gp transporters [4, 6] and the current study showed only modest (~ 20%) elevations in digoxin C max and AUCinf that were comparable to observations for DCV alone or in combination with ASV [4], and independent of BCV dose. Similarly, both DCV and ASV are OATP inhibitors [4, 6], but the increased plasma pravastatin with DCV/ASV/BCV (68% AUCinf increase, 101% C max increase) was slightly further increased by additional dosing of BCV (81% AUCinf increase, 130% C max increase), indicating a contribution by BCV to the overall effect.
Recent data have suggested that active hepatic transport of montelukast and decreased montelukast clearance under rifampin-induced OATP inhibition in rat and monkey models may confound the interpretation of studies that use montelukast as a probe of CYP2C8 activity [29]. In this study, montelukast exposure and clearance parameters were not appreciably affected by co-administration with DCV/ASV/BCV with or without additional BCV (Fig. 2 and Table 2). The lack of a BCV dose effect with montelukast, unlike with pravastatin, argues that OATP transport is not a significant influence on montelukast exposure in these subjects.
These data were obtained in non-Japanese subjects. Asunaprevir exposure has high variability across all populations but is approximately 1.6- to 2-fold higher in Japanese subjects with or without HCV infection than Caucasians for reasons that are not fully understood [6]. Exposure-safety models have not identified an increased risk of ASV hepatic AEs among Japanese patients [6]. Becalbuvir exposure is approximately 30–60% higher in Japanese than Caucasians, apparently owing largely to weight differences [25]. These racial differences are not considered clinically relevant and do not significantly impact the extrapolation of these study results to the Japanese population in which DCV/ASV/BCV is currently used.
5 Conclusion
The data indicate that for patients with HCV receiving treatment with the DCV/ASV/BCV fixed-dose co-formulation, a priori dose adjustments will not be required for concomitant agents metabolized by CYP1A2, CYP2C8, or CYP2C9, or for substrates of P-gp. The relatively modest inhibition of CYP2D6 and induction of CYP3A4 observed are unlikely to result in clinically relevant changes in exposure for most substrate drugs. Of note, the Japanese product information for DCV/ASV/BCV [30] contraindicates its use with a number of moderate/strong CYP3A4 inhibitors or inducers, including most antiretroviral agents, owing to their potential effects on DCV/ASV/BCV exposure rather than for concerns that the combination may act as a perpetrator of CYP3A4 interactions. However, in the absence of direct empirical data for individual agents, caution should be taken when co-administering DCV/ASV/BCV and CYP3A4- or CYP2D6-metabolized agents with narrow therapeutic or safety windows. Similar caution should be exercised with administering DCV/ASV/BCV with OATP transporter substrates with narrow therapeutic indices, as inhibition of influx transport could result in reduced drug concentrations in target tissues. Finally, the relatively high level of CYP2C19 induction observed would likely result in clinically significant reductions in systemic exposure to some sensitive substrates for this enzyme; co-administration of DCV/ASV/BCV with drugs primarily metabolized by CYP2C19 should be considered inadvisable in the absence of dose-adjustment data for specific agents.
References
Thrift AP, El-Serag HB, Kanwal F. Global epidemiology and burden of HCV infection and HCV-related disease. Nat Rev Gastroenterol Hepatol. 2017;14:122–32.
Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O’Boyle DR 2nd, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465:96–100.
McPhee F, Sheaffer AK, Friborg J, Hernandez D, Falk P, Zhai G, Levine S, Chaniewski S, Yu F, Barry D, Chen C, Lee MS, Mosure K, Sun LQ, Sinz M, Meanwell NA, Colonno RJ, Knipe J, Scola P. Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS-650032). Antimicrob Agents Chemother. 2012;56:5387–96.
Garimella T, You X, Wang R, Huang SP, Kandoussi H, Bifano M, Bertz R, Eley T. A review of daclatasvir drug–drug interactions. Adv Ther. 2016;33:1867–84.
Eley T, Sevinsky H, Huang SP, He B, Zhu K, Kandoussi H, Gardiner D, Grasela DM, Bertz R, Bifano M. The pharmacokinetics of daclatasvir and asunaprevir administered in combination in studies in healthy subjects and patients infected with hepatitis c virus. Clin Drug Investig. 2014;34:661–71.
Eley T, Garimella T, Li W, Bertz RJ. Asunaprevir: a review of preclinical and clinical pharmacokinetics and drug-drug interactions. Clin Pharmacokinet. 2015;54:1205–22.
Wang X, Li W, Huang S, He B, Chung E, Griffies A, Cooney E, Hughes EA, Kandoussi H, Sims K, Gardiner D, Bertz RJ, Elet T. Evaluation of pharmacokinetic drug-drug interaction (DDI) between BMS-791325, an NS5B non-nucleoside polymerase inhibitor, daclatasvir and asunaprevir in triple combination in HCV genotype 1-infected patients [abstract]. J Hepatol. 2013;58(Suppl. 1):S184.
Gentile I, Zappulo E, Buonomo AR, Maraolo AE, Borgia G. Beclabuvir for the treatment of hepatitis C. Expert Opin Investig Drugs. 2015;24:1111–21.
Lemm JA, Liu M, Gentles RG, Ding M, Voss S, Pelosi LA, Wang YK, Rigat KL, Mosure KW, Bender JA, Knipe JO, Colonno R, Meanwell NA, Kadow JF, Santone KS, Roberts SB, Gao M. Preclinical characterization of BMS-791325, an allosteric inhibitor of hepatitis C virus NS5B polymerase. Antimicrob Agents Chemother. 2014;58:3485–95.
Muir AJ, Poordad F, Lalezari J, Everson G, Dore GJ, Herring R, Sheikh A, Kwo P, Hezode C, Pockros PJ, Tran A, Yozviak J, Reau N, Ramji A, Stuart K, Thompson AJ, Vierling J, Freilich B, Cooper J, Ghesquiere W, Yang R, McPhee F, Hughes EA, Swenson ES, Yin PD. Daclatasvir in combination with asunaprevir and beclabuvir for hepatitis C virus genotype 1 infection with compensated cirrhosis. JAMA. 2015;313:1736–44.
Poordad F, Sievert W, Mollison L, Bennett M, Tse E, Brau N, Levin J, Sepe T, Lee SS, Angus P, Conway B, Pol S, Boyer N, Bronowicki JP, Jacobson I, Muir AJ, Reddy KR, Tam E, Ortiz-Lasanta G, de Ledinghen V, Sulkowski M, Boparai N, McPhee F, Hughes E, Swenson ES, Yin PD, UNITY-1 Study Group. Fixed-dose combination therapy with daclatasvir, asunaprevir, and beclabuvir for noncirrhotic patients with HCV genotype 1 infection. JAMA. 2015;313:1728–35.
Kao JH, Yu ML, Peng CY, Heo J, Chu CJ, Chang TT, Lee YJ, Hu TH, Yoon KT, Paik SW, Lim YS, Ahn SH, Isakov V, McPhee F, Hu W, Swenson ES, Yin PD, Treitel M. Daclatasvir/asunaprevir/beclabuvir, all-oral, fixed-dose combination for patients with chronic hepatitis C virus genotype 1. J Gastroenterol Hepatol. 2017;32(12):1998–2005.
Toyota J, Karino Y, Suzuki F, Ikeda F, Ido A, Tanaka K, Takaguchi K, Naganuma A, Tomita E, Chayama K, Fujiyama S, Inada Y, Yoshiji H, Watanabe H, Ishikawa H, Hu W, McPhee F, Linaberry M, Yin PD, Swenson ES, Kumada H. Daclatasvir/asunaprevir/beclabuvir fixed-dose combination in Japanese patients with HCV genotype 1 infection. J Gastroenterol. 2017;52:385–95.
Platt L, Easterbrook P, Gower E, McDonald B, Sabin K, McGowan C, Yanny I, Razavi H, Vickerman P. Prevalence and burden of HCV co-infection in people living with HIV: a global systematic review and meta-analysis. Lancet Infect Dis. 2016;16:797–808.
Cacoub P, Dabis F, Costagliola D, Almeida K, Lert F, Piroth L, Semaille C. Burden of HIV and hepatitis C co-infection: the changing epidemiology of hepatitis C in HIV-infected patients in France. Liver Int. 2015;35:65–70.
Smith BD, Morgan RL, Beckett GA, Falck-Ytter Y, Holtzman D, Teo CG, Jewett A, Baack B, Rein DB, Patel N, Alter M, Yartel A. Ward JW; Centers for Disease Control and Prevention. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep. 2012;61:1–32.
Donzelli M, Derungs A, Serratore MG, Noppen C, Nezic L, Krahenbuhl S, Haschke M. The basel cocktail for simultaneous phenotyping of human cytochrome P450 isoforms in plasma, saliva and dried blood spots. Clin Pharmacokinet. 2014;53:271–82.
Zgheib NK, Frye RF, Tracy TS, Romkes M, Branch RA. Validation of incorporating flurbiprofen into the Pittsburgh cocktail. Clin Pharmacol Ther. 2006;80:257–63.
Tye CK, Wang Z, Dockens RC, Vakkalagadda B, Wang C, Zhang Y, Su CC, Hageman MJ. Pre-absorption physicochemical compatibility assessment of 8-drug metabolic cocktail. Int J Pharm. 2016;514:364–73.
Jiang H, Kandoussi H, Zeng J, Wang J, Demers R, Eley T, He B, Burrell R, Easter J, Kadiyala P, Pursley J, Cojocaru L, Baker C, Ryan J, Aubry AF, Arnold ME. Multiplexed LC-MS/MS method for the simultaneous quantitation of three novel hepatitis C antivirals, daclatasvir, asunaprevir, and beclabuvir in human plasma. J Pharm Biomed Anal. 2015;107:409–18.
de Morais SM, Wilkinson GR, Blaisdell J, Nakamura K, Meyer UA, Goldstein JA. The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J Biol Chem. 1994;269:15419–22.
Novartis Pharmaceuticals Corporation. Lopressor® [US prescribing information]. East Hanover: Novartis Pharmaceuticals Corporation; 2012.
Concordia Pharmaceuticals Inc. Lanoxin® [US prescribing information]. St Michael: Concordia Pharmaceuticals Inc.; 2015.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for industry: drug interaction studies: study design, data analysis, implications for dosing, and labelling recommendations. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf. Accessed Feb 2017.
AbuTarif M, He B, Ding Y, Sims K, Zhu K, Rege B, Pursley J, Wind-Rotolo M, Li W, Bertz R. The effect of steady-state BMS-791325, a non-nucleoside HCV NS5B polymerase inhibitor, on the pharmacokinetics of midazolam, in healthy Japanese and Caucasian males [abstract]. Rev Antiviral Ther Infect Dis. 2014;2014(4):48–9. http://regist2.virology-education.com/abstractbook/2014_4.pdf. Accessed Oct 2017.
Hasegawa M, Tahara H, Inoue R, Kakuni M, Tateno C, Ushiki J. Investigation of drug-drug interactions caused by human pregnane X receptor-mediated induction of CYP3A4 and CYP2C subfamilies in chimeric mice with a humanized liver. Drug Metab Dispos. 2012;40:474–80.
Gentles RG, Ding M, Bender JA, Bergstrom CP, Grant-Young K, Hewawasam P, Hudyma T, Martin S, Nickel A, Regueiro-Ren A, Tu Y, Yang Z, Yeung KS, Zheng X, Chao S, Sun JH, Beno BR, Camac DM, Chang CH, Gao M, Morin PE, Sheriff S, Tredup J, Wan J, Witmer MR, Xie D, Hanumegowda U, Knipe J, Mosure K, Santone KS, Parker DD, Zhuo X, Lemm J, Liu M, Pelosi L, Rigat K, Voss S, Wang Y, Wang YK, Colonno RJ, Gao M, Roberts SB, Gao Q, Ng A, Meanwell NA, Kadow JF. Discovery and preclinical characterization of the cyclopropylindolobenzazepine BMS-791325, a potent allosteric inhibitor of the hepatitis C virus NS5B polymerase. J Med Chem. 2014;57:1855–79.
VandenBrink BM, Foti RS, Rock DA, Wienkers LC, Wahlstrom JL. Prediction of CYP2D6 drug interactions from in vitro data: evidence for substrate-dependent inhibition. Drug Metab Dispos. 2012;40:47–53.
Varma MV, Kimoto E, Scialis R, Bi Y, Lin J, Eng H, Kalgutkar AS, El-Kattan AF, Rodrigues AD, Tremaine LM. Transporter-mediated hepatic uptake plays an important role in the pharmacokinetics and drug-drug interactions of montelukast. Clin Pharmacol Ther. 2017;101:406–15.
Bristol-Myers Squibb KK. Ximency combination tablets (daclatasvir hydrochloride, asunaprevir, beclabuvir hydrochloride) [Japanese product information]. Tokyo: Bristol-Myers Squibb KK; 2016.
Acknowledgements
Assistance with the drafting and editing of the manuscript and figures was provided by Nick Fitch, PhD, CMPP, of ArticulateScience LLB (London, UK), with funding from Bristol-Myers Squibb.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was undertaken and funded by Bristol-Myers Squibb.
Conflict of interest
All authors are, or were at the time of the work described, employees of Bristol-Myers Squibb and may be stockholders thereof.
Ethics approval
The study was approved by the institutional review board/independent ethics committee at the study site and conducted in accordance with Good Clinical Practice and the ethical guidelines of the Declaration of Helsinki.
Informed consent
All subjects provided written informed consent.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Garimella, T., Tao, X., Sims, K. et al. Effects of a Fixed-Dose Co-Formulation of Daclatasvir, Asunaprevir, and Beclabuvir on the Pharmacokinetics of a Cocktail of Cytochrome P450 and Drug Transporter Substrates in Healthy Subjects. Drugs R D 18, 55–65 (2018). https://doi.org/10.1007/s40268-017-0222-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-017-0222-8