
Abstract
Neuropathology of hepatic encephalopathy (HE) in cirrhosis is primarily astroglial in nature characterized by Alzheimer type 2 astrocytosis together with activation of microglia indicative of neuroinflammation. Focal loss of neurons may also occur in the basal ganglia, thalamus and cerebellum. Pathophysiology of HE in cirrhosis is multifactorial, involving brain accumulation of ammonia and manganese, systemic and central inflammation, nutritional/metabolic factors and activation of the GABAergic neurotransmitter system. Neuroimaging and spectroscopic techniques reveal early deactivation of the anterior cingulate cortex in parallel with neuropsychological impairment. T1-weighted MR signal hyperintensities in basal ganglia resulting from manganese lead to a novel entity, ‘Parkinsonism in cirrhosis’. Elucidation of the pathophysiological mechanisms has resulted in novel therapeutic approaches to HE aimed at reduction of brain ammonia, reduction of systemic and central inflammation, and reduction of GABAergic tone via the discovery of antagonists of the neurosteroid-modulatory site on the GABA receptor complex.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Hepatic encephalopathy (HE) encompasses a wide range of cognitive, psychomotor and psychiatric disturbances resulting from acute or chronic severe liver diseases. The appearance of HE in cirrhosis heralds a poor prognosis with negative impact on health-related quality of life, liver transplant priority and patient survival. Consequently, effective therapies for HE are urgently required. The design of such therapies requires an evidence-based understanding of the mechanisms that underpin the pathophysiology of HE.
HE is subdivided into three major types:
- Type A:
-
HE resulting from acute liver failure.
- Type B:
-
HE resulting from portosystemic bypass/shunting.
- Type C:
-
HE resulting from cirrhosis.
Covert HE is a newly-defined sub-type of Type C HE that occurs in up to 80% of patients with cirrhosis and is diagnosed by well-established psychometric tests. The term ‘covert HE’ is the mildest form of HE, and it replaces the previous term ‘minimal HE’ (MHE), to which the addition of grade 1 ‘Overt HE’ (OHE) [1] is made, as shown in Table 1.
2 Cellular Neuropathology of Hepatic Encephalopathy (HE) In Cirrhosis
An effective understanding of the pathophysiological mechanisms relating to HE in cirrhosis requires an understanding of the nature and extent of cellular changes occurring in the brain as a consequence of the failing liver. In type C HE, pathological changes to cells of both the astroglial and neuronal lineages have been described.
2.1 Glial Cells
The cardinal neuropathological feature of HE in cirrhosis is a characteristic morphological alteration known as Alzheimer Type 2 astrocytosis, where astroglial cells (astrocytes) manifest nuclear pallor, swelling and margination of the chromatin pattern [2]. A second type of glial cell, the microglial cell, is also commonly implicated in HE in cirrhosis, and activation of microglia is indicative of a pro-inflammatory mechanism [3].
2.2 Neurons
Neuronal cell death, although less common, also occurs in HE [4]. Acquired Non-Wilsonian hepatocerebral degeneration (AHCD) occurs in cirrhosis often following multiple episodes of coma. Spongiform degeneration occurs in deep cortical layers, basal ganglia and cerebellum. Post-shunt myelopathy, like AHCD, occurs in patients with cirrhosis following multiple episodes of coma and, occasionally, following the TIPS procedure. Symptoms of spastic paresis or paralysis of the lower limbs are apparent, resulting from demyelination of direct and crossed corticospinal tracts.
Unsuspected Wernicke-type haemorrhagic lesions in thalamic nuclei resulting from thiamine deficiency occur in up to 30% of patients with end-stage cirrhosis of alcoholic aetiology. The higher incidence of Wernicke-type lesions in patients with cirrhosis probably relates to the fact that the liver is a major site for thiamine synthesis and storage in humans. Mild to severe cerebellar degeneration characterized by loss of Purkinje cells occurs in patients with cirrhosis [5]. There are no clear correlations between the incidence and extent of cerebellar degeneration, Wernicke-type thalamic lesions and Alzheimer Type 2 astrocytosis, suggesting distinct mechanisms.
Parkinsonism in cirrhosis is an entity characterized by extrapyramidal symptoms, with a reported prevalence as high as 21% [6]. The disorder has been attributed to dopaminergic neuronal deficits resulting from manganese deposition in the basal ganglia of the brain.
3 Pathophysiology
Major avenues of research into mechanisms implicated in the pathogenesis of HE in cirrhosis include:
-
1.
the effects of toxins on brain structural and functional integrity
-
2.
primary brain energy failure
-
3.
pro-inflammatory processes and
-
4.
central neurotransmission imbalance in favour of excess neuroinhibition mediated by GABA.
3.1 Ammonia
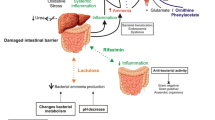
Removal of excess ammonia relies primarily on its conversion to urea via the urea cycle located in periportal hepatocytes together with the formation of glutamine via the enzyme glutamine synthetase (GS) localized in perivenous hepatocytes, as shown in Fig. 1. Patients with cirrhosis commonly develop portal-systemic shunting combined with reduced hepatocyte metabolic capacity resulting in hyperammonemia [2].

Schematic representation of the concept of inter-organ trafficking of ammonia under normal physiological conditions compared to a patient with cirrhosis and HE
However, impairment of hepatic ammonia removal in cirrhosis sets in motion an adaptive pathway involving activation of the gene coding for GS in skeletal muscle [7]. In this way, muscle provides an alternative pathway for ammonia removal as glutamine, as shown in Fig. 1.
Arterial blood and brain ammonia levels are increased several-fold in patients with cirrhosis, and dynamic 13NH3- positron emission tomography (PET) studies demonstrate significant increases of cerebral metabolic rate for ammonia in these patients [8].
Concentrations of ammonia equivalent to those reported in brain in acute and/or chronic liver failure are known to exert direct effects on both inhibitory and excitatory neurotransmission [9].
Ammonia is also a potent inhibitor of the rate-limiting tricarboxylic acid (TCA) cycle enzyme alpha-ketoglutarate dehydrogenase resulting in impaired glucose oxidation, lactate production and impending brain energy failure [10].
Removal of excess ammonia by brain depends exclusively on the synthesis of glutamine via GS. 1H-magnetic resonance spectroscopic studies of patients with cirrhosis reveal increased concentrations of brain glutamine that are correlated with the grade of HE [11].
3.2 Manganese
In a study of 51 patients with cirrhosis, 11 patients (21.6%) exhibited characteristic bilateral MRI signal hyperintensities in basal ganglia. The degree of signal hyperintensity was not correlated with the aetiology of cirrhosis, Child-Pugh scores or fasting blood ammonia, and no patients had OHE at the time of imaging. Extrapyramidal symptoms included a symmetric akinetic-rigid syndrome, tremor, stooped posture and gait impairment with rapid progression over months [6]. Blood manganese concentrations were elevated up to sevenfold in all of nine patients in whom measurements were made and cerebrospinal fluid (CSF) manganese concentrations were increased in all of three cases from whom CSF was available.
In a previous study, basal ganglia tissue from patients with cirrhosis who died in hepatic coma contained several-fold increased manganese concentrations as well as alterations of dopaminergic (DA) marker proteins and metabolite patterns characteristic of Parkinson’s Disease [12]. Liver transplantation results in normalization of MRI signal hyperintensities and circulating manganese levels resulting in improved CNS symptoms [6]. Treatment of two patients with cirrhosis-related Parkinsonism with L-DOPA resulted in substantial improvements of motor function [6].
3.3 Pro-inflammatory Mechanisms
Systemic inflammation resulting from infection and/or hepatocellular damage is common in cirrhosis, and the acquisition of a systemic inflammatory response (SIRS) is a major predictor of HE in these patients. SIRS results from the release into the circulation of pro-inflammatory cytokines such as tumour necrosis factor-alpha (TNFα) and the interleukins IL-1β and IL-6. Circulating levels of TNFα are invariably increased in patients with cirrhosis, and the increase correlates well with the grade of OHE [13].
Microglial activation indicative of inflammation of the brain has been identified in autopsied brain tissue samples from patients with cirrhosis who died in hepatic coma [14]. Activated microglia are known to express transcripts for the mitochondrial translocator protein (TLP), and PET studies with the selective TLP ligand 11C-PK11195 show activation of microglia in patients with cirrhosis and MHE. Particularly intense signals are observed in the anterior cingulate cortex, a brain structure associated with the control of attention in these patients [15].
Liver-to-brain pro-inflammatory signalling occurs in HE and, at the cellular level, human cerebrovascular endothelial cells exposed to TNFα manifest increased capacity for the transport of ammonia [16]. Astroglial cells exposed to combinations of ammonia and recombinant pro-inflammatory cytokines show increased expression of genes implicated in HE.
3.4 Cerebral Blood Flow and Metabolism
There is no convincing evidence to support the hypothesis that HE is primarily caused by brain energy failure. However, PET studies using the glucose transport ligand 18Fluorodeoxyglucose in patients with cirrhosis and covert HE reveal significant decreases in uptake in the anterior cingulate cortex [8]. Decreased brain glucose uptake in these patients was correlated with impaired performance on psychometric testing.
Ammonia inhibits the TCA cycle, an essential metabolic pathway involved in the maintenance of brain energy requirements [10]. Slowing of the cycle results in increased CSF lactate concentrations that correlate well with severity of HE in patients with cirrhosis [17].
Cerebral blood flow (CBF) in patients with cirrhosis is altered in a region-selective manner where flow to cerebral cortical regions is decreased while flow to basal ganglia, cerebellar and thalamic structures is significantly increased. Since this pattern of changes in CBF parallels the regional changes of brain glucose utilization, CBF auto-regulation (defined as the capacity of CBF to match brain activity independent of changes of systemic arterial pressure) appears to be preserved in patients with cirrhosis [18].
3.5 Inhibitory Neurotransmission
Activation of GABAergic transmission (‘increased GABAergic tone’) was proposed based on visual evoked response patterns in experimental animals with HE that were identical to patterns observed in normal animals treated with activators of the GABA receptor complex (GRC) [19]. These findings led to intense interest in activation of the GRC as a major factor implicated in the pathogenesis of HE, an interest that continues to this day. Initially, studies were focussed on the measurement of biochemical components of the GABA system. In all cases, these parameters were found to be present in normal amounts in material from patients who died in grade 4 HE [20].
The GRC has a number of allosteric modulatory sites that are targets for benzodiazepines and neurosteroids [21]. Occupation of these sites by their endogenous agonist ligands results in a magnification of the GABA signal and its neuroinhibitory response. Allopregnanolone (ALLO) is a neurosteroid modulator of the GRC and, hence, an activator of GABAergic neurotransmission. Significant increases of ALLO have been reported in brain tissue from patients with decompensated cirrhosis who died in stage 4 HE (coma) [22] (Fig. 2).

Increased brain concentrations of allopregnanolone in patients with cirrhosis who died in grade 4 HE, age-matched controls, patients with cirrhosis without encephalopathy (LD) and a case of uraemic coma (UC)
Furthermore, brain extracts from HE patients result in increased binding of the GABA agonist ligand 3H-muscimol to brain membrane preparations, thus satisfying the requirement that brain concentrations of ALLO observed in HE patients had the potential to result in ‘increased GABAergic tone’.
4 Translation of Pathophysiological Findings to the Clinic
The ultimate validation of a proposed mechanism for the pathogenesis of HE in cirrhosis relies on the results of clinical trials in which removal of the pathophysiological insult results in reduced severity of encephalopathy.
4.1 Agents Aimed at Lowering Ammonia Production
Agents shown to be effective in randomized clinical trials for the lowering of gut ammonia production leading to improved mental state include the non-absorbable disaccharide lactulose, the broad-spectrum antibiotic rifaximin and probiotics, all of which inhibit the production of ammonia in the gastrointestinal tract [23].
4.1.1 Agents Aimed at Increasing Ammonia Removal
l-ornithine l-aspartate (LOLA) has potent ammonia-lowering properties through mechanisms involving urea synthesis (l-ornithine is a urea cycle substrate) as well as removal of ammonia via GS by residual hepatocytes and skeletal muscle. Results of meta-analyses provide an evidence base for the efficacy of LOLA for the lowering of venous ammonia and improvement of mental status in all grades of HE in cirrhosis [24]. LOLA also has impressive hepatoprotective properties [25].
Benzoate and phenyl acetate have been successfully used for treatment of congenital hyperammonemias in children. Benzoate is also effective for the treatment of HE in cirrhosis [26], and studies in which phenyl acetate is combined with glycerol or L-ornithine are beginning to show promise.
4.1.2 GABA-Receptor Modulators
Agents aimed at decreasing ‘GABAergic tone’ in the brain of HE patients are still the subject of intense investigation. Antagonists of the benzodiazepine and neurosteroid modulatory sites on the GABA-A receptor are available and have been evaluated as possible treatments for HE in cirrhosis. Flumezenil, a potent antagonist of the benzodiazepine site, has proven efficacy in patients with OHE [27], although the effect is relatively short acting due to the short half-life of the agent. More recently, GR 3027, an antagonist of the neurosteroid site, was shown to improve spatial learning, circadian rhythm disturbances and motor coordination in an experimental animal model of chronic liver disease [28]. Clinical trials are currently ongoing.
5 Summary
The pathophysiology of HE in cirrhosis involves both astroglial and neuronal modifications caused by accumulation of ammonia and manganese, inflammation, nutritional/metabolic factors and neurosteroid-induced activation of the GABAergic system. Neuroimaging techniques reveal early deactivation of the anterior cingulate cortex and increased T1-weighted MR signal hyperintensities in basal ganglia resulting from manganese deposition, leading to Parkinsonism. These insights have resulted in novel therapeutic approaches to HE aimed at reduction of ammonia as well as antagonists of the neurosteroid-modulatory site on the GABA receptor complex.
References
Bajaj JS, Cordoba J, Mullen KD, Amodio P, Shawcross DL, Butterworth RF. Review article: the design of clinical trials in hepatic encephalopathy-an ISHEN consensus statement. Aliment Pharmacol Ther. 2011;33:739–47.
Butterworth RF, Giguere JF, Michaud J Lavoie J, Pomier Layrargues G. Ammonia: key factor in the pathogenesis of hepatic encephalopathy. Neurochem Pathol. 1987;6:1–12.
Butterworth RF. Hepatic encephalopathy: a central neuroinflammatory disorder? Hepatology. 2011;53:1372–6.
Butterworth RF. Neuronal cell death in hepatic encephalopathy. Metab Brain Dis. 2007;22:309–20.
Kri JJ, Butterworth RF. Diencephalic and cerebellar pathology in alcoholic and non-alcoholic patients with end-stage liver disease. Hepatology. 1997;26:837–41.
Burkhard PR, Delavelle J, Du Pasquier R, Spahr L. Chronic Parkinsonism associated with cirrhosis. Arch Neurol. 2003;60:521–8.
Desjardins P, Rao KV, Michalak A, Rose C, Butterworth RF. Effect of portacaval anastomosis on glutamine synthetase protein and gene expression in brain, liver and skeletal muscle. Metab Brain Dis. 1999;14:273–80.
Lockwood AH, Weissenborn K, Butterworth RF. An image of the brain in patients with liver disease. Curr Opin Neurol. 1997;10:525–33.
Felipo V, Butterworth RF. Neurobiology of ammonia. Prog Neurobiol. 2002;67:259–79.
Lai JCK, Cooper AJL. Brain α-ketoglutarate dehydrogenase: kinetic properties, regional distribution and effects of inhibitors. J Neurochem. 1986;47:1376–86.
Laubenberger J, Haeussinger D, Bayer S, Guffler H, Hennig J, Langer M. Protein magnetic resonance spectroscopy of brain in symptomatic and asymptomatic patients with liver cirrhosis. Gastroenterology. 1997;112:1610–6.
Butterworth RF, Spahr L, Fontaine S, Layrargues GP. Manganese toxicity, dopaminergic dysfunction and hepatic encephalopathy. Metab Brain Dis. 1995;10:259–67.
Odeh M. Pathogenesis of hepatic encephalopathy: the tumour necrosis factor-alpha theory. Eur J Clin Invest. 2007;37:291–304.
Zemtsova I, Goerg B, Keitel V, Bidmon HJ, Schror K, Haeussinger D. Microglial activation in hepatic encephalopathy in rats and humans. Hepatology. 2011;54:204–15.
Cagnin A, Taylor-Robinson SD, Forton DM, Banati RB. In vivo imaging of cerebral “peripheral benzodiazepine binding sites” in patients with hepatic encephalopathy. Gut. 2006;55:547–53.
Duchini A, Govindarajan S, Santucci M, Zampi G, Hofman FM. Effects of tumor necrosis factor-α and interleukin-6 on fluid-phase permeability in CNS-derived endothelial cells. J Invest Med. 1996;44:474–82.
Yao H, Sadoshima S, Nishimura Y, Fijii K Oshima M, Ishitsuka T et al. Cerebrospinal fluid lactate in patients with hepatic encephalopathy. Eur Neurol. 1987;27:182–7.
Larsen FS, Olsen KS, Ejlersen E, Hansen BA, Paulson OB, Knudsen GM. Cerebral blood flow autoregulation and transcranial Doppler sonography in patients with cirrhosis. Hepatology. 1995;22:730–6.
Jones EA. Ammonia, the GABA neurotransmitter system and hepatic encephalopathy. Metab Brain Dis. 2002;17:275–81.
Butterworth RF, Lavoie J, Giguere JF, Pomier Layrargues G. Affinities and densities of high-affinity [3H] muscimol (GABA-A) binding sites and of central benzodiazepine receptors are unchanged in autopsied brain tissue from cirrhotic patients with hepatic encephalopathy. Hepatology. 1988;8:1084–8.
Ahboucha S, Butterworth RF. Pathogenesis of hepatic encephalopathy: a new look at GABA from the molecular standpoint. Metab Brain Dis. 2004;19:331–43.
Ahboucha S, Pomier Layrargues G, Mamer O, Butterworth RF. Increased brain concentrations of a neuroinhibitory steroid in human hepatic encephalopathy. Ann Neurol. 2005;58:169–70.
Morgan MY, Blei AT, Gruengreiff K, Jalan R, Kircheis G, Marchesini G. The treatment of hepatic encephalopathy. Metab Brain Dis. 2007;22:389–405.
Butterworth RF, Kircheis G, Hilger N, McPhail MJW. Efficacy of l-ornithine l-aspartate for the treatment of hepatic encephalopathy in cirrhosis: systematic review and meta-analysis of randomized controlled trials. J Clin Exp Hepatol. 2018;8(3):301–13.
Butterworth RF, Gruengreiff K. l-ornithine l-aspartate for the treatment of hepatic encephalopathy in cirrhosis: evidence for novel hepatoprotective mechanisms. J Liver Clin Res. 2018;5(1):1044.
Sushma S, Dasarathy S, Tandon RK, Jain S, Gupta S, Bhist MS. Sodium benzoate in the treatment of hepatic encephalopathy: a double-blind randomized trial. Hepatology. 1992;16:138–44.
Pomier Layrargues G, Giguere JF, Lavoie J, Butterworth RF. Flumazenil in cirrhotic patients in hepatic coma: a randomized double-blind placebo-controlled crossover trial. Hepatology. 1994;19:32–7.
Johansson M, Agusti A, Llansola M, Montoliu J, Stromberg E, Malinina G. GR3027 antagonizes GABA A-receptor-potentiating neurosteroids and restores spatial learning and motor coordination in rats with chronic hyperammonemia and hepatic encephalopathy. Am J Physiol Gastrointest Liver Physiol. 2015;309:G400–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Funding for studies in the author’s research unit was provided by The Canadian Institutes for Health Research. This supplement to Drugs has been wholly funded by Merz Pharmaceuticals GmbH, Frankfurt, Germany.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Butterworth, R.F. Hepatic Encephalopathy in Cirrhosis: Pathology and Pathophysiology. Drugs 79 (Suppl 1), 17–21 (2019). https://doi.org/10.1007/s40265-018-1017-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-018-1017-0