
Abstract
Osteoarthritis (OA) is a chronic degenerative joint disease characterized by the progressive loss of articular cartilage, remodeling of the subchondral bone, and synovial inflammation. Mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase that controls critical cellular processes such as growth, proliferation, and protein synthesis. Recent studies suggest that mTOR plays a vital role in cartilage growth and development and in altering the articular cartilage homeostasis as well as contributing to the process of cartilage degeneration associated with OA. Both pharmacological inhibition and genetic deletion of mTOR have been shown to reduce the severity of OA in preclinical mouse models. In this review article, we discuss the roles of mTOR in cartilage development, in maintaining articular cartilage homeostasis, and its potential as an OA therapeutic target.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Aging is known to be one of the major risk factors precipitating the onset of osteoarthritis (OA) [1–3]. Although aging and OA are correlative, OA is not a predictable consequence of aging, since other factors such as obesity, joint injury, and genetics may come into play [1]. Among the early signs of OA is alteration in the extracellular matrix (ECM) composition within the articular cartilage. This elicits a chondrocytic, synthetic, and proliferative response that functions to maintain or restore the articular cartilage. With advancing age, there is a decline in the chondrocytic anabolic response, ultimately favouring matrix degradation [2–4]. The joint tissue is therefore unable to bear normal load or maintain homeostasis when stressed, which leads to matrix destruction and disease progression.
Destruction of the ECM of articular cartilage is a major indicator of OA [5]. With advanced age, chondrocytes exhibit reduced responsiveness to growth factors, abnormal accumulation of advanced glycation end products (AGE), mitochondrial dysfunction, and oxidative stress. As a result, cartilage homeostasis is disrupted and the ECM becomes more vulnerable to damage, leading to the onset of OA [6]. Chondrocytes are the one cell type present in the articular cartilage and are exclusively responsible for matrix turnover and maintenance [7]. They function to maintain cartilage homeostasis by maintaining a condition where the normal cartilage ECM composition deals with mechanical stress without structural or cellular damage [5].
With OA progression, excessive catabolic activity is largely mediated by proinflammatory cytokines and mediators such as matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) [6]. The breakdown of cartilage during OA pathogenesis is also a result of chondrocyte death as evidenced by the presence of apoptotic and non-apoptotic mechanisms in OA cartilage [8]. This excessive catabolic activity and loss of viable chondrocytes causes imbalance of cartilage homeostasis and cartilage matrix breakdown. There is also minimal proliferative activity in osteoarthritic chondrocytes, in contrast to essentially no proliferative activity in normal articular chondrocytes. In response to injury during OA progression, chondrocytes proliferate and form clusters, a characteristic feature of OA cartilage, in an attempt to repair cartilage lesions. In spite of attempts to salvage the cartilage degeneration, cartilage homeostasis cannot be maintained, as the biosynthetic anabolic activity becomes unable to compete with the degradative catabolic activity [9, 10].
Studies suggest that cellular processes such as autophagy and senescence could play a key role in determining the fate of chondrocytes within the articular cartilage. Mammalian cells maintain homeostatic functions such as the degradation and recycling of organelles via autophagy [11]. Autophagy is the process of cell degradation in a stressful or nutrient-deprived environment, consisting of formation of an isolation membrane, or autophagosome, around the contents to be degraded, which combines with a lysosome to form an autolysosome. This lysosomal degradation pathway is essential for homeostasis as well as survival, differentiation, and development [12]. Autophagy is particularly important to remove aggregate-prone or misfolded proteins and dysfunctional organelles in post-mitotic tissue, such as the articular cartilage, where a very low rate of cell turnover is experienced [13]. The molecular components that control the autophagy process, the Atg genes, were first identified in yeast. Among the Atg genes, the four major regulators of the autophagy pathway are Atg1, Atg6, Atg8 (ULK1, Beclin1, and LC3 in mammals, respectively) and Atg5 [14]. ULK1 is a serine/threonine kinase that functions as an intermediate in the transduction of proautophagic signals to autophagosome formation [15]. Beclin1 forms a complex with type II phosphatidylinositol 3-kinase (PI3K) and Vps34 allowing nucleation of the autophagic vesicle [16]. LC3 is present in two forms: LC3-I is located in the cytoplasm, while LC3-II is bound to the autophagosome membrane. During autophagy, LC3-I undergoes lipidation to be converted to LC3-II, resulting in the association of LC3-II with autophagy vesicles [17].
The reduced efficacy of articular cartilage repair that accompanies ECM degradation has also been proposed to be a result of chondrocyte phenotypic senescence [18]. As opposed to ‘replicative senescence’, which refers to the loss of the ability of mitotic cells to further divide in culture after a period of 30–40 population doublings (‘Hayflick limit’), phenotypic senescence develops well in advance of complete arrest of the cell cycle [1, 19]. Deterioration of chondrocyte function with age is characterized by a decline in their mitotic and synthetic activity, resulting in the synthesis of smaller proteoglycan aggregates. This is due to the shortening of aggrecan molecules, their chondroitin sulfate chains, and the reduction in the mean number of aggrecans in each aggregate. These age-related incidents may be a result of ECM proteoglycan degradation and/or alterations in proteoglycan synthesis [20]. Using three markers of cell senescence (beta-galactosidase expression, mitotic activity, and telomere length), studies conducted by Martin and Buckwalter [18] showed that beta-galactosidase expression increased with age, whereas mitotic activity (indicated by 3H-thymidine incorporation) and mean telomere length declined with age. Two mechanisms have been proposed to explain how the cells become senescent with age [18]. First, telomere erosion occurs as a result of incomplete replication of the telomeres during mitosis (end replication problem) such that a portion of the end of the telomere is lost with each cell division. As telomeres shorten, cells ultimately reach their replicative limit and progress into senescence [21]. While authors such as Loeser [1] argue that telomere erosion due to repeated cycles of cell division does not explain senescence in quiescent cells such as chondrocytes, Martin and Buckwalter [18] claim that even a slow rate of cell division would be sufficient to cause telomere erosion over many decades. Additionally, following cartilage injury, chondrocyte mitotic activity increases several fold; hence, repetitive joint stress could accelerate telomere erosion and eventual joint degeneration [22]. Stress-induced senescence due to oxidative damage is the second proposed mechanism of how cells approach senescence [23]. One causative factor for oxidative damage is the age-related degeneration of mitochondria, which protect cells from the toxic effects of free radicals and reactive oxygen species (ROS) as well as providing metabolic energy to support the homeostatic activities that chondrocytes require for tissue maintenance [18, 24]. Mitochondrial degeneration and loss is suggested to be associated with chondrocyte senescence. The altered activity and expression of regulatory proteins that control cell growth and proliferation in senescent cells can also result in the increased production of cytokines (e.g. interleukin [IL]-6 and IL-1), growth factors (e.g. epidermal growth factor), and MMPs. This ‘senescent secretory phenotype’ can also contribute to tissue aging through damage to the ECM [1]. While senescent cells undergo permanent cell cycle arrest, growth-promoting pathways are not inhibited [25]. The enlarged and flattened cell morphology is a distinct marker of cell senescence, a consequence of cell growth without cell divisions (i.e. cellular hypertrophy) [25, 26].
2 Mammalian Target of Rapamycin (mTOR)
The target of rapamycin (TOR) is a nutrient-sensing signaling cascade that is responsible for the regulation of growth and metabolism of cells, depending on the nutritional environment. The discovery of the TOR pathway was preceded by the discovery of the drug rapamycin. It was discovered on the island of Rapa Nui in the 1970s from the bacteria Streptomyces hygroscopicus and initially studied as an anti-fungal agent [27]. TOR is a 282 kDa protein composed of an evolutionarily conserved atypical serine-threonine kinase, part of the phosphatidylinositol kinase-related kinase (PIKK) family, also similar to the PI3K and 4-kinases (PI4Ks). It was originally discovered in Saccharomyces cerevisiae, where the immunosuppressant FK506, structurally similar to rapamycin, was found to bind to FK506-binding protein (FKBP12), a peptidyl-prolyl isomerase (PPIase), which is highly conserved across species and a very abundant protein in cells [28–31].
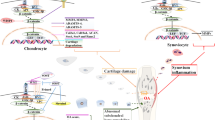
The TOR pathway can be inhibited by TOR inhibitors, which bind to FKBP12. This complex then targets and binds to TOR and inhibits it. Mutations in TOR prevent the binding of the FKBP12-rapamycin complex to it, depicting the role of TOR in the pathway [31]. The mammalian counterpart in mammals, mTOR (or FRAP, RAFT1 [32]), now known as the mechanistic target of rapamycin, is inhibited by the FKBP12–rapamycin complex [33]. mTOR comprises of two complexes that are similar in structure but not in function [34]: while the mTOR complex 1 (mTORC1) consists of mTOR, mLST8, and raptor; the mTOR complex 2 (mTORC2) consists of mTOR, mLST8, and rictor (not raptor). FKBP12–rapamycin complex binds to the raptor component of TORC1, not the rictor component of TORC2 in vivo, concluding that TORC1 is rapamycin sensitive while TORC2 is insensitive [35, 36]. mTORC1 is important for the regulation and promotion of cell growth, protein synthesis upstream (initiation, translation, elongation), ribosome biogenesis, cell cycle regulation via the S/G1 phase, inhibition of autophagy, accelerated aging and nutrient uptake and metabolism (Fig. 1). On the other hand, mTORC2 controls endocytosis, sphingolipid biosynthesis (ROS association), cell survival, and polarization of the cytoskeleton [37–40].

Some of the critical physiological and pathological roles of mammalian target of rapamycin. mTOR mammalian target of rapamycin, OA osteoarthritis
mTORC1 is a major negative regulator of autophagy (discussed in detail later) and is a downstream target of the PI3K and protein kinase B (Akt) pathway [41–43]. The PI3K/Akt/mTOR signaling cascade is activated by transmembrane tyrosine kinase growth factor receptors such as insulin-like growth factor (IGF)-1 [44]. When activated, it promotes cell growth, differentiation, survival, and downregulates apoptotic signals [45, 46]. AKT stimulates mTOR activity by phosphorylating the tuberous sclerosis complex (TSC2) and suppressing its repressive action [47]. TSC2 phosphorylation consequently promotes the activation of the Ras homolog enriched in brain (Rheb) and the activation of mTOR [48]. Activation of mTOR leads to the phosphorylation of p70 ribosomal S6 kinase (S6K) and eukaryotic initiation factor 4E-binding protein (4EBP), which are related to translational machinery, ribosomal biogenesis, and cell growth [49]. On the other hand, inhibition of mTOR by rapamycin induces the initiation of autophagy and the formation of autophagosomes [50]. mTORC1 inhibition by rapamycin also increases AKT phosphorylation by blocking the phosphorylation of S6K and insulin receptor substrate (IRS)-1, which are key factors of a negative feedback inhibition for PI3K/Akt signal transduction, consequently inducing survival and anti-apoptotic pathways [51, 52].
3 mTOR in Cartilage Growth and Development
Several studies suggest that mTOR plays a vital role in cartilage growth and development. Chen and Long [53] showed that disruption of mTORC1 signaling through deletion of either mTOR or raptor retards embryonic skeletal growth associated with severe delays in chondrocyte hypertrophy and bone formation [53]. This study also showed that growth reduction of cartilage is not due to changes in chondrocyte proliferation or survival, but is caused by a reduction in cell size and in the amount of cartilage matrix. It has also been shown that mechanical activation of mTOR is necessary for cell proliferation, chondrogenesis, and cartilage growth during bone development [54]. Rokutanda et al. [55] showed that the Akt–mTOR pathway positively and dominantly regulates chondrocyte proliferation, maturation and cartilage matrix production during skeletal development. Studies also show that mTOR signaling contributes to chondrocyte differentiation [56].
Rapamycin-treated animals have been shown to exhibit retarded growth and marked alterations in the growth plate associated with disruption of angiogenesis in the growth plate [57]. Direct infusion of rapamycin in growth plates by an osmotic minipump resulted in smaller hypertrophic and proliferative zones, decrease in the size of the growth plate, and inhibition of overall long bone growth in rabbits [58]. In young rats, treatment with rapamycin significantly decreased endochondral bone growth [59]. Treatment with rapamycin has also been shown to inhibit chondrogenesis of mesenchymal cells at the post-precartilage condensation stage [60]. It may seem like mTORC1 is essential for normal cartilage and skeletal development; however, recent reports suggest that mTORC2 also plays a key role in skeletal development. Chen et al. [61] showed that deletion of rictor resulted in shorter and narrower skeletal elements in both embryos and postnatal mice. Specifically, this study showed that deletion of rictor reduced the width but not the length of the initial cartilage anlage associated with a delay in chondrocyte hypertrophy, with no change in proliferation, apoptosis, cell size, or matrix production. Further, rictor-deficient mice exhibit impaired bone formation, resulting in thinner cortical bone postnatally. These studies strongly suggest that mTOR is a key regulator of chondrogenesis and cartilage development.
4 mTOR in Articular Cartilage Homeostasis
In recent years, a variety of studies have demonstrated a key role of mTOR signalling in chondrocyte metabolism, articular cartilage homeostasis, and OA pathophysiology [62]. Tchetina et al. [63] recently reported an upregulation in mTOR gene expression in the peripheral blood mononuclear cells (PBMCs) of OA patients and in the articular cartilage of all end-stage OA patients. They also showed that an increase in the expression of mTOR in PBMCs of OA patients related to disease activity, being associated with synovitis. Interestingly, this study also reported a “low mTOR expression subset” with significantly lower mTOR gene expression in PBMCs compared with healthy controls that correlated with pain upon walking, standing and increased total joint stiffness compared with the “High mTOR” subset.
It has also been shown that upregulation in the expression of mTOR in OA cartilage is accompanied with an increased rate of chondrocyte apoptosis [64]. Increase in mTOR expression and an increased rate of chondrocyte loss is not restricted to human OA cartilage, as mouse and dog cartilage subjected to OA surgical models also exhibit a significant increase in mTOR expression associated with enhanced articular chondrocyte loss [64].
Recent studies have shown that dysregulated autophagy could contribute to the pathophysiology of OA [4, 64–68]. Carames et al. [4] demonstrated that the expression of key autophagy markers ULK1 (most upstream autophagy inducer), Beclin1 (autophagy regulator), and LC3 (autophagosome formation) were significantly decreased in human aging, OA cartilage, chondrocytes, and aging-related and surgically induced OA mouse models. Furthermore, an increase in apoptotic cell death was observed via the increased expression of the apoptosis marker, poly ADP ribose polymerase (PARP) p85, in human aging and OA cartilage and chondrocytes as well as aging-related and surgically induced OA mouse models [4]. Data from autophagy polymerase chain reaction (PCR) array analysis also shows aberrant expression of several autophagy genes in human OA cartilage compared with normal human cartilage associated with increased expression of mTOR during OA [64].
Both pharmacological inhibition of mTOR as well as cartilage-specific deletion of mTOR have been shown to decrease the severity of experimental OA in mouse models [64, 69–71]. Inhibition of mTOR signaling by rapamycin and subsequent increase in autophagy was shown to be associated with a significant reduction in the severity of cartilage degeneration and synovitis associated with decrease in the expression of ADAMTS-5 and IL-1β in articular cartilage [71]. Further, cartilage-specific mTOR knock-out (KO) mice subjected to destabilisation of the medial meniscus in the OA model showed significant protection from surgery-induced OA, associated with increased autophagy and decreased articular chondrocyte cell death, elucidating a potential role for mTOR inhibition to restore homeostasis during OA [64]. Sasaki et al. [65] also showed that inhibition of autophagy caused OA-like gene expression changes, while the induction of autophagy by rapamycin reduced MMP13 and ADAMTS5 expression induced by IL-1β, a major pro-inflammatory cytokine implicated in OA [72]. Another study showed that glucocorticosteroids used in the short-term increase autophagy in human chondrocytes, but cause apoptosis in the long term. When rapamycin was given before treatment, cell viability and autophagy increased compared with dexamethasone-only cells [73].
It has also been shown that mTOR can be regulated via enhanced synthesis of endogenous n-3 polyunsaturated fatty acids (PUFAs) from n-6 PUFAs, thus promoting autophagy and chondrocyte cell survival and delaying the incidence of OA [74]. Genetic deletion of Von Hippel–Lindau (Vhl), a tumor-suppressor gene, in the articular cartilage results in increased phosphorylation of mTOR and increased MMP-13 expression associated with earlier dysregulation of cartilage homeostasis, increased chondrocyte apoptosis, compromised autophagy, and accelerated age-related and surgery-induced OA development [75]. Phosphoinositide-specific phospholipase γ1 (PLCγ1), a key regulator of cell metabolism, has been shown to regulate the ECM synthesis of human chondrocytes through triggering of the mTOR/P70S6K/S6 pathway [76]. In vitro and in vivo studies using glucosamine have also been shown to enhance autophagy associated with the inhibition of Akt/mTOR pathway, thus shedding light on possible disease-modifying or homeostatic properties exhibited by glucosamine [77].
mTOR modulation of autophagy in the articular cartilage seems to be partly regulated through the ULK1/AMPK pathway. ULK1 is the most upstream autophagy inducer. mTORC1 has been shown to associate with the ULK1-Atg13-FIP200 complex, whereby it phosphorylates both ULK1 and Atg13, which represses ULK1 kinase activity [3, 78, 79]. Therefore, when mTOR is active in conditions such as the presence of abundant amino acids, autophagy is repressed as a consequence of ULK1 inhibition by mTORC1-mediated phosphorylation [80]. Indeed, studies performed in cartilage-specific mTOR KO mice and human patient OA cells treated with rapamycin showed that the ability of mTOR to modulate ULK1/AMPK autophagy pathway may be partly responsible for the imbalance between catabolic and anabolic processes and decreased chondroprotection and ultimately cartilage destruction observed during OA.
Since mTOR is a complex mediator, it may affect other cellular processes independently of the autophagy pathway in articular cartilage. For example, mTOR plays a role in growth-promoting pathways and remains active during senescence [81]. The significance of active growth-promoting pathways in cellular senescence was shown by Demidenko et al. [82]. p21 induction caused cell-cycle arrest in HT1080 human fibrosarcoma cells, as well as a senescent morphology (beta-galactosidase staining, cell hypertrophy, increased levels of cyclin D1, active mTOR) in the presence of serum growth factors. However, the addition of rapamycin decelerated cellular senescence and partially reduced beta-galactosidase staining. Still, suppression of the permanent loss of proliferative potential by rapamycin is only achievable if the cell cycle inhibitors are removed [82]. Hence, while rapamycin does not reverse cell cycle arrest, it does suppress inappropriate growth stimulation via mTOR. These findings have yet to be examined in the context of chondrocyte senescence.
Since mTOR is active in senescent cells, and mTOR is also a major repressor of autophagy, one would assume autophagy to be inhibited in senescent cells. Interestingly, Narita and colleagues [83, 84] observed that autophagy can be activated in senescent cells. Their research uncovered a unique cellular compartment, the TOR-autophagy spatial coupling compartment (TASCC), which forms during Ras-induced senescence. The authors discovered a large accumulation of mTORC1-studded lysosomes together with autophagosomes in a well-defined perinuclear location. According to their proposed model, autophagosome-derived amino acids reinforce mTOR enrichment and activity, leading to the synthesis of senescence-associated secretory proteins. Blocking the localization of mTORC1 to the TASCC resulted in a strong reduction in the synthesis of two key SASP components, IL-6 and IL-8. Similarly, blocking autophagy (i.e. cutting off amino acid supply) leads to a complete loss of mTORC1 from the TASCC [83, 85]. Thus, there is increasing evidence that the progressive loss of cell function with age contributes to age-related changes in articular cartilage structure and function and increases the risk of the articular cartilage degeneration associated with OA. Furthermore, mTOR has been shown to have significant roles in both autophagy and senescence, as well as its recently discovered role facilitating the mass synthesis of secretory proteins [83].
5 mTOR as a Potential Therapeutic Target in Osteoarthritis
Recent pre-clinical studies have shown a decrease in the severity of OA in animal models subjected to pharmacological inhibition and genetic deletion of mTOR. These results depict mTOR as a potential therapeutic target in OA. Specifically, the ability of mTOR to regulate chondrocyte survival, cell death, and senescence is promising. Furthermore, the effect of mTOR modulation on lifespan has been examined in yeast, C. elegans, Drosophila and most importantly, mice [86–88]. Harrison et al. [89] reported a significant increase in lifespan in male and female mice following rapamycin treatment at 600 days of age. It would be interesting to track how this treatment results in the aging of the joints in these mice and whether treatment with rapamycin protects articular cartilage from degeneration and maintains articular cartilage integrity.
Genetic or pharmacological manipulation of mTOR signalling is time sensitive, since mTOR is essential for the early stages of cartilage growth and development. Premature modulation of the mTOR-signaling pathway has dire consequences on skeletal growth in vivo. In recent years, inhibition of the mTOR pathway has received substantial attention as an anti-cancer approach, leading to the clinical development of rapamycin analogs (rapalogs) (Table 1) [90]. The mTOR pathway has also been shown to be involved in the development of vascular lesions associated with antiphospholipid syndrome, an autoimmune disease [91]. Clinical studies in patients with antiphospholipid syndrome nephropathy who required transplantation showed that treatment with sirolimus resulted in no recurrence of vascular lesions and decreased vascular proliferation on biopsy as compared with patients with antiphospholipid antibodies who were not receiving sirolimus. In rheumatic diseases, rapamycin has been shown to reduce the disease activity in patients with systemic lupus erythematosus [92]. In a case study involving a single patient with scleroderma, treatment with rapamycin decreased skin tightness and increased the range of motion of fingers and arms [93]. The beneficial effects of rapamycin have also been reported in a patient with dermatomyositis [94]. In animal models of systemic sclerosis, treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models [95] .
Despite these advances, the disease-modifying effects of rapamycin and its rapalogs have not been tested in OA clinical studies. As previously discussed, mTOR has a negative feedback inhibition on the activity of the PI3K/Akt pathway, and the inhibition of mTOR results in increased activity of the PI3K/Akt/nuclear factor (NF)-κB pathway, which may increase MMP secretion by chondrocytes. Chen et al. [96] propose that a dual inhibition of PI3K and mTOR may be a potential therapeutic approach in OA (Fig. 2). Besides this prospect of dual inhibition, targeting autophagy genes to upregulate autophagy, impart chondroprotection, and promote articular cartilage homeostasis can also be tested.

The PI3K/Akt/mTOR signaling cascade regulating autophagy. Upon activation of PI3K, Akt is phosphorylated, which promotes the activation of mTORC1. Activated mTORC1 inhibits autophagy by associating with the ULK1-Atg13-FIP200 complex, thus inhibiting autophagosome formation. Inhibition of mTOR and the subsequent increase in autophagic activity may restore homeostasis in articular cartilage chondrocytes. Further, by simultaneously targeting the PI3K/Akt/NF-κB pathway, a dual inhibition of PI3K and mTOR may be a potential approach to limit cartilage degeneration. Alternatively, upregulation of ULK1 may also impart chondroprotection by enhancement of autophagic activity. Red lines signify potential points of therapeutic intervention. Akt protein kinase B, mTOR mammalian target of rapamycin, NF nuclear factor, PI3K phosphatidylinositol 3-kinase
The side effects observed with mTOR inhibitors may be a limitation of their use. However, this may be resolved by preventive and management measures during the course of treatment with combination therapy. In addition, intra-articular delivery of mTOR inhibitors in affected OA joints could reduce the risk of systemic side effects. As further validation of rapamycin or other mTOR inhibitors is conducted in pre-clinical studies, it will be essential to dissect the mTOR-dependent, mTOR-independent, and other off-target effects of mTOR inhibitors. Overall, the outcomes of preclinical studies of mTOR modulation in OA, as well as clinical studies conducted using mTOR inhibitors for the treatment of cancer and other diseases give credibility to the use of mTOR inhibitors as a potential and promising therapeutic target for OA patients.
References
Loeser RF. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthritis Cartilage. 2009;17(8):971–9.
Grogan SP, D’Lima DD. Joint aging and chondrocyte cell death. Int J Clin Rheumtol. 2010;5(2):199–214.
Lotz MK, Carames B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat Rev Rheumatol. 2011;7(10):579–87.
Carames B, et al. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62(3):791–801.
Lotz M, Loeser RF. Effects of aging on articular cartilage homeostasis. Bone. 2012;51(2):241–8.
Li Y, Wei X, Zhou J, et al. The age-related changes in cartilage and osteoarthritis. Biomed Res Int. 2013;2013:916530.
García-Carvajal ZY, Garciadiego-Cazares D, Parra-Cid C, et al. Cartilage tissue engineering: the role of extracellular matrix (ECM) and novel strategies. In: Andrades JA, editor. Regenerative medicine and tissue engineering. Croatia: InTech; 2013. pp. 365–97.
Almonte-Becerril M, Navarro-Garcia F, Gonzalez-Robles A, et al. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis. 2010;15(5):631–8.
Lotz MK, Otsuki S, Grogan SP, et al. Cartilage cell clusters. Arthritis Rheum. 2010;62(8):2206–18.
Sandell LJ, Aigner T. Articular cartilage and changes in arthritis. An introduction: cell biology of osteoarthritis. Arthritis Res. 2001;3(2):107–13.
Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell. 2008;15(3):344–57.
Mizushima, N. Physiological Functions of Autophagy. In: Levine B, Yoshimori T, Deretic V, editors. Autophagy in infection and immunity. Berlin: Springer; 2009. pp. 71–84.
Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–96.
Chan EY, Tooze SA. Evolution of Atg1 function and regulation. Autophagy. 2009;5(6):758–65.
Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282(35):25464–74.
Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, AUTOPHAGY, and tumor suppressor function. Autophagy. 2005;1(1):46–52.
Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–8.
Martin JA, Buckwalter JA. The role of chondrocyte senescence in the pathogenesis of osteoarthritis and in limiting cartilage repair. J Bone Joint Surg Am. 2003; 85-A (Suppl 2):106–10.
Hayflick L. Intracellular determinants of cell aging. Mech Ageing Dev. 1984;28(2–3):177–85.
Martin JA, Buckwalter JA. Telomere erosion and senescence in human articular cartilage chondrocytes. J Gerontol A Biol Sci Med Sci. 2001;56(4):B172–9.
Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–40.
Buckwalter JA, Mankin HJ. Articular cartilage: degeneration and osteoarthritis, repair, regeneration, and transplantation. Instr Course Lect. 1998;47:487–504.
Toussaint O, Medrano EE, von Zglinicki T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp Gerontol. 2000;35(8):927–45.
Golden TR, Melov S. Mitochondrial DNA mutations, oxidative stress, and aging. Mech Ageing Dev. 2001;122(14):1577–89.
Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7(21):3355–61.
Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192(4):547–56.
Vézina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo). 1975;28(10):721–6.
Siekierka JJ, Wiederrecht G, Greulich H, Boulton D, Hung SH, Cryan J, Hodges PJ, Sigal NH. The cytosolic-binding protein for the immunosuppressant FK-506 is both a ubiquitous and highly conserved peptidyl-prolyl cis-trans isomerase. J Biol Chem. 1990;265(34):21011–5.
Wiederrecht G, Hung S, Chan HK, et al. Characterization of high molecular weight FK-506 binding activities reveals a novel FK-506-binding protein as well as a protein complex. J Biol Chem. 1992;267(30):21753–60.
Heitman J, Movva NR, Hiestand PC, Hall MN. FK 506-binding protein proline rotamase is a target for the immunosuppressive agent FK 506 in saccharomyces cerevisiae. Proc Natl Acad Sci. 1991;88:1948–52.
Lorenz MC, Heitman J. TOR mutations confer rapamycin resistance by preventing interaction with FKBP12-rapamycin. J Biol Chem. 1995;270(46):27531–7.
Chiu MI, Katz H, Berlin V. RAPT1, a mammalian homolog of yeast TOR, interacts with the FKBP12/rapamycin complex. Proc Natl Acad Sci. 1994;91:12574–8.
Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, Abraham RT. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270(2):815–22.
Helliwell SB, Wagner P, Kunz J, et al. TOR1 and TOR2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Mol Biol Cell. 1994;5(1):105–18.
Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, Have distinct roles in cell growth control. Mol Cell. 2002;10:457–68.
Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–302.
Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–45.
Dobashi Y, Watanabe Y, Miwa C, et al. Mammalian target of rapamycin: a central node of complex signaling cascades. Int J Clin Exp Pathol. 2011;4(5):476–95.
Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–84.
Cutler NS, Pan X, Heitman J, et al. The TOR signal transduction cascade controls cellular differentiation in response to nutrients. Mol Biol Cell. 2001;12(12):4103–13.
Cuyàs E, Coromnas-Faja, Joven J, et al. Cell cycle regulation by the nutrient-sensing mammalian target of rapamycin (mTOR) pathway. In: Noguchi E, Gadaleta MC, editors. Cell cycle control: mechanisms and protocols. Springer; New York; 2014. pp. 113–144.
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–74.
Heras-Sandoval D, Perez-Rojas JM, Hernandez-Damian J, et al. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014;26(12):2694–701.
Dudek H, Datta SR, Franke TJ, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275(5300):661–5.
Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K–Akt signaling pathway. Curr Opin Neurobiol. 2001;11(3):297–305.
Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT—a major therapeutic target. Biochimica et Biophysica Acta. 2004;1697(1–2):3–16.
Li Y, Inoki K, Yeung R, Guan KL. Regulation of TSC2 by 14-3-3 binding. J Biol Chem. 2002;277(47):44593–6.
Long X, Lin Y, Ortiz-Vega S, et al. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15(8):702–13.
Corradetti MN, Guan KL. Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene. 2006;25(48):6347–60.
Nazio F, Strappazzon F, Antonioli M, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15(4):406–16.
Sato T, Nakashima A, Guo L, et al. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene. 2010;29(18):2746–52.
Harrington LS, Findlay GM, Gray A, et al. The TSC1-2 tumor suppressor controls insulin–PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166(2):213–23.
Chen J, Long F. mTORC1 signaling controls mammalian skeletal growth through stimulation of protein synthesis. Development. 2014;141(14):2848–54.
Guan Y, Yang X, Yang W, et al. Mechanical activation of mammalian target of rapamycin pathway is required for cartilage development. FASEB J. 2014;28(10):4470–81.
Rokutanda S, et al. Akt regulates skeletal development through GSK3, mTOR, and FoxOs. Develop Biol. 2009;328:78–93. doi:10.1016/j.ydbio.2009.01.009.
Phornphutkul C, Wu KY, Augeung V, et al. mTOR signaling contributes to chondrocyte differentiation. Dev Dyn. 2008;237(3):702–12.
Alvarez-Garcia O, Garcia-Lopez E, Loredo V, et al. Rapamycin induces growth retardation by disrupting angiogenesis in the growth plate. Kidney Int. 2010;78(6):561–8.
Phornphutkul C, Lee M, Voigt C, et al. The effect of rapamycin on bone growth in rabbits. J Orthop Res. 2009;27(9):1157–61.
Sanchez CP, He YZ. Bone growth during rapamycin therapy in young rats. BMC Pediatr. 2009;9:3.
Oh CD, Kim SJ, Ju JW, et al. Immunosuppressant rapamycin inhibits protein kinase C alpha and p38 mitogen-activated protein kinase leading to the inhibition of chondrogenesis. Eur J Pharmacol. 2001;427(3):175–85.
Chen J, Holguin N, Shi Y, et al. mTORC2 signaling promotes skeletal growth and bone formation in mice. J Bone Miner Res. Epub 4 Sep 2014.
Tchetina EV. The importance of the mTOR regulatory network in chondrocyte biology and osteoarthritis. EMJ Rheumatol. 2014;1:84–95.
Tchetina EV, Poole AR, Zaitseva EM, et al. Differences in mammalian target of rapamycin gene expression in the peripheral blood and articular cartilages of osteoarthritic patients and disease activity. Arthritis. 2013;2013:461486.
Zhang Y, Vasheghani F, Li YH, et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann Rheum Dis. 2014. doi:10.1136/annrheumdis-2013-204599.
Sasaki H, Takayama K, Matsushita T, et al. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2012;64(6):1920–8.
Carames B, Taniguchi N, Seino D, et al. Mechanical injury suppresses autophagy regulators and pharmacologic activation of autophagy results in chondroprotection. Arthritis Rheum. 2012;64(4):1182–92.
Wu C, Zheng J, Yao X, et al. Defective autophagy in chondrocytes with Kashin-Beck disease but higher than osteoarthritis. Osteoarthritis Cartilage. 2014;22(11):1936–46.
Chang J, Wang W, Zhang H, et al. The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis. Int J Mol Med. 2013;32(6):1311–8.
Matsuzaki T, Matsushita T, Tabata Y, et al. Intra-articular administration of gelatin hydrogels incorporating rapamycin-micelles reduces the development of experimental osteoarthritis in a murine model. Biomaterials. 2014;35(37):9904–11.
Takayama K, Kawakami Y, Kobayashi M, et al. Local intra-articular injection of rapamycin delays articular cartilage degeneration in a murine model of osteoarthritis. Arthritis Res Ther. 2014;16(6):482.
Carames B, Hasegawa A, Taniguchi N, et al. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis. 2012;71(4):575–81.
Kapoor M, et al. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7(1):33–42.
Liu N, et al. Autophagy in human articular chondrocytes is cytoprotective following glucocorticoid stimulation. Mol Med Rep. 2014;9(6):2166–72.
Huang MJ, et al. Enhancement of the synthesis of n-3 PUFAs in fat-1 transgenic mice inhibits mTORC1 signalling and delays surgically induced osteoarthritis in comparison with wild-type mice. Ann Rheum Dis. 2014;73(9):1719–27.
Weng T, et al. Loss of Vhl in cartilage accelerated the progression of age-associated and surgically induced murine osteoarthritis. Osteoarthritis Cartilage. 2014;22(8):1197–205.
Zeng G, et al. Disruption of phosphoinositide-specific phospholipases Cgamma1 contributes to extracellular matrix synthesis of human osteoarthritis chondrocytes. Int J Mol Sci. 2014;15(8):13236–46.
Carames B, et al. Glucosamine activates autophagy in vitro and in vivo. Arthritis Rheum. 2013;65(7):1843–52.
Hosokawa N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–91.
Lee JW, et al. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5(11):e15394.
Dunlop EA, et al. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy. 2011;7(7):737–47.
Blagosklonny MV. Hypoxia, MTOR and autophagy: converging on senescence or quiescence. Autophagy. 2013;9(2):260–2.
Demidenko ZN, Zubova SG, Bukreeva EI, et al. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8(12):1888–95.
Narita M, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332(6032):966–70.
Young AR, Narita M, Narita M. Spatio-temporal association between mTOR and autophagy during cellular senescence. Autophagy. 2011;7(11):1387–8.
Zoncu R, Sabatini DM. Cell biology. The TASCC of secretion. Science. 2011;332(6032):923–5.
Powers RW 3rd, et al. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20(2):174–84.
Vellai T, et al. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426(6967):620.
Kapahi P, et al. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14(10):885–90.
Harrison DE, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–5.
Seto B. Rapamycin and mTOR: a serendipitous discovery and implications for breast cancer. Clin Transl Med. 2012;1(1):29.
Canaud G, et al. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N Engl J Med. 2014;371(4):303–12.
Fernandez D, et al. Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54(9):2983–8.
Fried L, et al. Efficacy of rapamycin in scleroderma: a case study. Lymphat Res Biol. 2008;6(3–4):217–9.
Nadiminti U, Arbiser JL. Rapamycin (sirolimus) as a steroid-sparing agent in dermatomyositis. J Am Acad Dermatol. 2005;52(2, Supplement):S17–9.
Yoshizaki A, et al. Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis Rheum. 2010;62(8):2476–87.
Chen J, Crawford R, Xiao Y. Vertical inhibition of the PI3K/Akt/mTOR pathway for the treatment of osteoarthritis. J Cell Biochem. 2013;114(2):245–9.
US National Institutes of Health. A pilot study evaluating the use of mTor inhibitor sirolimus in children and young adults with desmoid-type fibromatosis. 2010. http://clinicaltrials.gov/ct2/show/NCT01265030?term=mtor&rank=8. Accessed 20 Dec 2014.
US National Institutes of Health. Gemcitabine plus rapamycin versus gemcitabine to treat advanced soft tissue sarcoma. 2010. http://clinicaltrials.gov/ct2/show/NCT01684449?term=mTOR&rank=61. Accessed 20 Dec 2014.
US National Institutes of Health. Pulsed Oral Sirolimus in Autosomal Dominant Polycystic Kidney Disease (RAP). 2014. http://clinicaltrials.gov/ct2/show/NCT02055079?term=mTOR&rank=64. Accessed 20 Dec 2014.
US National Institutes of Health. Controlled level EVERolimus in acute coronary syndromes (CLEVER-ACS). 2012. https://clinicaltrials.gov/ct2/show/NCT01529554. Accessed 20 Dec 2014.
US National Institutes of Health. Prospective study of rapamycin for the treatment of SLE (Rapamune). 2008. https://clinicaltrials.gov/ct2/show/NCT00779194. Accessed 20 Dec 2014.
US National Institutes of Health. A phase II study of EVEROLIMUS in patients with primary or relapsed chondrosarcomas (CHONRAD). 2013. http://clinicaltrials.gov/ct2/show/NCT02008019?term=rapamycin&rank=68. Accessed 20 Dec 2014
US National Institutes of Health. Everolimus and lenalidomide in treating patients with relapsed or refractory non-Hodgkin or Hodgkin lymphoma. 2010. https://clinicaltrials.gov/ct2/show/NCT01075321. Accessed 20 Dec 2014.
US National Institutes of Health. Daily everolimus in combination with trastuzumab and vinorelbine in HER2/Neu positive women with locally advanced or metastatic breast cancer (BOLERO-3). 2009. http://clinicaltrials.gov/ct2/show/NCT01007942?term=NCT01007942&rank=1. Accessed 20 Dec 2014.
US National Institutes of Health. Neoadjuvant rapamycin in patients undergoing radical cystectomy. 2013. https://clinicaltrials.gov/ct2/show/NCT01827618. Accessed 20 Dec 2014.
Hudes G, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356(22):2271–81.
Rangwala R, et al. Combined MTOR and Autophagy Inhibition: PHASE I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10(8):9–20.
Yee AJ, et al. Outcomes in patients with relapsed or refractory multiple myeloma in a phase I study of everolimus in combination with lenalidomide. Br J Haematol. 2014;166(3):401–9.
Bagatell R, et al. Phase 1 trial of temsirolimus in combination with irinotecan and temozolomide in children, adolescents and young adults with relapsed or refractory solid tumors: a Children’s Oncology Group Study. Pediatr Blood Cancer. 2014;61(5):833–9.
Acknowledgments
This work is supported by Dr. Mohit Kapoor’s Canadian Institutes of Health Research Operating Grant MOP: 201003.
Conflict of interest
The authors declare no conflict of interest with respect to this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Bandna Pal and Helal Endisha share equal first author contribution.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Pal, B., Endisha, H., Zhang, Y. et al. mTOR: A Potential Therapeutic Target in Osteoarthritis?. Drugs R D 15, 27–36 (2015). https://doi.org/10.1007/s40268-015-0082-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-015-0082-z