
Abstract
Maralixibat (Livmarli™) is an orally-administered, small-molecule ileal bile acid transporter (IBAT) inhibitor being developed by Mirum Pharmaceuticals for the treatment of rare cholestatic liver diseases including Alagille syndrome (ALGS), progressive familial intrahepatic cholestasis (PFIC) and biliary atresia. Maralixibat received its first approval on 29 September 2021, in the USA, for use in the treatment of cholestatic pruritus in patients with ALGS 1 year of age and older. Maralixibat is also under regulatory review for ALGS in Europe, and clinical development for cholestatic liver disorders including ALGS in patients under 1 year of age, PFIC and biliary atresia is continuing in several other countries. This article summarises the milestones in the development of maralixibat leading to this first approval for ALGS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Digital Features for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.17019878 |
A small-molecule IBAT inhibitor is being developed by Mirum Pharmaceuticals for the treatment of cholestatic liver diseases |
Received its first approval on 29 September 2021 in the USA |
Approved for use in the treatment of cholestatic pruritus in patients 1 year of age and older with Alagille syndrome (ALGS) |
1 Introduction
Maralixibat (Livmarli™) is an orally-administered, small-molecule ileal bile acid transporter (IBAT) inhibitor being developed by Mirum Pharmaceuticals for the treatment of rare cholestatic liver diseases including Alagille syndrome (ALGS), progressive familial intrahepatic cholestasis (PFIC) and biliary atresia [1, 2]. After surgical interruption of enterohepatic circulation was shown to have some success in reducing pruritus in patients with ALGS, pharmacological interruption of bile acid transport was proposed as an alternative to surgical intervention to manage pruritus [3]. Due to its key role in bile acid reabsorption, IBAT was seen an ideal target for pharmacological interruption of bile acid transport [4].
Maralixibat received its first approval on 29 September 2021, from the US Food and Drug Administration (FDA), for use in the treatment of cholestatic pruritus in patients 1 year of age and older with ALGS [5]. Maralixibat is available as an oral solution and should be taken 30 min before the first meal of the day at a recommended dosage of 380 µg/kg once daily [1]. Treatment should be initiated at a dosage of 190 µg/kg once daily with an increase to 380 µg/kg once daily after 1 week, as tolerated (maximum daily dose of 28.5 mg) [1].
Maralixibat is also under regulatory review for ALGS in Europe [6], and clinical development for cholestatic liver disorders including ALGS, PFIC and biliary atresia is continuing in several other countries [2]. Maralixibat has Orphan Drug designation for the treatment of ALGS, PFIC and biliary atresia from both the US FDA and the European Medicines Agency. Clinical development of the drug for the treatment of hypercholesterolaemia, primary biliary cholangitis (PBC) and primary sclerosing cholangitis has been discontinued. A randomised phase II study in adults with PBC found that maralixibat did not significantly improve pruritus compared with placebo [7].

Key milestones in the development of maralixibat. ALGS Alagille syndrome, BA biliary atresia, CLD cholestatic liver disease, NDA New Drug Application, PBC primary biliary cholangitis, PFIC progressive familial intrahepatic cholestasis, PSC primary sclerosing cholangitis
1.1 Company Agreements
Maralixibat was originally developed by Pfizer (previously Pharmacia Corp.) for the treatment of hypercholesterolaemia before being licensed to Lumena Pharmaceuticals in June 2012 [8]. Lumena was subsequently acquired by Shire in May 2014 [9]. In November 2018, Mirum Pharmaceuticals entered into an agreement with Shire for the exclusive global rights to develop and market maralixibat [10].
In October 2020, Mirum Pharmaceuticals entered into a distribution agreement with Eversana for maralixibat, under which Mirum will use Eversana’s commercialisation platform in the US market [11]. Mirum entered into exclusive licensing agreements with CANbridge Pharmaceuticals (in April 2021) [12], GC Pharma (in June 2021) [13] and Takeda Pharmaceutical Company (in September 2021) [14] to develop and commercialise maralixibat in Greater China (China, Hong Kong, Macau and Taiwan), South Korea and Japan, respectively.
2 Scientific Summary
2.1 Pharmacodynamics
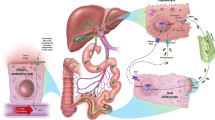
In cholestatic liver diseases, maralixibat acts through the inhibition of the IBAT, resulting in decreased reabsorption of bile acids (primarily the salt forms) from the terminal ileum and increased excretion of bile acids in the faeces [1, 2]. This results in the lowering of systemic levels of bile acids, potentially reducing bile acid-mediated liver damage and related complications [1, 2]. Reductions from baseline in serum bile acid (sBA) levels have been observed during treatment with maralixibat in clinical trials in patients with cholestatic liver diseases (Sect. 2.3).
2.2 Pharmacokinetics
Maralixibat is minimally absorbed and pharmacokinetic parameters cannot be reliably calculated at doses less than 20 mg [1]. In paediatric patients with ALGS treated with maralixibat (Sect. 2.3.1), plasma concentrations were below the limit of quantification (0.25 ng/mL) in the majority of samples [1].

Chemical structure of maralixibat chloride
Maralixibat exposure was approximately dose proportional following single oral administrations of maralixibat 30 mg, 45 mg and 100 mg under fasting conditions [1]. In vitro, maralixibat is highly (91%) bound to human plasma proteins. Maralixibat undergoes minimal metabolism and appears to have low potential for clinically relevant drug-drug interactions. Following a single oral dose of maralixibat 30 mg in healthy adult subjects, maralixibat had a mean plasma half-life of 1.6 h. The vast majority of maralixibat excretion occurs through the faecal route, with 94% of faecal excretion as unchanged maralixibat [1].
Features and properties of maralixibat
Alternative names | Maralixibat chloride; Livmarli™; CAN 108; lopixibat; lopixibat chloride; LUM-001; SD-5613; SHP 625 |
Classes | Amines; anti-inflammatories; antihyperlipidaemics; azabicyclo compounds; benzothiepins; dioxoles; hepatoprotectants; onium compounds; small molecules |
Mechanism of action | Sodium-bile acid cotransporter inhibition |
Route of administration | Oral |
Pharmacodynamics | Inhibits the ileal bile acid transporter, resulting in decreased reabsorption of bile acids from the terminal ileum and reductions in serum bile acid levels; may reduce absorption of fat-soluble vitamins |
Pharmacokinetics | Minimal absorption; approximately dose-proportional exposure; 91% bound to human plasma proteins in vitro; limited metabolism; mean half-life of 1.6 h; excretion is predominantly (73%) through the faecal route, mostly as unchanged maralixibat |
Most common adverse events | Diarrhoea, abdominal pain, vomiting, fat-soluble vitamin deficiency, transaminases increased, gastrointestinal bleeding |
ATC codes | |
WHO ATC code | A05A-X04 (maralixibat chloride) |
EphMRA ATC code | A5A (Bile Therapy and Cholagogues) |
Chemical name | 1-{[4-({4-[(4R,5R)-3,3-dibutyl-7-(dimethylamino)-4-hydroxy-1,1-dioxo-2,3,4,5-tetrahydro-1H-1λ6-benzothiepin-5-yl]phenoxy}methyl)phenyl]methyl}-1,4-diazabicyclo[2.2.2]octan-1-ium chloride |
2.3 Therapeutic Trials
2.3.1 Alagille Syndrome
Maralixibat was associated with significant reductions in sBA levels and pruritus in children with ALGS in the randomised, placebo-controlled phase IIb ICONIC trial [15]. Thirty-one paediatric ALGS patients (all with genotyped mutations within JAG1) with cholestasis (sBA level > 3 × the upper limit of normal) and intractable pruritus were enrolled, with the trial consisting of an 18-week open-label treatment run-in period; a 4-week randomised, double-blind, placebo-controlled drug-withdrawal period; a subsequent 26-week open-label stable-dosing treatment period; and an optional open-label long-term extension period. The run-in period consisted of a 6-week dose-escalation period followed by a 12-week stable-dosing period where all patients were administered maralixibat 380 µg/kg once daily. Patients who completed the run-in period (n = 29; median age of 5 years) were then randomised to continue treatment with maralixibat or receive matching placebo during the 4-week drug-withdrawal period (weeks 19–22). The primary endpoint, evaluated in the responder population (i.e. patients achieving an sBA reduction of ≥ 50% from baseline to week 12 or week 18), was the mean change in sBA levels during the drug-withdrawal period [15].
The primary endpoint of the trial (evaluated per protocol) was met, with a significant least-squares mean difference in sBA levels (− 117 μmol/L) between maralixibat (n = 5) and placebo (n = 10) recipients in the responder population during the drug-withdrawal period (p-value not given) [15]. In the overall population, mean sBA levels decreased by 88 µmol/L (31%) from baseline (283 μmol/L) to week 18. During the randomised drug-withdrawal period, mean sBA levels in the placebo group (n = 16) returned to baseline levels (+ 94 µmol/L change) whereas sBA levels continued to fall (− 17 µmol/L change) in the group continuing maralixibat (n = 13), with a significant between-group difference. With all patients again receiving maralixibat after the end of the drug-withdrawal period, there was a significant reduction from baseline in sBA levels among patients still receiving study drug treatment at week 48 (n = 27; − 96 µmol/L change from baseline) and week 204 (n = 15; − 181 µmol/L change from baseline) [15].
The effect of maralixibat treatment on pruritus followed a similar pattern [15]. In the trial, pruritus was measured using the Itch Reported Outcome (ItchRO) instrument, in which pruritus symptoms are assessed on a 5-point ordinal response scale with scores ranging from 0 (no pruritus) to 4 (very severe). For randomised patients, the mean weekly morning average of observer-reported ItchRO [IcthRO(Obs)] pruritus severity scores improved significantly from baseline (2.9) to week 18 (− 1.7-point change). Following the randomised drug-withdrawal period, mean ItchRO(Obs) scores were 1.4 in patients continuing maralixibat and 2.8 in the placebo group (after maralixibat withdrawal), with a significant least-squares mean between-group difference (− 1.5 points). Furthermore, significant improvements from baseline in ItchRO(Obs) scores were observed at week 48 (− 2.3-point change from baseline) and week 204 (− 2.4-point change from baseline) among patients continuing to receive maralixibat. Patients treated with maralixibat in ICONIC also had significant improvements in xanthomas, height and health-related quality of life [15].
Improvements in sBA levels and pruritus were also observed in a proportion of patients treated with maralixibat in the phase II IMAGO and ITCH trials [3] and IMAGINE [16] and IMAGINE-II [17] long-term extension studies, although findings were inconsistent overall. In addition, an analysis was performed using novel techniques to compare real-world evidence from the ALGS global clinical research database GALA with a cohort of maralixibat-treated ALGS patients (n = 84) [18]. With up to 6 years of data for maralixibat-treated patients, the analysis suggested the potential for maralixibat to improve event-free survival (defined as the time to first event of hepatic decompensation, surgical biliary diversion, liver transplantation or death) in patients with ALGS [18].
2.3.2 Progressive Familial Intrahepatic Cholestasis
Maralixibat reduces sBA and pruritus in a proportion of patients with PFIC associated with bile salt export pump (BSEP) deficiency (PFIC type 2) based on the findings of the open-label, single-arm phase II INDIGO trial [19,20,21]. Twenty-five patients (median age of 4 years) with PFIC due to BSEP deficiency were enrolled and received maralixibat 280 µg/kg once daily (with an increase to maralixibat 280 µg/kg twice daily permitted in the extension, after 48 weeks). Mean change in sBA levels was − 29 µmol/L at week 13 (primary endpoint) and − 59 µmol/L at week 48 [21]. Overall, responses (defined as a ≥ 1-point reduction in ItchRO score and ≥ 70% reduction in sBA) were reported in six patients at week 48 with one further patient achieving a response after increasing maralixibat to twice-daily dosing [20].
With long-term follow-up, all seven responders remained on treatment at week 237, having a mean sBA level of 44.2 µmol/L (234.4 µmol/L reduction from baseline) [19]. Improvements in liver biochemistry and growth parameters were also observed, and none of the responders were listed for liver transplant after > 4.5 years of maralixibat treatment [19].
Key clinical trials of maralixibat (Mirum Pharmaceuticals Inc.)
Identifier(s) | Indication | Phase | Drug(s) | Location(s) | Status |
|---|---|---|---|---|---|
NCT02160782; ICONIC | Alagille syndrome | II | Maralixibat; placebo | Australia, Belgium, France, Poland, Spain, UK | Completed |
NCT01903460; IMAGO | Alagille syndrome | II | Maralixibat; placebo | UK | Completed |
NCT02047318; IMAGINE | Alagille syndrome | II | Maralixibat | UK | Completed |
NCT02057692; ITCH | Alagille syndrome | II | Maralixibat; placebo | Canada, USA | Completed |
NCT02117713; IMAGINE-II | Alagille syndrome | II | Maralixibat | Canada, USA | Completed |
NCT02057718; INDIGO | PFIC | II | Maralixibat | France, Poland, UK, USA | Completed |
NCT03905330; MARCH-PFIC | PFIC | III | Maralixibat; placebo | Multinational | Recruiting |
NCT04185363; MARCH-ON | PFIC | III | Maralixibat | Multinational | Enrolling by invitation |
NCT04168385; MERGE | Cholestatic liver disease | II | Maralixibat | Australia, Belgium, Canada, France, Poland, Spain, UK, USA | Enrolling by invitation |
NCT04729751: RISE | Cholestatic liver disease | II | Maralixibat | Belgium, France, Poland, UK, USA | Recruiting |
NCT04524390; EMBARK | Biliary atresia | II | Maralixibat; placebo | Germany, Poland, UK, USA | Recruiting |
2.4 Adverse Events
Based on data from 86 patients who received maralixibat (66.5–760 µg/kg/day; median duration of exposure, 32.3 months) in the ALGS clinical development program (comprised of three phase II trials and their long-term extensions), the most commonly reported adverse reactions were diarrhoea (reported in 55.8% of patients; 41.6 events per 100 patient-years), abdominal pain (53.5%; 38.6 events), vomiting (40.7%; 19.8 events), fat-soluble vitamin deficiency (25.6%; 11.1 events), transaminases increased (18.6%; 6.9 events) and gastrointestinal bleeding (10.4%; 3.8 events) [1]. Serious adverse events (SAEs) were experienced by 28 patients (32.6%), including three patients (3.5%) having a treatment-related SAE [22]. The majority of gastrointestinal events were reported in the first 4 weeks of treatment, were mild to moderate in severity, and resolved within 1 week [23]. No patients discontinued maralixibat because of gastrointestinal adverse events [23].
While noting that patients in the ALGS clinical development program had abnormal liver tests at baseline, treatment-emergent elevations or worsening of liver tests (mostly transaminases or bilirubin) were observed during maralixibat treatment [1]. Increases in ALT and AST, respectively, to > 3 × baseline levels were observed in 24% and 14% of maralixibat recipients, and increases to > 5 × baseline levels were observed in 2% and 1%. Most cases of liver test elevations improved or resolved either spontaneously or following maralixibat dose modification or discontinuation [1].
2.5 Ongoing Clinical Trials
Maralixibat clinical development is ongoing. The efficacy and safety of maralixibat in paediatric patients (aged 1–17 years) with PFIC is being evaluated in the randomised, double-blind, placebo-controlled phase III MARCH-PFIC trial (NCT03905330), with patients who complete MARCH-PFIC being eligible to enter an open-label extension study (MARCH-ON; NCT04185363). The long-term safety of maralixibat is also being investigated in the open-label phase II MERGE trial (NCT04168385) which is open to patients who have previously participated in a study evaluating maralixibat in the treatment of cholestatic liver disease. In addition, the single-arm, open-label phase II RISE trial (NCT04729751) is assessing the safety and tolerability of maralixibat in infants < 12 months of age with ALGS or PFIC. There is also a double-blind, placebo-controlled, phase II trial (EMBARK; NCT04524390) underway that is aiming to evaluate the efficacy and safety of maralixibat in infants with biliary atresia after hepatoportoenterostomy. Finally, there is also an Expanded Access Program ongoing for patients outside the USA with cholestatic pruritus associated with ALGS.
3 Current Status
Maralixibat received its first approval on 29 September 2021, in the USA, for use in the treatment of cholestatic pruritus in patients 1 year of age and older with ALGS [5].
Change history
06 December 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40265-021-01659-y
References
US FDA. LivmarliTM (maralixibat): US prescribing information. 2021. https://www.fda.gov. Accessed 22 Oct 2021.
Mirum Pharmaceuticals. Mirum Pharmaceuticals presents analyses from its rare liver disease programs at the EASL International Liver Congress 2021 [media release]. 2021. https://www.mirumpharma.com. Accessed 22 Oct 2021.
Shneider BL, Spino C, Kamath BM, et al. Placebo-controlled randomized trial of an intestinal bile salt transport inhibitor for pruritus in Alagille syndrome. Hepatol Commun. 2018;2(10):1184–98.
Neimark E, Chen F, Li X, et al. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology. 2004;40(1):149–56.
US FDA. Corrected NDA approval. 2021. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/214662Orig1s000Corrected_ltr.pdf. Accessed 22 Oct 2021.
Mirum Pharmaceuticals. Mirum Pharmaceuticals announces European Medicines Agency validation of the Marketing Authorization Application for maralixibat in patients with PFIC2 [media release]. 2020. http://www.mirumpharma.com. Accessed 22 Oct 2021.
Mayo MJ, Pockros PJ, Jones D, et al. A randomized, controlled, phase 2 study of maralixibat in the treatment of itching associated with primary biliary cholangitis. Hepatol Commun. 2019;3(3):365–81.
Lumena Pharmaceuticals. Lumena Pharmaceuticals announces $23 million Series A financing. 2013. http://www.lumenapharma.com. Accessed 22 Oct 2021.
Shire. Shire adds to rare disease portfolio with acquisition of Lumena Pharmaceuticals, bringing late stage compounds for rare GI/hepatic conditions [media release]. 2014. https://www.takeda.com. Accessed 22 Oct 2021.
Mirum Pharmaceuticals. Mirum Pharmaceuticals secures $120 million in Series A financing to develop phase 3-ready maralixibat for rare liver diseases [media release]. 2018. https://mirumpharma.com. Accessed 22 Oct 2021.
Mirum Pharmaceuticals. Mirum Pharmaceuticals announces partnership with EVERSANA to support launch and commercialization of maralixibat for Alagille syndrome in the United States [media release]. 2020. https://mirumpharma.com. Accessed 22 Oct 2021.
Mirum Pharmaceuticals. Mirum Pharmaceuticals and CANbridge Pharmaceuticals enter into exclusive licensing agreement to develop and commercialize maralixibat in Greater China for rare liver diseases [media release]. 2021. https://mirumpharma.com. Accessed 22 Oct 2021.
Mirum Pharmaceuticals. Mirum Pharmaceuticals and GC Pharma enter into exclusive licensing agreement to develop and commercialize maralixibat for rare liver diseases in South Korea [media release]. 2021. https://mirumpharma.com. Accessed 22 Oct 2021.
Mirum Pharmaceuticals. Mirum Pharmaceuticals and Takeda enter into exclusive licensing agreement to develop and commercialize maralixibat for rare pediatric liver diseases in Japan [media release]. 2021. https://mirumpharma.com. Accessed 22 Oct 2021.
Gonzales E, Hardikar W, Stormon M, et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. Lancet. 2021;398(10311):1581–92.
Baker A, Kelly DA, Gu J, et al. A long-term phase 2 safety and efficacy study of the apical sodium-dependent bile acid transporter inhibitor maralixibat in children with Alagille syndrome: preliminary results from the IMAGINE study. Hepatology. 2017;66(6):1255A-6A.
Shneider BL, Spino C, Kamath BM, et al. Preliminary analysis of ITCH and IMAGINE II—outcome of long-term administration of maralixibat in children with Alagille syndrome [abstract no. L05]. In: The Liver Meeting (AASLD). 2020.
Hansen BE, Vandriel SM, Vig P, et al. Application of real-world evidence analytics: a 6-year event-free survival analysis in Alagille syndrome of the GALA clinical research database and maralixibat treated patients [abstract no. LO7]. In: The Liver Meeting (AASLD). 2021.
Thompson R, Kelly D, Miethke A, et al. Serum bile acid control in long-term maralixibat-treated patients is associated with native liver survival in children with progressive familial intrahepatic cholestasis due to bile salt export pump deficiency. J Pediatr Gastroenterol Nutr. 2021;72(Suppl 1):783–4.
Thompson RJ, Jaecklin T, Peetz C, et al. Genotype and dose-dependent response to maralixibat in patients with bile salt export pump deficiency. Hepatology. 2019;70(Suppl 1):56A.
Thompson R, Kelly D, Rajwal S, et al. Growth analysis in children with progressive familial intrahepatic cholestasis treated with the apical sodium-dependent bile acid transporter inhibitor maralixibat. J Hepatol. 2019;70(1 Suppl):e131–2.
Raman RK, Garner W, Vig P, et al. An integrated analysis of long-term clinical safety in maralixibat-treated participants with Alagille syndrome [abstract no. P0-1285 plus poster]. In: EASL International Liver Congress. 2021.
Kamath BM, Raman RK, Garner W, et al. Gastrointestinal tolerability of maralixibat in patients with Alagille syndrome: an integrated analysis of short-and long-term treatment. J Pediatr Gastroenterol Nutr. 2021;72(Suppl 1):868.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from comments received were made by the author on the basis of scientific completeness and accuracy. Matt Shirley is a salaried employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
The original article has been revised due to retrospective open choice order.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Shirley, M. Maralixibat: First Approval. Drugs 82, 71–76 (2022). https://doi.org/10.1007/s40265-021-01649-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01649-0