
Abstract
Efgartigimod (efgartigimod alfa-fcab, Vyvgart™) is a first-in-class neonatal Fc receptor antagonist being developed by argenx for the treatment of autoimmune diseases including myasthenia gravis. In December 2021, intravenous efgartigimod received its first approval in the USA for the treatment of generalized myasthenia gravis in adults who are anti-acetylcholine receptor (AChR) antibody positive. Intravenous efgartigimod has also been evaluated for generalized myasthenia gravis in various other countries, with the agent subsequently approved in Japan in January 2022 for generalized myasthenia gravis patients regardless of antibody status and in preregistration stage in the EU. Several clinical studies of intravenous and subcutaneous formulation of efgartigimod are also being investigated for other autoimmune diseases including bullous pemphigoid, chronic inflammatory demyelinating polyradiculoneuropathy, immune thrombocytopenia, autoimmune myositis and pemphigus. This article summarizes the milestones in the development of efgartigimod leading to this first approval for generalized myasthenia gravis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Digital Features for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.19067744. |
A neonatal Fc receptor antagonist is being developed by argenx for the treatment of autoimmune diseases including myasthenia gravis. |
Received its first approval on 17 December 2021 in the USA. |
Approved for use in the treatment of generalized myasthenia gravis in adults who are anti-AChR antibody positive. |
1 Introduction
Myasthenia gravis is a chronic autoimmune neuromuscular disorder that causes localized or general voluntary muscle weakness [1]. The pathogenesis of myasthenia gravis includes the binding of immunoglobulin G (IgG) antibodies to postsynaptic acetylcholine receptors (AChRs) or other components at the neuromuscular junction, resulting in impaired neuromuscular transmission by inhibiting acetylcholine-dependent signaling and inducing accelerated internalization and degradation of AChRs [1]. Recently, targeted immunotherapies have emerged as promising therapeutic approaches for myasthenia gravis that may overcome some limitations (e.g. inadequate symptom relief and undesirable adverse events) associated with traditional therapeutic approaches, such as corticosteroids and nonsteroidal immunosuppressive therapies (NSISTs) [2].
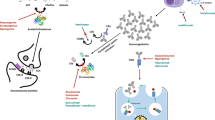
The neonatal Fc receptor plays a key role in prolonging the life-span of IgG as it protects them from lysosomal degradation by recycling them back into the circulation [3]. Targeting the neonatal Fc receptor may offer a novel therapeutic opportunity for myasthenia gravis where inhibition of this receptor causes IgG catabolism, leading to reduced overall IgG and pathological autoantibody levels [3]. Efgartigimod (efgartigimod alfa-fcab, Vyvgart™) is a first-in-class neonatal Fc receptor antagonist being developed by argenx for the treatment of myasthenia gravis and other autoimmune diseases.

Key milestones in the development of intravenous efgartigimod for generalized myasthenia gravis. BLA Biologics License Application, MAA Market Authorisation Application, PAA pre-approval access
Intravenous efgartigimod received its first approval in 17 December 2021 in the USA for the treatment of generalized myasthenia gravis in adults who are anti-AChR antibody positive [4]. On 20 January 2022, efgartigimod was subsequently approved in Japan for the treatment of generalized myasthenia gravis in adults who do not have sufficient response to steroids or NSISTs [5]. The recommended dosage of efgartigimod is 10 mg/kg (or 1200 mg for patients weighing ≥ 120 kg) administered as a 1 h intravenous infusion once weekly for 4 weeks as one treatment cycle; the solution must be diluted with 0.9% sodium chloride injection to a total of 125 mL prior to administration [6]. Patients should be monitored for signs and symptoms of hypersensitivity reactions during infusion and for 1 h thereafter. Subsequent cycles are administered based on clinical evaluation; the safety of administering efgartigimod sooner than 50 days after previous cycle was not studied in patients with generalized myasthenia gravis. As efgartigimod causes transient decrease in IgG levels, immunization with live or live-attenuated vaccines is not recommended during treatment [6]. Preliminary evidence with these non-live vaccines, including influenza, pneumococcal, mRNA COVID-19 vaccines, suggests that the ability to mount an immune response is not impaired by efgartigimod treatment [7].
Intravenous efgartigimod is undergoing regulatory review for the treatment of generalized myasthenia gravis in the EU. The agent is also undergoing phase III clinical development for immune thrombocytopenia worldwide. In addition to the intravenous formulation, argenx is developing a recombinant human hyaluronidase-based subcutaneous formulation of efgartigimod, using ENHANZE® technology (licensed from Halozyme Therapeutics). Several clinical studies of subcutaneous efgartigimod are underway in healthy volunteers and in patients with autoimmune diseases, including bullous pemphigoid, chronic inflammatory demyelinating polyradiculoneuropathy, immune thrombocytopenia, myasthenia gravis, autoimmune myositis and pemphigus.
1.1 Company Agreements
In July 2011, arGEN-X (now renamed to argenx) signed a non-exclusive license agreement with BioWa Inc to further increase the therapeutic potency of their developing monoclonal antibodies, combining argenx’s proprietary SIMPLE antibody™ platform with BioWa Inc’s POTELLIGENT® technology [8]. In 2012, arGEN-X was granted a license to ABDEG™ technology from the University of Texas Southwestern Medical Center [9]. In November 2013, arGEN-X raised funding from PMV, a Flemish investment company, as an extension of its series B financing round to accelerate the clinical development of efgartigimod [10].
In February 2015, Lonza and arGEN-X entered into a multi-product GS Xceed™ license agreement, which secures the latter’s long-term access to Lonza’s proprietary GS Xceed™ system for the development and manufacture of both current and future therapeutic antibodies [11].
In February 2019, argenx entered into a global collaboration and license agreement with Halozyme Therapeutics to gain access to the ENHANZE® subcutaneous delivery technology to develop multiple subcutaneous therapeutics, including products that target the human neonatal Fc receptor (e.g. efgartigimod) and up to two additional targets [12]. The agreement was expanded in October 2020 to include up to six additional targets [13]. Halozyme is entitled to upfront, target nomination, future milestone and royalty payments [12].
In November 2020, argenx entered into a purchase agreement with Bayer Healthcare Pharmaceuticals Inc to acquire an US FDA priority review voucher from the latter [14]. Argenx expects to redeem the priority review voucher for a future marketing application for efgartigimod to reduce the target review time [14].
In January 2021, argenx entered into an exclusive license agreement with Zai Lab under which the latter acquired the exclusive rights for the development and commercialisation of efgartigimod in Greater China, including mainland China, Hong Kong, Taiwan and Macau [15]. Under the terms of agreement, argenx will receive an upfront payment, guaranteed non-creditable, non-refundable development cost-sharing payment, as well as an additional milestone payment if efgartigimod is approved in the USA. Argenx is also eligible to receive tiered royalties based on sales levels of efgartigimod in Greater China.
In March 2021, argenx entered into a preclinical research collaboration with Hansa Biopharma AB to evaluate the therapeutic potential of combining efgartigimod and imlifidase [16]. Under the terms of agreement, both companies will contribute equally in terms of resource allocation and will share all intellectual property and data developed through the collaboration, while maintaining exclusive rights to their respective technologies and products [16].
In April 2021, argenx entered into a global license and collaboration agreement with Elektrofi to gain access to Elektrofi’s high-concentration, low-volume delivery technology for the development of subcutaneous formulations of the human neonatal Fc receptor directed antibodies, including efgartigimod, and up to one additional target [17]. Under the terms of agreement, argenx will make an upfront payment, as well as future milestone payments and sales-based royalties to Elektrofi [17].
In October 2021, argenx signed an exclusive partnership agreement with Medison to commercialize efgartigimod for generalized myasthenia gravis in Israel [18]. Medison is responsible to seek requisite regulatory approvals for efgartigimod in Israel [18].
2 Scientific Summary
2.1 Pharmacodynamics
Efgartigimod is a humanized IgG1 Fc fragment that has been engineered by argenx’s proprietary ABDEG™ technology, of which ABDEG mutations increase its affinity for Fc receptor at neutral and acidic pH [19]. Efgartigimod binds to the neonatal Fc receptor and inhibits its interaction with IgG, thereby reducing IgG recycling and increasing degradation of IgG and pathological autoantibodies, without altering other immunoglobulins and albumin levels. Intravenous efgartigimod rapidly reduced serum IgG levels in cynomolgus monkeys [19] and myasthenic mice [20]. In a mouse model for muscle-specific kinase myasthenia gravis, muscle weakness and fatigability were also improved in efgartigimod-treated mice relative to control mice [20].
Features and properties of efgartigimod
Alternative names | ARGX-113; Efgartigimod–argenx; Efgartigimod alfa-fcab–argenx; efgartigimod PH20; efgartigimod PH20 SC; Vyvgart |
Class | Anti-inflammatories; immunoglobulin Fc fragments; monoclonal antibodies |
Mechanism of action | Neonatal Fc receptor antagonist |
Route of administration | Intravenous; subcutaneous |
Pharmacodynamics | A humanized IgG1 Fc fragment that binds to the neonatal Fc receptor and blocks its interaction with IgG, thereby reducing IgG levels |
Pharmacokinetics | Intravenous formulation displays linear pharmacokinetics and drug exposure increases proportionally after doses up to 50 mg/kg, volume of distribution 15–20 L and terminal half-life 80–120 h; SC formulation has comparable half-life to that of IV formulation with a bioavailability of ≈ 50% |
Most frequent adverse events | Headache, upper respiratory tract infection, urinary tract infection |
ATC codes | |
WHO ATC code | L04A-A58 (efgartigimod alfa) |
EphMRA ATC code | L4 (Immunosuppressants) |
Chemical name | Immunoglobulin G1, anti-(fcrn receptor) (human monoclonal ARGX-113 fc fragment) |
Key clinical trials of efgartigimod (argenx)
Drug(s) | Indication | Phase | Status | Location(s) | Identifier |
|---|---|---|---|---|---|
Efgartigimod (IV), placebo | Myasthenia gravis in adults | III | Completed | Multinational | NCT03669588, EudraCT2018-002132-25, JapicCTI195083, ARGX113-1704; ADAPT |
Efgartigimod (IV) | Myasthenia gravis in adults | III | Ongoing | Multinational | NCT03770403, EudraCT2018-002133-37, JapicCTI205144, ARGX113-1705, ADATPT+ |
Efgartigimod (IV) and (SC) | Myasthenia gravis in adults | III | Ongoing | Multinational | NCT04735432l, EudtaCT2020-004085-19, ARGX-113-2001, ADAPTsc |
Efgartigimod (IV) | Myasthenia gravis in adults | II/III | Recruiting | Multinational | NCT04980495, EudraCT2021-002504-12, ARGX113-2003, ADAPT NXT |
Efgartigimod (IV) | Myasthenia gravis in children | II/III | Recruiting | Multinational | NCT04833894, EudraCT2020-005841-18, ARGX113-2006, P097/2020, ADAPT JR |
Efgartigimod (IV), placebo | Myasthenia gravis in adults | II | Completed | Multinational | NCT02965573, EudraCT2016-002938-73, ARGX113-1602 |
Efgartigimod (SC), placebo | Pemphigus in adults | III | Recruiting | Multinational | NCT04598451, EudraCT2020-002915-23, ARGX113-1904, ADDRESS |
Efgartigimod (SC), placebo | Pemphigus in adults | III | Recruiting | Multinational | NCT04598477, EudraCT2020-002917-16, ARGX113-1905, ADDRESS+ |
Efgartigimod (IV) | Pemphigus in adults | II | Completed | Multinational | NCT03334058, EudraCT2017-002333-40, ARGX113-1701 |
Efgartigimod (IV), placebo | ITP in adults | III | Ongoing | Multinational | NCT04188379, EudraCT2019-002100-41, JapicCTI205112, ARGX113-1801, ADVANCE |
Efgartigimod (IV) | ITP in adults | III | Recruiting | Multinational | NCT04225156, EudraCT2019-002101-21, JapicCTI205113, ARGX113-1803, ADVANCE+ |
Efgartigimod (SC), placebo | ITP in adults | III | Recruiting | Multinational | NCT04687072, EudraCT2020-004032-21, ARGX113-2004,ADVANCE SC |
Efgartigimod (SC) | ITP in adults | III | Recruiting | Multinational | NCT04812925, EudraCT2020-004033-20, ARGX113-2005, ADVANCE SC+ |
Efgartigimod (IV), placebo | ITP in adults | II | Completed | Multinational | NCT03102593, EudraCT2016-003038-26, ARGX113-1603 |
Efgartigimod (SC), placebo | CIDP in adults | II | Recruiting | Multinational | NCT04281472, EudraCT2020-002915-23, ARGX113-1904, ADHERE |
Efgartigimod (SC) | CIDP in adults | II | Recruiting | Multinational | NCT04280718, EudraCT2019-003107-35, ARGX113-1902, ADHERE+ |
In healthy volunteers, single and multiple ascending doses of intravenous efgartigimod rapidly reduced total IgG levels by up to 50% and 85%, respectively, from baseline [19]. Efgartigimod did not alter serum levels of other immunoglobulins (e.g. IgA or IgM) or albumin and the total IgG levels returned to baseline ≈ 8 weeks after the last intravenous administration [19]. Similarly, intravenous efgartigimod rapidly decreased the total IgG levels from baseline in patients with myasthenia gravis and other autoimmune diseases, with AChR autoantibody levels following a similar pattern to that seen with total IgG levels. For instance, in patients with generalized myasthenia gravis and who are AChR antibody positive, there were mean maximum reductions from baseline of 61.3% and 57.6% in total IgG and AChR antibody levels, respectively, 1 week after the last intravenous infusion of efgartigimod in the first treatment cycle [21]. The total IgG levels returned to baseline levels 9 weeks after the last infusion in the first treatment cycle [21]. Intravenous efgartigimod also induced rapid reduction of total IgG levels in patients with immune thrombocytopenia (up to 63.7% from baseline) [22] or pemphigus vulgaris and foliaceus (up to 74% from baseline) [23]. In patients with pemphigus vulgaris and foliaceus, IgG autoantibodies targeting desmoglein-1 and -3 levels also decreased over time, with a median reduction from baseline of 70% and 42%, respectively, at the end of the treatment-free follow-up [23]. Overall, the reductions in IgG levels were generally similar regardless of IgG subtypes [21,22,23].
In an open-label phase I study in healthy volunteers, initial intravenous administration of efgartigimod followed by weekly subcutaneous administration of efgartigimod 300 mg (2 mL) provided sufficient drug exposure to maintain IgG reduction of ≈ 50% at a steady state [24].
2.2 Pharmacokinetics
Efgartigimod exhibits linear pharmacokinetics and efgartigimod exposure increases proportionally after doses up to 50 mg/kg (5 times the recommended dosage) [6]. The volume of distribution is 15–20 L and the terminal half-life is 80–120 h. Following a single intravenous dose of efgartigimod 10 mg/kg in healthy volunteers, < 0.1% of the dose was recovered in urine [6]. Of note, in a phase I open-label clinical trial in healthy volunteers, after a single dose of subcutaneous efgartigimod , the drug had a comparable half-life to that of intravenous efgartigimod, with a bioavailability of ≈ 50% [24].
According to a population pharmacokinetic analysis, efgartigimod exposures do not appear to be impacted to any clinically significant extent by patient age, sex and race [6]. No dedicated pharmacokinetic studies have been performed in patients with kidney or hepatic impairment; however, a population pharmacokinetic analysis based on clinical study data indicated that patients with mild kidney impairment had a 22% increase in efgartigimod exposure compared with those with normal kidney function [6].
Given that efgartigimod is not metabolized by CYP450 enzymes, drug-drug interactions between efgartigimod and drugs that are substrates, inducers or inhibitors of CYP450 enzymes are unlikely [6]. Efgartigimod may decrease systemic exposure to drugs that bind to the human neonatal Fc receptor, such as immunoglobulin products, monoclonal antibodies or antibody derivatives containing the human Fc domain of the IgG subclass; patients should be closely monitored for reduced effectiveness. If long-term concomitant use of such drugs is essential, consider discontinuing efgartigimod and using alternative treatments [6].
2.3 Therapeutic Trials
2.3.1 Generalized Myasthenia Gravis
Efgartigimod rapidly improved generalized myasthenia gravis symptoms and daily living activities in a 26-week multinational, randomized, double-blind, placebo-controlled phase III ADAPT trial (NCT03669588) [21]. Patients with generalized myasthenia gravis who had a Myasthenia Gravis Activities of Daily Living (MG-ADL) score of ≥ 5 (> 50% non-ocular) and were receiving stable doses of ≥ 1 myasthenia gravis treatment (e.g. acetylcholinesterase inhibitors, corticosteroids and/or NSISTs) were randomized to receive intravenous efgartigimod 10 mg/kg (n = 84) or placebo (n = 83) administered as weekly infusions over four weeks per cycle. After the first cycle, patients could receive subsequent cycles based on individual clinical evaluation, with the cycle not commencing for ≥ 8 weeks from initiation of the previous cycle. During the study, patients were allowed to receive non-live vaccines if administered ≥ 48 h prior to, or after efgartigimod infusion. Patients who participated the study were eligible to roll over to an open-label extension ADAPT+ trial (NCT03770403; Sect. 2.5) [21].
Patients treated with efgartigimod received an average of two treatment cycles, with the mean time from the first infusion in the first treatment cycle to the second treatment cycle being 94 days [6]. In the first treatment cycle, the MG-ADL responder rate in AChR antibody positive patients was significantly (p < 0.0001) higher with efgartigimod than with placebo [68% vs 30%; odds ratio (OR) 4.95, 95% CI 2.21–11.53; primary endpoint]. MG-ADL responder was defined as a patient who had a ≥ 2-point reduction in MG-ADL score for ≥ 4 consecutive weeks, with the first improvement occurring no later than 1 week after the last infusion of the cycle. The Quantitative Myasthenia Gravis (QMG) responder rate (i.e. a ≥ 3-point reduction in the total QMG score for ≥ 4 consecutive weeks, with the first improvement occurring no later than 1 week after the last infusion of the cycle) was also significantly (p < 0.0001) higher with efgartigimod than with placebo (63% vs 14%; OR 10.84, 95% CI 4.18–31.20). Furthermore, improvements in Myasthenia Gravis Composite (MGC) score and MG–Specific Quality of Life 15-item scale (MG-QOL15r) questionnaire were more favourable with efgartigimod than with placebo. Overall, efgartigimod recipients achieved up to 9-point reductions in MG-ADL score and 10-point reductions in QMG score, with a significantly higher proportion of efgartigimod than placebo recipients attaining a MG-ADL score of 0 or 1 (minimal symptom expression) in the first treatment cycle (40% vs 11%; p < 0.0001) [21]. The beneficial effects of efgartigimod over placebo were reproducible after a second and third treatment cycle and were consistently seen regardless of gender, age, affected muscle domain (bulbar, ocular, respiratory and limb/gross motor) or concomitant medications (NSISTs, steroids and/or acetylcholinesterase inhibitors) [18, 21, 25].
In a randomized, double-blind, placebo-controlled proof-of-concept phase II trial in patients with generalized myasthenia gravis and anti-AChR antibody positivity who were on stable standard-of-care therapy (NCT02965573; n = 24), a significantly greater proportion of patients achieved a clinically meaningful improvement in MG-ADL scores (i.e. a ≥ 2-point reduction from baseline) with efgartigimod (four weekly doses of 10 mg/kg intravenous infusions over a 3-week period) compared with placebo (75% vs 25%; p = 0.0391), with the improvement persisting for ≥ 6 consecutive weeks [26]. Rapid and strong clinical improvements associated with efgartigimod relative to placebo were also seen when assessed by QMG score, MGC score and MG-QOL15r questionnaire. For all efficacy and quality of life scales, initial improvements were seen as early as 7 days after the first infusion [26]. Further analyses revealed that 50% of 12 efgartigimod-treated patients maintained clinically meaningful improvement of MG-ADL score 8 weeks after the last infusion of efgartigimod [27].
2.3.2 Other Autoimmune Diseases
In patients with immune thrombocytopenia inadequately controlled with standard-of-care therapy, clinically meaningful platelet responses (i.e. platelet counts of ≥ 50 × 109/L for ≥ 2 visits) were achieved in 46% of efgartigimod recipients (four weekly 5 mg/kg or 10 mg/kg intravenous infusions over 3 weeks) versus 25% of placebo recipients during a randomized, double-blind, placebo-controlled proof-of-concept phase II trial (NCT03102593; n = 38) [22]. A significantly higher proportion of efgartigimod than placebo recipients maintained clinically meaningful platelet responses for ≥ 10 cumulative days (38% vs 0%; p = 0.03). The beneficial effects of efgartigimod was sustained among patients who entered the open-label extension period and received additional cycle of four weekly intravenous infusions of efgartigimod 10 mg/kg [22].
In patients with mild-to-moderate pemphigus vulgaris or foliaceus, most (90%) patients achieved early disease control (i.e. no new lesions and established lesions starting to heal) with intravenous efartigimod after a median of 17 days of treatment during a multicenter, open-label feasibility phase II trial (NCT03334058; n = 34) [23]. Furthermore, an optimized, prolonged treatment with efartigimod in combination with prednisone led to complete clinical remission (i.e. no new lesions and established lesions completely healed) in 64% of patients within 2–41 weeks. Efgartigimod was administered as a 10 mg/kg intravenous infusion once weekly for four weeks (induction phase), which was followed by various maintenance dosing regimens, including one efgartigimod intravenous infusion at weeks 2 and 6, biweekly for 8 weeks or biweekly for 12 weeks. During the induction phase, some patients received intravenous efgartigimod 25 mg/kg once weekly until achieving end of consolidation (i.e. no new lesions for ≥ 2 weeks and ≈ 80% of lesions healed), after which they received one efgartigimod intravenous infusion biweekly for up to 34 weeks. Efgartigimod was administered as monotherapy or as add-on therapy to low-dose oral prednisone [23].
2.4 Adverse Events
Intravenous efgartigimod was generally well tolerated in patients with generalized myasthenia gravis during the ADAPT study [21]. The most common adverse events occurring in ≥ 10% of efgartigimod recipients (n = 84) and at a higher incidence than in placebo recipients (n = 83) included headache (29% vs 28%), upper respiratory tract infection (11% vs 5%) and urinary tract infection (10% vs 5%). Most of reported infections were mild to moderate in severity. Serious adverse events and treatment discontinuation due to adverse events occurred in 5% and 4% of efgartigimod recipients versus 8% and 4% of placebo recipients, respectively. There were no clinically relevant changes in laboratory parameters (including albumin levels which are also recycled by the neonatal Fc receptor via a distinct binding site), vital signs or electrocardiograms related to efgartigimod [21].
The tolerability profile of intravenous efgartigimod in patients with immune thrombocytopenia [22] or pemphigus vulgaris or foliaceus [23] was generally similar to that observed in patients with generalized myasthenia gravis.
As with all therapeutic proteins, efgartigimod has a potential for immunogenicity. In the ADAPT study with up to 26 weeks of treatment, anti-drug antibodies (ADAs) were detected in 20% of efgartigimod recipients, with 7% of patients developing neutralizing antibodies [6]. The available data are too limited to assess the impact of ADAs on pharmacokinetics, efficacy and safety of efgartigimod [6].
2.5 Ongoing Clinical Trials
An ongoing multicentre open-label extension phase III trial (NCT03770403; ADAPT+) is assessing the long-term safety and tolerability of intravenous efgartigimod in patients with generalized myasthenia gravis who had participated in the ADAPT study. Recruitment is underway in an open-label phase II/III trial to evaluate the efficacy of different dosing regimens of intravenous efgartigimod in patients with generalized myasthenia gravis (NCT04980495; ADAPT NXT). Also recruiting is an open-label, phase II/III trial that will evaluate the pharmacokinetics, pharmacodynamics, efficacy and safety of intravenous efgartigimod in children aged 2 to < 18 years with generalized myasthenia gravis (NCT04833894; ADAPT JR). In addition, a multicentre randomized, double-blind placebo-controlled, phase III trial in patients with immune thrombocytopenia is currently ongoing to evaluate the efficacy and safety of intravenous efgartigimod versus placebo (NCT04188379; ADVANCE) and a multicentre, open-label extension of ADVANCE study is currently recruiting (NCT04225156; ADVANCE+).
A randomized, open-label phase III clinical trial is ongoing to compare the pharmacodynamics, pharmacokinetics, efficacy and safety of subcutaneous efgartigimod versus intravenous efgartigimod in patients with generalized myasthenia gravis (NCT04735432; ADAPTsc). Recruitment is underway in several phase III clinical trials to compare the efficacy and safety of subcutaneous efgartigimod versus placebo in patients with immune thrombocytopenia (NCT04687072; ADVANCE SC) or pemphigus vulgaris or foliaceous (NCT04598451; ADDRESS), with each trial having a subsequent open-label extension phase III trial to assess the long-term efficacy and safety of the drug [NCT04812925 (ADVANCE SC+), NCT04598477 (ADDRESS+)]. Also recruiting is a phase II trial designed to evaluate the efficacy and safety of subcutaneous efgartigimod versus placebo in patients with chronic inflammatory demyelinating polyneuropathy (NCT04281472; ADHERE), which has a subsequent open-label extension phase II trial to assess the long-term efficacy and safety of the drug (NCT04280718; ADHERE+). Lastly, clinical study designs to evaluate subcutaneous efgartigimod use in patients with bullous pemphigoid (BALLAD) or with myositis (ALKIVIA) are being finalized [28].
3 Current Status
Intravenous efgartigimod received its first approval on 17 December 2021 for the treatment of generalized myasthenia gravis in adults who are anti-AChR antibody positive in the USA [4]. Subsequently, efgartigimod was approved in Japan on 20 January 2022 for the treatment of generalized myasthenia gravis in adults who do not have sufficient response to steroids or NSISTs [5].
Change history
30 March 2022
A Correction to this paper has been published: https://doi.org/10.1007/s40265-022-01712-4
References
Lunemann JD. Getting specific: targeting Fc receptors in myasthenia gravis. Nat Rev Neurol. 2021;17(10):597–8.
Dalakas MC. Immunotherapy in myasthenia gravis in the era of biologics. Nat Rev Neurol. 2019;15(2):113–24.
Dalakas MC, Spaeth PJ. The importance of FcRn in neuro-immunotherapies: from IgG catabolism, FCGRT gene polymorphisms, IVIg dosing and efficiency to specific FcRn inhibitors. Ther Adv Neurol Disord. 2021;14:1756286421997381.
argenx. FDA approves new treatment for myasthenia gravis [media release]. 17 Dec 2021. https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-myasthenia-gravis.
argenx. argenx announces VYVGART™ approval in Japan for the treatment of generalized myasthenia gravis [media release]. 20 Jan 2022. https://www.argenx.com/.
argenx. Vyvgart™ (efgartigimod alfa-fcab) injection, for intravenous use: US highlights of prescribing information. 2021. https://argenx.com/product/vyvgart-prescribing-information.pdf. Accessed 13 Jan 2022.
Guptill J, Sleasman J, Steeland S, et al. Effect of efgartigimod, a neonatal fc receptor blocker, on humoral vaccine responses in autoimmune patients [abstract]. Muscle Nerve. 2021;64(Suppl 1):S91.
arGEN-X. arGEN-X announces non-exclusive license with BioWa for POTELLIGENT® technology [media release]. 26 Jul 2011. http://www.biowa.com.
arGEN-X. arGEN-X advances ARGX-113 into preclinical development for autoimmune disorders [media release]. 24 Apr 2013. http://www.argen-x.com.
arGEN-X. arGEN-X raises EUR 5 million from PMV in extension of EUR 32.5 million series B [media release]. 4 Nov 2013. http://www.arGEN-X.com.
Lonza, arGEN-X. Lonza and arGEN-X announce a multi-product GS XceedTM license agreement for therapeutic antibodies [media release]. 18 Feb 2015. http://www.lonza.com.
argenx, Halozyme Therapeutics. argenx and Halozyme enter global collaboration and license agreement for ENHANZE® technology [media release]. 4 Feb 2019. http://www.argenx.com.
Halozyme Therapeutics, argenx. Halozyme announces expansion of collaboration and license agreement with argenx for ENHANZE® technology [media release]. 5 Oct 2020. http://www.halozyme.com.
argenx. argenx enters into agreement to acquire Priority Review Voucher [media release]. 23 Nov 2020. http://www.argenx.com.
argenx, ZAI Lab. argenx and Zai Lab announce strategic collaboration for efgartigimod in Greater China [media release]. 6 Jan 2021. http://www.argenx.com.
Hansa Biopharma AB, argenx. Hansa Biopharma enters preclinical research collaboration with argenx [media release]. 29 Mar 2021. http://www.hansabiopharma.com.
Elektrofi, argenx. Elektrofi enters global collaboration and license agreement with argenx to explore new subcutaneous product formulations [media release]. 20 Apr 2021. http://www.elektrofi.com.
argenx. argenx reports third quarter 2021 financial results and provides business update [media release]. 28 Oct 2021. http://www.argenx.com.
Ulrichts P, Guglietta A, Dreier T, et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J Clin Invest. 2018;128(10):4372–86.
Huijbers MG, Plomp JJ, van Es IE, et al. Efgartigimod improves muscle weakness in a mouse model for muscle-specific kinase myasthenia gravis. Exp Neurol. 2019;317:133–43.
Howard JF Jr, Bril V, Vu T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526–36.
Newland AC, Sanchez-Gonzalez B, Rejto L, et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am J Hematol. 2020;95(2):178–87.
Goebeler M, Bata-Csorgo Z, De Simone C, et al. Treatment of pemphigus vulgaris and foliaceus with efgartigimod, a neonatal Fc receptor inhibitor: a phase II multicentre, open-label feasibility trial. Br J Dermatol. 2021;05:05.
argenx. argenx announces data from phase 1 study of efgartigimod (ARGX-113) subcutaneous formulation demonstrating comparable characteristics to intravenous formulation [media release]. 14 Jun 2018. http://www.argenx.com.
Karam C, Vu T, Bril V, et al. Efgartigimod treatment of patients with generalized myasthenia gravis demonstrates consistent improvements across all muscle subgroups and regardless of background immunosuppressive therapy [abstract]. Muscle Nerve. 2021;64(Suppl 1):S92.
Howard JF Jr, Bril V, Burns TM, et al. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology. 2019;92(23):e2661–73.
Howard J, Bril V, Mantegazza R, et al. Additional analyses of the phase 2 efgartigimod study in myasthenia gravis [abstract no. 4484]. Neurology. 2020;94(15 Suppl):4484.
argenx. argenx highlights strategic priorities for 2022 [media release]. 7 Jan 2022. http://www.argenx.com.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Young-A Heo is a salaried employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
The original article has been revised due to retrospective open choice order.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Heo, YA. Efgartigimod: First Approval. Drugs 82, 341–348 (2022). https://doi.org/10.1007/s40265-022-01678-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-022-01678-3