
Abstract
Thyroid hormones have important effects on cellular development, growth, and metabolism and are necessary for the healthy function of almost all tissues. Hyperthyroid patients with excess thyroid hormone levels experience tachycardia, fatigue, muscle wasting, and osteoporosis. However, although high thyroid hormone levels have adverse effects, efforts have been made to harness the beneficial effects, such as reduced serum low-density lipoprotein (LDL) cholesterol levels, elevated basal metabolic rate, and weight loss. Thyroid hormones interact with nuclear thyroid hormone receptors (TRs), and cholesterol levels are reduced through TRβ, whereas extrahepatic adverse actions are primarily connected to TRα. Thus, to develop a useful compound for clinical use, efforts have been focusing on developing compounds with isomer-specific functions based on the structure of thyroid hormones, i.e., thyromimetics that are liver and/or TRβ specific. In this short review, we discuss the development of the early thyromimetics that enabled, through modern molecular techniques, the progress towards improved design of TRβ-selective thyromimetics. We also address the early promise shown in human clinical trials and the current status of these drugs and other emerging compounds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Associations between thyroid hormone status and plasma low-density lipoprotein (LDL) cholesterol levels has led to the investigation of thyroid hormone mimetics, or thyromimetics, as lipid-lowering agents. |
New molecules with very high thyroid hormone receptor (TR)-β affinity and hepatic selectivity have been developed. However, the safety of these potent molecules must be carefully assessed in humans, as it is still unclear whether the failure of eprotirome was due to molecule- or class-related adverse effects. |
1 Introduction
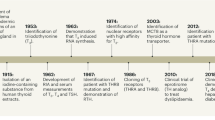
Thyroid hormones have important effects on cellular development, growth, and metabolism and are necessary for the healthy function of almost all tissues [1]. There are two main types of thyroid hormones; L-triiodothyronine (T3) and L-thyroxine (T4). The synthesis and secretion of thyroid hormones is regulated by a negative feedback loop that involves the hypothalamus, pituitary, and thyroid glands (hypothalamic/pituitary/thyroid [HPT] axis). The hypothalamus secretes thyrotropin-releasing hormone (TRH), which signals the pituitary, which subsequently releases thyroid-stimulating hormone (TSH), which stimulates the thyroid gland to start synthesizing thyroid hormones. For instance, high thyroid hormone levels will inhibit TRH production, decrease TSH secretion from the pituitary, and hence decrease thyroid hormone synthesis [2]. Other factors may also influence the HPT axis: thyroid hormone secretion is negatively regulated by endogenous signals such as glucocorticoids, somatostatin, and dopamine [3, 4], whereas, for instance, estrogen or exposure to cold will increase TSH and TRH secretion and TSH and thyroid hormone levels, respectively [5, 6]. Fasting and food deprivation has also been shown to induce pronounced changes in the HPT axis [7].
Only a small fraction of thyroid hormones are available in free form in plasma; most are bound to thyroxine-binding globulin (TBG), thyroxine-binding prealbumin (TBPA), and albumin [8]. Thyroid hormone is predominately secreted from the thyroid gland in the form of T4, and this is also the main form in plasma, with a plasma concentration 40-fold higher than that of T3 [1]. T4 is converted to the primary active form T3 through the action of deiodinases 1 and 2 (D1 and D2) or to the inactive metabolite reverse T3 (rT3) by D3 [9]. Thyroid hormones can be further deiodinated in the liver and metabolized by sulfatation and glucuronidation to increase water solubility and facilitate excretion via bile or urine [10]. The main route of thyroid hormone excretion is via urine as thyroid hormones undergo enterohepatic recirculation in the intestine.
The cellular availability of thyroid hormones is also regulated by the cellular uptake, mediated by membrane transporters, including monocarboxylate transporter (MCT) 8 and 10, L-type amino acid transporters (LATs), and organic anion-transporting polypeptide 1C1 (OATP1C1) [11]. These membrane transporters are differentially expressed and vary in substrate preference, which in turn leads to tissue selectivity of different thyroid hormones. Reduced nicotinamide adenine dinucleotide phosphate (NADPH)-dependent cytosolic T3-binding protein (CTBP), also known as μ-crystallin (CRYM), can sequester thyroid hormones in the cytosol and thereby control the local availability of T3, giving an additional level of control [12].
As mentioned, thyroid hormones are needed for a plethora of functions in nearly all tissues, for instance bone development and growth, intermediate metabolism, as well as development and function of the adipose tissue and the brain [1, 11]. The importance of thyroid hormones for metabolic homeostasis, the diverse functions of thyroid hormones, and the need for tight regulation are apparent in states of excess or reduced thyroid hormone levels. Hypothyroidism is linked to reduced brain development, weight gain, reduced body temperature, reduced heart rate, and elevated plasma total and low-density lipoprotein (LDL) cholesterol. Conversely, hyperthyroid patients suffer from increased tachycardia, arrhythmia, hyperthermia, fatigue and anxiety, muscle wasting, osteoporosis, and short stature in children [1, 11, 13]. However, despite the adverse effects of high thyroid hormone levels, efforts have been made to develop molecules that harness the beneficial effects, such as reduced serum LDL cholesterol levels, elevated basal metabolic rate, and weight loss. The reduction of LDL cholesterol, which is a strong risk factor for arteriosclerosis, has strong clinical value, and the causal relationship between hypothyroidism and atherosclerosis is well known since first being described in 1883 by Theodore Kosher, as reported by Cappola and Ladenson [14].
Thyroid hormones interact with nuclear thyroid hormone receptors (TRs). The TRs were first discovered in the 1980s and are members of the nuclear receptor (NR) superfamily [15,16,17]. The two primary forms of TRs, TRα and TRβ, are transcribed from THRA (NR1A1, chromosome 17) and THRB (NR1A2, chromosome 3), respectively. Alternating the splicing and transcription start sites results in several isoforms that lack the ligand-binding domain (LBD) or the DNA-binding domain (DBD) [18,19,20]. The TRs, like other NRs, share a common structure. The amino-terminal domain varies among the TR isoforms, whereas the centrally located DBD is highly conserved [1]. T3 binds with tenfold higher affinity to TRs than T4 and is the transcriptionally active and more potent thyroid hormone for TRs [21]. Thyroid hormone-bound TRs act as homodimers or heterodimers with retinoid X receptor (RXR) and bind to thyroid hormone response elements (TREs) on DNA. The sequence and arrangement of TREs varies, with different binding specificity to homodimers, heterodimers, and monomers. TRs have also been shown to bind TREs in the unliganded form. In hypothyroidism, or other instances of low thyroid hormone levels, unliganded TRs may bind to TREs and repress basal transcription of genes positively regulated by thyroid hormones [22, 23]. The different isoforms and splice variants may influence gene transcription by competing for binding of thyroid hormones or to the TREs [24,25,26]. TRα is nearly ubiquitously expressed, with the highest expression in the heart, brain, and lungs, whereas TRβ is mainly expressed in the liver, kidneys, pituitary gland, and brain. There are isoform- and splice-variant specific as well as overlapping functions that can depend on tissue-/cell-type distribution or on specific receptor properties. This needs to be further elucidated for more accurate and targeted drug development [27]. TRβ is the predominant receptor in the liver, hence the reduction of cholesterol levels is primarily mediated through this isoform, whereas the extrahepatic adverse actions are primarily connected to TRα. Thus, to develop a useful compound to be used in clinic, much effort has been made to develop molecules with isomer-specific functions based on the structure of thyroid hormones, i.e., thyromimetics that are liver and/or TRβ specific (Table 1).
2 Rationale for the Development of Thyroid Receptor β Agonists
The association between thyroid hormone status and plasma LDL cholesterol levels has led to the investigation of thyroid hormone mimetics, or thyromimetics, as lipid-lowering agents. However, in the 1950s, Strisower and colleagues [28,29,30] found it was not possible to directly use thyroid hormones to lower plasma cholesterol levels because they also had deleterious effects on the heart, bone, and muscle. The beneficial effects of thyroid hormone supplement were initially shown in experiments in the late 1950s in which administration of dried thyroid hormone was shown to reduce plasma LDL cholesterol [31]. In the 1960s, this was followed up with an analogue, dextrothyroxine (D-enantiomer of thyroxine) [32], which again showed a positive effect on LDL cholesterol levels.
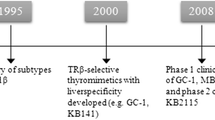
Development of thyroid hormone analogues started in the 1980s, and these compounds primarily targeted the liver. Two compounds, L-94901 and CG-23425, successfully reduced cholesterol levels without unfavorable effects on the heart in rats. CG-23425 was later confirmed to be TRβ selective [33]. In addition to the aforementioned compounds, a T2 analog, DITPA (3,5-diiodothyropropionic acid), originally proposed as therapy for heart failure [34], showed promising LDL cholesterol-lowering effects. However, DITPA was not well tolerated in humans and incurred adverse effects, such as increased markers of bone turnover and fatigue [35, 36]. In the late 1980s and early 1990s, novel molecular techniques were developed that enabled further studies of the TRs using different models. Knockout mice models dissected the phenotypic outcomes and physiological functions, including differences, of the receptors [37]. In addition, crystal structure studies of the ligand-binding pocket of the TRs revealed that these subtypes differed only in one amino acid residue (Asn331, TRβ and Ser277, TRα) and that the selectivity originated from this difference [38,39,40]. Collectively, these discoveries made it possible to improve the design of TRβ1-selective thyromimetics, and a number of different ligands were subsequently developed, including KB141 [41], sobetirome (GC-1) [42], eprotirome (KB2115) [43], MB07811 (VK2809) [44], and—most recently—MGL3196 [45].
3 Hepatic Action and Lipid Regulation of Thyromimetics
Various animal experiments have revealed that most thyromimetics regulate cholesterol metabolism in the liver via different pathways than those used by statins. Statins affect the rate-limiting enzyme in cholesterol synthesis, 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA), whereas thyromimetics do not. Thyromimetics and statins both increase the hepatic expression of LDL receptors, resulting in plasma clearance of LDL cholesterol [33]. However, studies of sobetirome and eprotirome in LDL receptor-deficient mice demonstrated a significant reduction in serum cholesterol [46]. Consequently, LDL receptors are not entirely prerequisite for the thyromimetic-dependent cholesterol reduction in rodents. Sobetirome, eprotirome, and MB07811 also inhibit the expression of sterol regulatory element-binding transcription factor 1 (SREBP-1c) in different rodent models, thus potentially reducing hepatic fatty acid, triglyceride synthesis, and very low-density lipoprotein (VLDL) assembly [47,48,49]. Moreover, the rate-limiting enzyme in the conversion of cholesterol into bile acids, cholesterol 7 alpha-hydroxylase (CYP7A1), is induced by eprotirome and MB07811, promoting a depletion of hepatic cholesterol, although data are conflicting as to whether CYP7A1 is actually induced in humans [50,51,52]. Thyromimetics also increase the expression of high-density lipoprotein (HDL) receptor SR-B1 in the liver and stimulate reverse cholesterol transport (RCT) in mouse and rabbit models [47, 53, 54]. Although RCT differs in humans, the aforementioned data suggest that thyromimetics might have the potential to prevent the development of atherosclerosis. Collectively, these data indicate that thyromimetics regulate lipid parameters through numerous associated pathways, such as bile acid synthesis, RCT, and cholesterol reabsorption via LDL receptors.
While structure–activity relationship studies have facilitated the development of modern TRβ-specific ligands, targeting the liver is most likely equally important to improve selectivity and circumvent side effects such as disturbance of the HPT axis and the heart. Since TRβ is predominantly expressed in the liver, these intersecting effects of most recent liver-selective thyromimetics certainly contribute to a mutual hepatic gene-expressing profile. The tissue selectivity is not completely understood, but it appears that first-pass liver uptake and cellular uptake is important. For instance, MB07811, a highly liver-specific drug that is converted in the liver to the active TRβ ligand MB07344, displays no extrahepatic effects except a mild decrease of total T4, most probably through increased expression of D1 in the liver. Moreover, studies in rats have shown that administration of the non-liver-specific thyromimetic KB141 and T3 at equal doses results in gene expression changes in extra-hepatic tissues; this was not evident with MB07811 [49].
Modern thyromimetics bind to TRβ with different affinities. Sobetirome displays up to tenfold selectivity for TRβ over TRα and with affinity similar to that of T3 [42], whereas MB07344 displays around 12-fold selectivity for TRβ over TRα and approximately half of the affinity of T3 [44, 45]. The most recently developed drug, MGL3196, displays a 28-fold TRβ selectivity in functional assays [45]. Of note, two known thyromimetics (sobetirome and KB141), reported to be selective in binding assays, failed to display selectivity towards TRβ in this functional assay [45]. As mentioned, most recent thyromimetics evidently activate a common set of target genes in the liver, such as the LDL receptor and cytochrome P450 (CYP)7A1. Although not shown for TRβ, one could speculate that different thyromimetics, with different affinities, would also potentially display gene specificity. Modulation of the ligand-binding pocket induces different conformational changes of the receptor and, potentially, recruitment of different sets of co-regulators. Although data are limited for most thyromimetics, a study carried out in mouse liver and human cell lines showed an almost identical gene expression profile for sobetirome and T3 [55], suggesting that gene specificity is predisposed by selectivity and concentration of the drug. However, since sobetirome and T3 display the same affinity for TRβ, future studies exploiting other thyromimetics are required to confirm this.
4 Thyromimetics That Reached Clinical Testing
4.1 Sobetirome (GC-1)
Sobetirome (3,5-Dimethyl-4(4′-hydroxy-3′-isopropylbenzyl) phenoxy) acetic acid) was developed in the late 1990s and was initially shown in hypothyroid mice to reduce cholesterol levels back to normal euthyroid levels [42]. This well studied TRβ-specific thyromimetic accumulates selectively in the liver and has been studied in different rodent models and in monkeys [56]. Collectively, sobetirome displayed all required beneficial effects, including the ability to lower cholesterol in mice and to reduce LDL cholesterol in monkeys, without negative cardiac effects [47, 57]. Recent studies have also shown that sobetirome reduced atherosclerosis in apolipoprotein E (ApoE)-deficient mice by increased hepatic bile acid synthesis via increased Cyp7a1 expression [53]. As previously mentioned, the beneficial effects on plasma cholesterol levels are partly independent of the LDL receptors in the liver [46].
Although the data regarding sobetirome are clearly encouraging, it has only completed one phase I clinical trial, in 2008. A randomized, double-blind, placebo-controlled study found lipid-lowering effects with both single (32 subjects) and multiple (24 subjects) doses. LDL cholesterol levels decreased by up to 22% in patients receiving a single dose of sobetirome. Furthermore, in the 2-week multiple-dose study, LDL cholesterol decreased up to 41%. No extrahepatic effects were observed in either study [56]. To the best of our knowledge, no future clinical trials are scheduled.
4.2 Eprotirome (KB2115)
Eprotirome (3-[[3,5-dibromo-4-[4-hydroxy-3-(1-methylethyl)-phenoxy]-phenyl]-amino]-3-oxopropanoic acid) is a modestly TRβ-selective thyromimetic displaying hepatic-selective uptake. Data from early studies investigating eprotirome for the treatment of hyperlipidemia in humans were encouraging. The first was a randomized, double-blind, single-dose and repeated-dose clinical study in which 24 moderately overweight subjects with elevated plasma cholesterol levels received eprotirome for 2 weeks. Eprotirome treatment was found to reduce serum cholesterol (up to 40%) and ApoB without side effects. Importantly, no negative cardiac effects were observed [43]. The favorable data from the initial short-term studies were followed-up in clinical phase II trials in which eprotirome was administered either as monotherapy or as a supplement to statin therapy [58, 59]. Patients (n = 189) with hypercholesterolemia receiving statins (simvastatin and atorvastatin) also received eprotirome in a randomized, placebo-controlled, double-blinded study. This 12-week study revealed that eprotirome in conjunction with statin therapy promoted further reductions in LDL cholesterol and ApoB. Again, no significant adverse effects (e.g., cardiotoxicity or hypothyroidism) were linked to eprotirome. However, a mild transient increase in alanine aminotransferase (ALT) levels was observed [59]. As mentioned, eprotirome was also administered as monotherapy in a multicenter, randomized, placebo-controlled, double-blinded study to 98 subjects with primary hypercholesterolemia. In this study, serum LDL cholesterol was reduced by 23% (eprotirome 100 µg) and 30% (eprotirome 200 µg), whereas serum HDL and ApoA-1 were unchanged. Low-grade increases in liver enzymes were evident in most patients [58]. In both studies, serum concentrations of ApoB and lipoprotein(a) (Lp(a)) were significantly reduced. The Lp(a) levels were unchanged in the initial short-term studies [43] but were reduced in both the monotherapy and combination (eprotirome and statins) 12-week studies. These data suggest that eprotirome monotherapy is sufficient to reduce Lp(a) levels, and the decrease is not a consequence of the combination of eprotirome and statins. In all three studies, administration of eprotirome reduced free and total T4, but free and total T3 were unchanged. The decreased free T4 level is possibly because eprotirome decreased the endogenous production of TSH but upregulated the expression of hepatic deiodinase enzyme D1.
Given the encouraging data from trials investigating eprotirome (e.g., no extrahepatic toxic effects, robust lowering of LDL cholesterol), a phase III clinical trial (the AKKA trial) was conducted in patients with familial hypercholesterolemia to further assess efficacy and safety [60]. This double-blind, placebo-controlled, parallel-group study enrolled 236 patients, 62% of whom received statins, and 38% of whom received both a statin and ezetimibe. As in previous studies, plasma LDL cholesterol was reduced by 22% (eprotirome 100 µg) and 12% (eprotirome 50 µg). In addition, ApoB, triglycerides, and Lp(a) levels also decreased significantly. Unfortunately, eprotirome also displayed the potential to induce liver injury: aspartate aminotransferase (AST) and ALT levels increased significantly, by 114 and 189%, respectively. Moreover, in a parallel study in dogs, administration of eprotirome for 12 months revealed unfavorable consequences on cartilage [61]. Consequently, the AKKA trial was prematurely terminated after 6 weeks, and further development of eprotirome ceased [60].
4.3 Mb07811 (vk2809)
MB07811 (2R, 4S)-4-(3-chlorophenyl)-2-[(3,5-dimethyl-4-(4′-hydroxy-3′- isopropylbenzyl)phenoxy) methyl]-2-oxido-[1,2,3]-dioxaphosphonane is a pro-drug that undergoes first-pass hepatic extraction. It is subsequently converted via a CYP3A-catalysed reaction to generate the negatively charged phosphonate-containing thyromimetic MB07344 (3,5-dimethyl- 4-(4′-hydroxy-3′-isopropylbenzyl)phenoxy) methylphosphonic acid. MB07344 is rapidly eliminated in bile to escape enterohepatic recirculation, and the production is limited to tissues expressing CYP3A, i.e., primarily the liver [44]. The activated form has greater affinity for TRβ (Ki = 3 nM) than for TRα (Ki = 35 nM), and preclinical studies of MB07811 showed a reduction of total plasma cholesterol and in hepatic and plasma triglycerides in hyperlipidemic and normal rodent models [49]. These intriguing discoveries were followed-up in two studies: MB07811 as monotherapy and in combination with atorvastatin in rabbits, dogs, and monkeys [62], and in rodent models of hepatic steatosis [48]. In the first study, administration of MB07811 decreased total plasma cholesterol in all models. Moreover, combination treatment resulted in an even greater decrease. In the second study, MB07811 reduced hepatic steatosis, plasma triglycerides and free fatty acids (FFAs). Clearance of liver lipids was evidenced by increased hepatic mitochondrial respiration rates. Importantly, given the liver specificity, MB07811 displayed no severe cardiac or thyroid hormone axis (THA) effects [44, 49]. MB07811 reached human trials and was evaluated in a phase I clinical trial in 2006 and a second phase Ib trial between 2007 and 2008. The second was a randomized double-blind trial in which 56 subjects with mild hypercholesterolemia received oral MB07811 once daily for 14 days. MB07811 appeared to be safe and well tolerated at all doses tested (0.25–40 mg). No cardiac or thyroid dysfunctions were observed in the MB07811 and placebo groups, apart from a mild dose-dependent reduction in TSH levels and increased ALT levels with increasing doses. Importantly, MB07811 decreased LDL cholesterol (41%), triglyceride (78%), ApoB, and Lp(a) levels [63].
In 2009, Metabasis Therapeutics, Inc., the developer of MB07811, was acquired by Ligand Pharmaceutical. VK2809 (MB07811) is now being evaluated in a phase II clinical trial by Viking Therapeutics (ClinicalTrials.gov identifier: NCT02927184). This is a randomized, double-blind, placebo-controlled study enrolling approximately 80 patients with elevated LDL cholesterol and non-alcoholic fatty liver disease. Patients will receive VK2809 or placebo for 12 weeks, and the primary endpoint will evaluate the effect on LDL cholesterol. Results are expected in late 2017 [64].
4.4 Mgl3196 (via3196)
MGL3196 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-1,6- dihydropyridazin-3-yloxy)phenyl]-3,5-dioxo-2,3,4,5- tetrahydro[1, 2, 4]triazine-6-carbonitrile is a liver-targeting, highly TRβ-selective agonist (28-fold over TRα). Initial studies in different rodent models, including diet-induced obese mice and hypercholesterolemic rabbits and mice, demonstrated encouraging anti-dyslipidemic effects. MGL3196 reduced non-HDL cholesterol and liver triglycerides without affecting TSH levels. No significant change in heart size or bone mineral density was observed. Additionally, combination treatment with atorvastatin in the rabbit model displayed an indicative additive lipid-lowering effect [45].
The first human studies of MGL3196 were a single-dose ascending study and a 2-week multiple-dose study, both randomized, double blind, and placebo controlled [65]. The data from those studies revealed no adverse events, such as extrahepatic toxicity, or elevated liver enzymes. LDL cholesterol was reduced up to 30% and triglycerides up to 60%. No effects on TSH and free T3 were observed at any doses. Given the moderate efficacy of statins in reducing triglyceride and Lp(a) levels and the indicative effect on lipid levels in rabbits using MGL3196 in combination with atorvastatin, two clinical phase I studies were completed recently: one study of MGL3196 with rosuvastatin and/or simvastatin (ClinicalTrials.gov identifier: NCT02542969), and one of MGL3196 with atorvastatin (ClinicalTrials.gov identifier: NCT02749578). In addition, two phase II studies are currently underway: in the first, patients with non-alcoholic steatohepatitis will receive MGL3196 for 12 weeks and the primary endpoint will evaluate hepatic fat content (ClinicalTrials.gov Identifier: NCT02912260); the second aims to investigate the effect on LDL cholesterol of MGL-3196 once daily for 12 weeks in patients with heterozygous familial hypercholesterolemia (HeFH) (ClinicalTrials.gov identifier: NCT03038022).
5 Future Use of Thyromimetics in Hyperlipidemia
The development of drugs that target hyperlipidemias, cardiovascular disease, and of complete statins, to reduce cardiovascular events remains real and desirable. Mechanistically, TRβ agonists show great potential, but no compounds have yet reached phase III trials or the clinic. Specifically targeting TRβ (and nuclear receptors in general) has been more difficult than anticipated because detailed tissue and cellular distribution is not fully available [66], so the risk of undesirable side effects or unexpected effects on other physiological processes remains. Although TRβ agonists do not, or minimally, affect the hypothalamic-pituitary axis, long-term treatment with these compounds may determine a state of hyperthyroidism in TRβ-dependent tissues and a state of normo- or hypothyroidism in TRα-dependent tissues. This is a peculiar condition for which knowledge is currently lacking on the resulting effects on general homeostasis. The development of monoclonal antibodies against proprotein convertase subtilisin/kexin type 9 (PCSK9) and their ability to achieve extremely low levels of circulating LDL cholesterol without apparent adverse effects (for review, see Seidah et al. [67] and Greig and Deeks [68]) raises the bar for the competitiveness of the new TRβ agonists. Although the cost effectiveness of currently available PCSK9 inhibitors is still under debate [69], it was recently shown that evolocumab in addition to statin therapy reduced the percent atheroma volume of plaques [70] and cardiovascular morbidity in patients with clinically evident atherosclerotic cardiovascular disease [71]. Furthermore, the capacity of PCSK9 inhibitors to greatly decrease Lp(a) levels [72] poses a direct challenge to thyromimetic candidates. New thyromimetic molecules with very high TRβ affinity and hepatic selectively are being developed (i.e., SKL-13784 [73]), and they may have a place in the future landscape because of their ability to positively affect cholesterol and lipid metabolism independent of LDL cholesterol or the LDL receptor [74]. However, the safety of these potent molecules must first be assessed in humans, especially as it is unclear whether the failure of eprotirome was due to molecule- or class-related adverse effects.
6 Conclusion
We believe TRβ agonists still have potential for the treatment of hyperlipidemia and cardiometabolic diseases, mainly because of their LDL cholesterol- and LDL receptor-independent effects. Nevertheless, targeting nuclear receptors has been more complex than expected because of the involvement of these receptors in several cellular processes and because some of the hypotheses generated in preclinical models have not translated to use in humans (i.e., see the failure of liver X receptor [LXR] agonist in humans [75]). The successful development of new, effective and thus far safe monoclonal antibodies against the PCSK9 enzyme in the treatment of hyperlipidemia has led some to question whether pursuing the targeting of TRβ as a strategy to treat hyperlipidemia remains worthwhile.
References
Yen PM. Physiological and molecular basis of thyroid hormone action. Physiol Rev. 2001;81(3):1097–142.
Jackson IMD. Thyrotropin-releasing hormone. N Engl J Med. 1982;306(3):145–55.
Samuels MH, Henry P, Ridgway EC. Effects of dopamine and somatostatin on pulsatile pituitary glycoprotein secretion. J Clin Endocrinol Metab. 1992;74(1):217–22.
Wilber JF, Utiger RD. The effect of glucocorticoids on thyrotropin secretion. J Clin Invest. 1969;48(11):2096–103.
Eastman CJ, Ekins RP, Leith IM, Williams ES. Thyroid hormone responses to prolonged cold exposure in man. J Physiol. 1974;241(1):175–81.
Yarwood NJ, Gurr JA, Sheppard MC, Franklyn JA. Estradiol modulates thyroid hormone regulation of the human glycoprotein hormone alpha subunit gene. J Biol Chem. 1993;268(29):21984–9.
Boelen A, Wiersinga WM, Fliers E. Fasting-induced changes in the hypothalamus-pituitary-thyroid axis. Thyroid. 2008;18(2):123–9.
Schussler GC. The thyroxine-binding proteins. Thyroid. 2000;10(2):141–9.
Germain DLS, Hernandez A, Schneider MJ, Galton VA. Insights into the role of deiodinases from studies of genetically modified animals. Thyroid. 2005;15(8):905–16.
Visser WE, Friesema ECH, Visser TJ. Minireview: thyroid hormone transporters: the knowns and the unknowns. Mol Endocrinol. 2011;25(1):1–14.
Mullur R, Liu Y-Y, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev. 2014;94(2):355–82.
Suzuki S, Mori J-i, Hashizume K. μ-crystallin, a NADPH-dependent T3-binding protein in cytosol. Trends Endocrinol Metab. 2007;18(7):286–9.
Oppenheimer JH, Schwartz HL. Molecular basis of thyroid hormone-dependent brain development. Endocr Rev. 1997;18(4):462–75.
Cappola AR, Ladenson PW. Hypothyroidism and atherosclerosis. J Clin Endocrinol Metab. 2003;88(6):2438–44.
Hodin RA, Lazar MA, Wintman BI, Darling DS, Koenig RJ, Larsen PR, et al. Identification of a thyroid hormone receptor that is pituitary-specific. Science. 1989;244(4900):76–9.
Sap J, Munoz A, Damm K, Goldberg Y, Ghysdael J, Leutz A, et al. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature. 1986;324(6098):635–40.
Weinberger C, Thompson CC, Ong ES, Lebo R, Gruol DJ, Evans RM. The c-erb-A gene encodes a thyroid hormone receptor. Nature. 1986;324(6098):641–6.
Williams GR. Cloning and characterization of two novel thyroid hormone receptor beta isoforms. Mol Cell Biol. 2000;20(22):8329–42.
Mitsuhashi T, Tennyson GE, Nikodem VM. Alternative splicing generates messages encoding rat c-erbA proteins that do not bind thyroid hormone. Proc Natl Acad Sci USA. 1988;85(16):5804–8.
Plateroti M, Gauthier K, Domon-Dell C, Freund JN, Samarut J, Chassande O. Functional interference between thyroid hormone receptor alpha (TRalpha) and natural truncated TRDeltaalpha isoforms in the control of intestine development. Mol Cell Biol. 2001;21(14):4761–72.
Oetting A, Yen PM. New insights into thyroid hormone action. Best Pract Res Clin Endocrinol Metab. 2007;21(2):193–208.
Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14(2):121–41.
Zhang JS, Lazar MA. The mechanism of action of thyroid hormones. Annu Rev Physiol. 2000;62:439–66.
Shibusawa N, Hollenberg AN, Wondisford FE. Thyroid hormone receptor DNA binding is required for both positive and negative gene regulation. J Biol Chem. 2003;278(2):732–8.
Williams GR, Zavacki AM, Harney JW, Brent GA. Thyroid hormone receptor binds with unique properties to response elements that contain hexamer domains in an inverted palindrome arrangement. Endocrinology. 1994;134(4):1888–96.
O’Shea PJ, Williams GR. Insight into the physiological actions of thyroid hormone receptors from genetically modified mice. J Endocrinol. 2002;175(3):553–70.
Flamant F, Gauthier K. Thyroid hormone receptors: The challenge of elucidating isotype-specific functions and cell-specific response. Biochim Biophys Acta. 2013;1830(7):3900–7.
Strisower B, Gofman JW, Galioni EF, Almada AA, Simon A. Effect of thyroid extract on serum lipoproteins and serum cholesterol. Metab Clin Exp. 1954;3(3):218–27.
Strisower B, Gofman JW, Galioni E, Rubinger JH, O’Brien GW, Simon A. Effect of long-term administration of desiccated thyroid on serum lipoprotein and cholesterol levels. J Clin Endocrinol Metab. 1955;15(1):73–80.
Strisower B, Elmlinger P, Gofman JW, Delalla O. The effect of l-thyroxine on serum lipoprotein and cholesterol concentrations. J Clin Endocrinol Metab. 1959;19(1):117–26.
Galioni EF, Gofman JW, Guzvich P, Pouteau J, Rubinger JH, Strisower B. Long-term effect of dried thyroid on serum-lipoprotein and serum-cholesterol levels. Lancet. 1957;272(6960):120–3.
The coronary drug project. Findings leading to further modifications of its protocol with respect to dextrothyroxine. The coronary drug project research group. JAMA. 1972;220(7):996–1008.
Baxter JD, Webb P. Thyroid hormone mimetics: potential applications in atherosclerosis, obesity and type 2 diabetes. Nat Rev Drug Discov. 2009;8(4):308–20.
Pennock GD, Raya TE, Bahl JJ, Goldman S, Morkin E. Cardiac effects of 3,5-diiodothyropropionic acid, a thyroid hormone analog with inotropic selectivity. J Pharmacol Exp Ther. 1992;263(1):163–9.
Ladenson PW, McCarren M, Morkin E, Edson RG, Shih MC, Warren SR, et al. Effects of the thyromimetic agent diiodothyropropionic acid on body weight, body mass index, and serum lipoproteins: a pilot prospective, randomized, controlled study. J Clin Endocrinol Metab. 2010;95(3):1349–54.
Goldman S, McCarren M, Morkin E, Ladenson PW, Edson R, Warren S, et al. DITPA (3,5-Diiodothyropropionic Acid), a thyroid hormone analog to treat heart failure: phase II trial veterans affairs cooperative study. Circulation. 2009;119(24):3093–100.
Flamant F, Samarut J. Thyroid hormone receptors: lessons from knockout and knock-in mutant mice. Trends Endocrinol Metab. 2003;14(2):85–90.
Wagner RL, Apriletti JW, McGrath ME, West BL, Baxter JD, Fletterick RJ. A structural role for hormone in the thyroid hormone receptor. Nature. 1995;378(6558):690–7.
Ribeiro RC, Apriletti JW, Wagner RL, Feng W, Kushner PJ, Nilsson S, et al. X-ray crystallographic and functional studies of thyroid hormone receptor. J Steroid Biochem Mol Biol. 1998;65(1–6):133–41.
Wagner RL, Huber BR, Shiau AK, Kelly A, Cunha Lima ST, Scanlan TS, et al. Hormone selectivity in thyroid hormone receptors. Mol Endocrinol. 2001;15(3):398–410.
Grover GJ, Mellstrom K, Ye L, Malm J, Li YL, Bladh LG, et al. Selective thyroid hormone receptor-beta activation: a strategy for reduction of weight, cholesterol, and lipoprotein (a) with reduced cardiovascular liability. Proc Natl Acad Sci USA. 2003;100(17):10067–72.
Trost SU, Swanson E, Gloss B, Wang-Iverson DB, Zhang H, Volodarsky T, et al. The thyroid hormone receptor-beta-selective agonist GC-1 differentially affects plasma lipids and cardiac activity. Endocrinology. 2000;141(9):3057–64.
Berkenstam A, Kristensen J, Mellstrom K, Carlsson B, Malm J, Rehnmark S, et al. The thyroid hormone mimetic compound KB2115 lowers plasma LDL cholesterol and stimulates bile acid synthesis without cardiac effects in humans. Proc Natl Acad Sci USA. 2008;105(2):663–7.
Fujitaki JM, Cable EE, Ito BR, Zhang BH, Hou J, Yang C, et al. Preclinical pharmacokinetics of a HepDirect prodrug of a novel phosphonate-containing thyroid hormone receptor agonist. Drug Metab Dispos. 2008;36(11):2393–403.
Kelly MJ, Pietranico-Cole S, Larigan JD, Haynes NE, Reynolds CH, Scott N, et al. Discovery of 2-[3,5-dichloro-4-(5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yloxy)phenyl]-3,5-dio xo-2,3,4,5-tetrahydro[1,2,4]triazine-6-carbonitrile (MGL-3196), a Highly Selective Thyroid Hormone Receptor beta agonist in clinical trials for the treatment of dyslipidemia. J Med Chem. 2014;57(10):3912–23.
Lin JZ, Martagon AJ, Hsueh WA, Baxter JD, Gustafsson JA, Webb P, et al. Thyroid hormone receptor agonists reduce serum cholesterol independent of the LDL receptor. Endocrinology. 2012;153(12):6136–44.
Johansson L, Rudling M, Scanlan TS, Lundasen T, Webb P, Baxter J, et al. Selective thyroid receptor modulation by GC-1 reduces serum lipids and stimulates steps of reverse cholesterol transport in euthyroid mice. Proc Natl Acad Sci USA. 2005;102(29):10297–302.
Cable EE, Finn PD, Stebbins JW, Hou J, Ito BR, van Poelje PD, et al. Reduction of hepatic steatosis in rats and mice after treatment with a liver-targeted thyroid hormone receptor agonist. Hepatology. 2009;49(2):407–17.
Erion MD, Cable EE, Ito BR, Jiang H, Fujitaki JM, Finn PD, et al. Targeting thyroid hormone receptor-beta agonists to the liver reduces cholesterol and triglycerides and improves the therapeutic index. Proc Natl Acad Sci USA. 2007;104(39):15490–5.
Lammel Lindemann JA, Angajala A, Engler DA, Webb P, Ayers SD. Thyroid hormone induction of human cholesterol 7 alpha-hydroxylase (Cyp7a1) in vitro. Mol Cell Endocrinol. 2014;388(1–2):32–40.
Drover VA, Agellon LB. Regulation of the human cholesterol 7alpha-hydroxylase gene (CYP7A1) by thyroid hormone in transgenic mice. Endocrinology. 2004;145(2):574–81.
Bonde Y, Breuer O, Lutjohann D, Sjoberg S, Angelin B, Rudling M. Thyroid hormone reduces PCSK9 and stimulates bile acid synthesis in humans. J Lipid Res. 2014;55(11):2408–15.
Kannisto K, Rehnmark S, Slatis K, Webb P, Larsson L, Gafvels M, et al. The thyroid receptor beta modulator GC-1 reduces atherosclerosis in ApoE deficient mice. Atherosclerosis. 2014;237(2):544–54.
Tancevski I, Wehinger A, Demetz E, Hoefer J, Eller P, Huber E, et al. The thyromimetic T-0681 protects from atherosclerosis. J Lipid Res. 2009;50(5):938–44.
Yuan C, Lin JZ, Sieglaff DH, Ayers SD, Denoto-Reynolds F, Baxter JD, et al. Identical gene regulation patterns of T3 and selective thyroid hormone receptor modulator GC-1. Endocrinology. 2012;153(1):501–11.
Scanlan TS. Sobetirome: a case history of bench-to-clinic drug discovery and development. Heart Fail Rev. 2010;15(2):177–82.
Grover GJ, Egan DM, Sleph PG, Beehler BC, Chiellini G, Nguyen NH, et al. Effects of the thyroid hormone receptor agonist GC-1 on metabolic rate and cholesterol in rats and primates: selective actions relative to 3,5,3’-triiodo-L-thyronine. Endocrinology. 2004;145(4):1656–61.
Angelin B, Kristensen JD, Eriksson M, Carlsson B, Klein I, Olsson AG, et al. Reductions in serum levels of LDL cholesterol, apolipoprotein B, triglycerides and lipoprotein(a) in hypercholesterolaemic patients treated with the liver-selective thyroid hormone receptor agonist eprotirome. J Intern Med. 2015;277(3):331–42.
Ladenson PW, Kristensen JD, Ridgway EC, Olsson AG, Carlsson B, Klein I, et al. Use of the thyroid hormone analogue eprotirome in statin-treated dyslipidemia. N Engl J Med. 2010;362(10):906–16.
Sjouke B, Langslet G, Ceska R, Nicholls SJ, Nissen SE, Ohlander M, et al. Eprotirome in patients with familial hypercholesterolaemia (the AKKA trial): a randomised, double-blind, placebo-controlled phase 3 study. Lancet Diabetes Endocrinol. 2014;2(6):455–63.
Lammel Lindemann J, Webb P. Sobetirome: the past, present and questions about the future. Expert Opin Ther Targets. 2016;20(2):145–9.
Ito BR, Zhang BH, Cable EE, Song X, Fujitaki JM, MacKenna DA, et al. Thyroid hormone beta receptor activation has additive cholesterol lowering activity in combination with atorvastatin in rabbits, dogs and monkeys. Br J Pharmacol. 2009;156(3):454–65.
Lian B, Hanley R, Schoenfeld S. A phase 1 randomized, double-blind, placebo-controlled, multiple ascending dose study to evaluate safety, tolerability and pharmacokinetics of the liver-selective TR-beta agonist VK2809 (MB07811) in hypercholesterolemic subjects. J Am Coll Cardiol. 2016;67(13_S, April):1932.
Viking Therapeutics. Viking Therapeutics Doses First Patient in Phase 2 Clinical Trial of VK2809 in Patients with Hypercholesterolemia and Non-Alcoholic Fatty Liver Disease [media release]. 4 Oct 2016. http://ir.vikingtherapeutics.com/press-releases.
Taub R, Chiang E, Chabot-Blanchet M, Kelly MJ, Reeves RA, Guertin MC, et al. Lipid lowering in healthy volunteers treated with multiple doses of MGL-3196, a liver-targeted thyroid hormone receptor-beta agonist. Atherosclerosis. 2013;230(2):373–80.
Elbers LP, Kastelein JJ, Sjouke B. Thyroid hormone mimetics: the past, current status and future challenges. Curr Atheroscler Rep. 2016;18(3):14.
Seidah NG, Abifadel M, Prost S, Boileau C, Prat A. The Proprotein convertases in hypercholesterolemia and cardiovascular diseases: emphasis on proprotein convertase subtilisin/kexin 9. Pharmacol Rev. 2017;69(1):33–52.
Greig SL, Deeks ED. Alirocumab: a review in hypercholesterolemia. Am J Cardiovasc Drugs. 2016;16(2):141–52.
Kazi DS, Moran AE, Coxson PG, Penko J, Ollendorf DA, Pearson SD, et al. Cost-effectiveness of PCSK9 inhibitor therapy in patients with heterozygous familial hypercholesterolemia or atherosclerotic cardiovascular disease. JAMA. 2016;316(7):743–53.
Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ, et al. Effect of evolocumab on progression of coronary disease in statin-treated patients: the GLAGOV randomized clinical trial. JAMA. 2016;316(22):2373–84.
Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713–22.
Desai NR, Kohli P, Giugliano RP, O’Donoghue ML, Somaratne R, Zhou J, et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy: an analysis from the LDL-C assessment with proprotein convertase subtilisin kexin type 9 monoclonal antibody inhibition combined with statin therapy (LAPLACE)-thrombolysis in myocardial infarction (TIMI) 57 trial. Circulation. 2013;128(9):962–9.
Takahashi N, Asano Y, Maeda K, Watanabe N. In vivo evaluation of 1-benzyl-4-aminoindole-based thyroid hormone receptor beta agonists: importance of liver selectivity in drug discovery. Biol Pharm Bull. 2014;37(7):1103–8.
Romney JS, Chan J, Carr FE, Mooradian AD, Wong NC. Identification of the thyroid hormone-responsive messenger RNA spot 11 as apolipoprotein-A1 messenger RNA and effects of the hormone on the promoter. Mol Endocrinol. 1992;6(6):943–50.
Kirchgessner TG, Sleph P, Ostrowski J, Lupisella J, Ryan CS, Liu X, et al. Beneficial and adverse effects of an LXR agonist on human lipid and lipoprotein metabolism and circulating neutrophils. Cell Metab. 2016;24(2):223–33.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflict of interest
Tomas Jakobsson, Lise-Lotte Vedin, and Paolo Parini have no conflicts of interest that might be relevant to the contents of this manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jakobsson, T., Vedin, LL. & Parini, P. Potential Role of Thyroid Receptor β Agonists in the Treatment of Hyperlipidemia. Drugs 77, 1613–1621 (2017). https://doi.org/10.1007/s40265-017-0791-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0791-4