
Abstract
Carbapenem-resistant Enterobacteriaceae are amongst the most feared pathogens due to severely limited treatment options. In response to this threat, three novel β-lactamase inhibitors have been developed in an attempt to reinvigorate and sustain our current antimicrobial therapies. Avibactam, vaborbactam, and relebactam are inhibitor agents with high affinity to Ambler class A and C β-lactamases and favorable outcomes in current clinical trials. However, although they do possess key similarities, these agents have unique differences which may have important clinical implications. The microbiologic spectrum, pharmacokinetics, and key clinical trials for each of these novel agents are reviewed. A proposed role in therapy and potential novel combinations are examined.
Similar content being viewed by others


Three novel β-lactamase inhibitors are discussed in this review: avibactam, vaborbactam, and relebactam. |
Current clinical studies have focused primarily on their co-formulation with β-lactams; ceftazidime–avibactam, meropenem–vaborbactam, and imipenem–relebactam. |
These β-lactamase inhibitors have activity against class A and C β-lactamases, including carbapenemases of the Klebsiella pneumoniae carbapenemase (KPC) family. |
Most clinical studies with results to date have involved non-pathogen-specific common infectious syndromes such as complicated urinary tract infection and complicated intra-abdominal infection. |
1 Introduction
The evolution of bacterial resistance to antimicrobials has been a problem since the earliest days of penicillin. This escalation has only intensified with the widespread consumption of antimicrobials as part of agricultural, veterinary, and healthcare practice.
β-lactam antibiotics constitute 60% of worldwide antibiotic usage by weight and are among the most effective agents for treatment [1]. Therefore, β-lactam-hydrolyzing enzymes (β-lactamases), are considered the most important and clinically relevant mechanism of bacterial resistance. Genes encoding β-lactamases may be present on the bacterial chromosome, and are also often found on mobile genetic elements such as plasmids and transposons. This results in the potential for rapid spread through horizontal as well as vertical genetic transmission.
There are two major classification schemes of β-lactamases. The Ambler classification is based on amino-acid sequence similarities and the Bush classification is based on functionality. The Ambler Classification divides β-lactamases into four molecular classes, A–D, with classes A, C, and D utilizing a serine moiety, and class B consisting of a metalloenzymatic zinc ion at its active site. The Bush schema is based on enzymatic functionality and results in three major groups: Group 1 cephalosporinases, Group 2 serine β-lactamases, and Group 3 metallo-β-lactamases (MBL), with each group encompassing further subdivisions [2]. Notably, by the end of 2009, over 890 unique protein sequences of β-lactamases were reported by Bush and Jacoby [2]. In a more recent estimate, >2000 unique β-lactamases have been described to date [3].
The first clinically used β-lactamase inhibitors, such as clavulanic acid, sulbactam, and tazobactam were discovered in the mid-to-late 1980s. A conserved similarity with these inhibitors is the shared β-lactam backbone. In essence, these inhibitors form stable intermediates with β-lactamases, thereby allowing their companion β-lactam to effectively bind to a target penicillin binding protein [4]. These agents were a revolutionary advance allowing for the reinvigoration and expansion of both the spectrum and longevity of then available antimicrobials. However, the ever-increasing number of clinically important β-lactamases has spurred a demand for novel inhibitor agents.
Resistance to carbapenems in Gram-negative bacteria is of particular concern, as carbapenems are still regarded the first-line therapeutic option for many Gram-negative, multi-drug resistant (MDR) bacterial infections. Globally, enzymatic carbapenem resistance in Enterobacteriaceae is most often conveyed by carbapenemases in the family of serine-based Klebsiella pneumoniae carbapenemases (KPC). Other important carbapenemases include New Delhi metallo-β-lactamases (NDM), Verona integron-encoded metallo-β-lactamases (VIM) and oxacillinase-48-like (OXA-48) carbapenemases. Carbapenemases have a global distribution with substantial regional variability [5]. KPC-producing Enterobacteriaceae predominate in the USA, Italy, Greece, Italy and South America, while MBL are most commonly found within the Indian subcontinent and in particular specific countries in Europe including Romania, Denmark, Spain, and Hungary [5]. The distribution of OXA-48 is centered in Turkey and its surrounding counties [5]. This global epidemiology underscores the widespread occurrence, and the risk of regional spread due to increasing worldwide interconnectivity, utilization of intensive care units, and the rising availability of medical tourism.
Carbapenemase-producing organisms (CPO) are often resistant to other drug classes as well and few therapeutic options remain for patients infected with CPO. Salvage agents such as polymyxins and tigecycline may display high toxicity and low efficacy. Mortality rated in invasive carbapenem-resistant Enterobacteriaceae (CRE) infections are estimated between 32 and 44% [6].
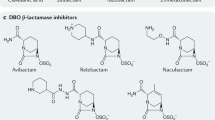
In this review, we will discuss three novel inhibitors. These three novel compounds—avibactam (formerly AVE1330A and NXL104), relebactam (formerly MK7655), and vaborbactam, (formerly RPX7009). Each have unique pharmacokinetic and pharmacodynamic properties (see chemical structures in Fig. 1). Whereas avibactam is currently available in co-formulation with ceftazidime in the USA, and has been approved by the European Medicines Agency (June 2016), none of the other agents are currently available outside of studies. The most obvious benefit of these newer agents over older-generation β-lactamase inhibitors, such as tazobactam, clavulanate, and sulbactam, is their ability to inhibit certain extended-spectrum β-lactamases and carbapenemases.

Chemical structures of avibactam, vaborbactam and relebactam
For this non-systematic, narrative review, we have reviewed the following databases up to 2/23/2017: Pubmed, clinicaltrials.gov, Google Scholar. In addition, we have used reference list from published articles and we have searched abstract banks of national meetings and press releases where applicable.
2 Avibactam
2.1 Avibactam: Introduction
Avibactam (previously NXL104, AVE1330A) is a synthetic diazabicyclooctane non-β-lactam inhibitor, which inhibits Ambler class A and C and some class D β-lactamases. It was the first novel inhibitor to receive FDA approval as the combination of ceftazidime–avibactam. Ceftazidime–avibactam received approval in February, 2015 for the treatment of complicated urinary tract infections (cUTI) and for the treatment of complicated intra-abdominal infection (cIAI) when used in combination with metronidazole.
Ceftazidime, as the active companion agent, is a β-lactam which inhibits peptidoglycan synthesis via the inhibition of penicillin-binding proteins (PBPs). The inhibition of PBPs leads to cell wall instability and subsequent lysis of bacterial cells.
2.2 Mechanism of Avibactam
Avibactam has been shown to exert its inhibition properties via a two-stage process. There is an initial non-covalent association with a susceptible β-lactamase binding site followed by a covalent acylation at the β-lactamase serine residue [7, 8]. A unique feature of avibactam in comparison to earlier-generation inhibitor agents is that avibactam binds reversibly to β-lactamases allowing for re-cyclization and inhibition of additional β-lactamase molecules. Additionally, in vitro studies have found that avibactam did not appreciably induce Enterobacter cloacae chromosomal AmpC production [8].
2.3 Microbiologic Spectrum of Avibactam
The crucial advantage of avibactam over older β-lactamase inhibitors is its ability to inhibit a wide range of β-lactamases, including the extended spectrum (ESBL), AmpC, and Class A and D carbapenemases, in particular KPC and OXA-48 (Table 1) [9, 10].
In a large in vitro study of clinical US Enterobacteriaceae, only 11 isolates of >20,000 isolates had a ceftazidime–avibactam MIC > 8 µg/mL. Two of the 11 resistant isolates expressed an MBL, which are intrinsically resistant to avibactam inhibition [9]. This highlights a key limitation of avibactam, in that it is not active against class B MBLs. Furthermore, Livermore et al. also showed that avibactam also was able to restore the activity of ceftazidime against bacterial strains with the OXA-48 enzyme [11].
These studies highlighted the finding that non-MBL mechanisms of resistance exist for avibactam. Variants of SHV-1 and KPC-2 possessing a single point mutation were able to confer avibactam resistance [12]. Experimentally, three AmpC de-repressed Enterobacter cloacae isolates had a significantly elevated MIC to the combination of ceftaroline and avibactam. One isolate had an OmpC and OmpF deficiency and in the remaining two, there were point mutations in the AmpC gene [8]. These findings also highlighted that resistance to ceftazidime–avibactam was identified in selected Enterobacter strains and postulated to have occurred due to a combination of impermeability and an overproduction of AmpC overwhelming inhibitor capacity [11]. Importantly, avibactam resistance in clinical isolates after treatment of patients with ceftazidime–avibactam has been reported [13]. Various bla KPC-3 mutations that rendered avibactam inactive against the resulting KPC-3 enzyme were identified. Of concern, these mutated genes were found to be plasmid-borne. Some of these mutations decreased the carbapenemase activity of KPC-3 and led to carbapenem-susceptible isolates [14].
In addition to its role in MDR Enterobacteriaceae, ceftazidime–avibactam is also active against certain MDR Pseudomonas aeruginosa strains [9]. In a 4-year US study, 81% of ceftazidime-resistant P. aeruginosa strains were susceptible to ceftazidime–avibactam [15]. In contrast, the addition of avibactam to ceftazidime does not improve its activity against Acinetobacter baumannii.
While the combination of ceftazidime–avibactam did exhibit improved activity for Bacteroides fragilis, Clostridium perfringens, and both Prevotella and Porphyromonas species, ceftazidime–avibactam does not exhibit reliable anaerobic activity [8, 16]. Furthermore, anti-staphylococcal and anti-streptococcal activity is limited as well.
2.4 Pharmacokinetics/Pharmacodynamics of Avibactam
In two cohorts of eight subjects administered doses of ceftazidime–avibactam at a dose of 1000/250 and 2000/500 mg, the pharmacokinetics of both agents combined were not significantly altered when compared to administration alone [8]. Merdjan et al. conducted early Phase 1 studies on the safety and tolerability of ceftazidime–avibactam administered at a 4:1 ratio. These studies confirmed that avibactam plasma concentrations consistently were related to renal function, and that accordingly, avibactam exposure increased with increasing severity of renal impairment [17]. Additional Phase 1 data in healthy volunteers found avibactam pharmacokinetics were linear for doses ranging from 50–2000 mg. Following infusion, avibactam has rapid distribution, a half-life of 1.7–2.1 h, and is primarily (95%) renally cleared with rate dependent on creatinine clearance [8]. In six anuric patients on renal replacement therapy, 54% of avibactam was removed during dialysis [8]. As such, the preliminary data suggest that avibactam pharmacokinetics and clearance is similar to that of its companion agent, ceftazidime.
Based on Phase 1 data, the recommended dose for patients with normal renal function is 2000 mg/500 mg ceftazidime–avibactam administered every 8 h. The recommended dose adjustment for patients are: 1.25 g every 8 h for creatinine clearance 31–50 mL/min, 0.94 g every 12 h for CrCL 16–30 mL/min, 0.94 g every 24 h for CrCL 6–15 mL/min, and 0.94 g every 48 h for CrCL ≤ 5 mL/min (both ceftazidime and avibactam are hemodialyzable; thus the package insert recommends to administer after hemodialysis on hemodialysis days). When penetration into the epithelial lining fluid (ELF) of the lung was evaluated in a Phase 1 study on healthy adults, both ceftazidime and avibactam were found to penetrate into ELF dose proportionally, with ELF exposure to both drugs ∼30% of plasma exposure [18].
2.5 Avibactam: Clinical Studies
Clinical studies are summarized in Table 2. Two Phase 2 trials were performed to evaluate the potential efficacy of ceftazidime–avibactam. The first, by Vazquez et al. enrolled 137 patients, randomized in a 1:1 fashion to receive either ceftazidime–avibactam or imipenem for the treatment of cUTI. The microbiologic response was comparable; the response of 19/27 (70.4%) in the ceftazidime–avibactam and 25/35 (71.4%) in the imipenem groups [19]. Notably, the renal dose adjustment protocol used differed significantly from the current FDA label. The dosing studied in this trial was 500 mg of ceftazidime and 125 mg of avibactam every 8 h, a dose that was 4 times lower than the current package insert (2 g/500 mg ceftazidime–avibactam every 8 h) for patients with normal renal function. A second Phase 2 trial by Lucasti et al. studied the efficacy of ceftazidime/avibactam plus metronidazole compared with meropenem in the treatment of cIAI. A favorable response was observed in 91.2% (62/68) and 93.4% (71/76) of patients, respectively [20].
Two key Phase 3 clinical trials form the foundation of clinical data. The RECAPTURE study, evaluated the efficacy of ceftazidime–avibactam compared to doripenem for the treatment of cUTI. Patients were randomized in a 1:1 fashion to receive either ceftazidime–avibactam or doripenem [21]. The microbiological modified intent-to-treat population consisted of 393 and 417 patients in the ceftazidime–avibactam and doripenem groups, respectively. Non-inferiority was met for the co-primary endpoints of patient-reported symptomatic resolution at day 5 [276 of 393 (70.2%) vs. 276 of 417 (66.2%) patients] and the combined symptomatic resolution/microbiological eradication at test of cure [280 of 393 (71.2%) vs. 269 of 417 (64.5%)] patients. The microbiologic response at end-of-treatment was similar for both groups at 95.2 and 94.7% in the ceftazidime–avibactam compared to the doripenem group. When examining susceptibility of baseline pathogens, ceftazidime–avibactam and doripenem were active against 311/400 (77.8%) and 297/419 (70.9%) of organisms, respectively. Of interest, in the subset of patients with bacterial isolates that were non-susceptible to ceftazidime, microbiological cure occurred in 47/75 (62.7%) of the ceftazidime–avibactam-treated patients and 51/84 (60.7%) of doripenem-treated patients, respectively [21]. In this study, renal impairment did not affect clinical outcome, although patients with a creatinine clearance ≤ 30 mL/min or on dialysis were excluded.
Mazuski et al. performed a Phase 3 randomized double-blind study to determine the efficacy of ceftazidime–avibactam with metronidazole compared to meropenem in the treatment of cIAI [22]. As previously, patients with a creatinine clearance ≤30 mL/min were excluded. An additional limitation is that the study enrolled primarily a high proportion of patients with appendicitis as their primary diagnosis, and in general all patients were not critically ill with over 80% of patients having an APACHE II score of ≤10, and a low incidence of bacteremia (4.2% ceftazidime–avibactam and metronidazole group vs. 2.7% meropenem group). Of particular interest, in patients with ceftazidime-resistant Gram-negative infections, ceftazidime–avibactam plus metronidazole and meropenem resulted in clinical cure rates of 83.0% (39/47) and 85.9% (55/64), respectively [22]. Mortality rates were also similar at 2.5% (13/520) and 1.5% (8/523) between the ceftazidime–avibactam and meropenem groups. Overall, ceftazidime–avibactam plus metronidazole was non-inferior to meropenem in this trial [22]. In subgroup analysis, patients with moderate renal impairment (estimated creatinine clearance between 30 and 50 mL/min) treated with ceftazidime/avibactam plus metronidazole (45%) had lower cure rates as compared patients with moderate renal impairment treated with meropenem (74%). The reason for this observation may have been an observed delay in dose readjustment to full dosing in those patients who recovered renal function [23]. Of note, in these Phase 3 cIAI trials, patients with moderate renal failure were given 1000/250 mg ceftazidime/avibactam every 12 h. The current package insert recommendations are to give 1000/250 mg ceftazidime/avibactam every 8 h to patients with moderate renal failure.
The REPRISE study compared ceftazidime–avibactam versus best available therapy (BAT) in patients with cUTI or cIAI infections caused by ceftazidime-resistant Enterobacteriaceae or P. aeruginosa in a randomized, open-label trial. Of note, the authors stated that “Because of the unfeasibility of recruiting large numbers of patients infected with resistant Gram-negative pathogens, we did not do any formal power calculations for this study, or any formal statistical comparisons between the treatment groups. Rather, we used the corresponding CIs for the efficacy of best available therapy to provide a context for descriptive estimates of ceftazidime–avibactam efficacy.” Using this methodology, 154 ceftazidime–avibactam-treated patients (of whom 144 had cUTI) were compared to 148 patients treated with BAT. Numerically, clinical responses were similar and microbiologic responses were somewhat higher in the ceftazidime–avibactam group [24].
A study comparing ceftazidime–avibactam versus meropenem in the treatment of hospital-acquired bacterial pneumonia (HABP) and ventilator-associated bacterial pneumonia (VABP) has recently been completed (clinicaltrials.gov identifier NCT01808092), and results should be available in the near future.
Post-marketing clinical experience with ceftazidime–avibactam from observational studies is starting to be reported. These data are important as patients with infections caused by the target organisms (e.g. carbapenemase-producing Enterobacteriaceae) are often not included in registrational trials. At IDweek 2016, a case series of 60 patients with CRE infections treated with ceftazidime–avibactam was presented [25]. In this cohort, 51% had a microbiologic cure, 66% had clinical success as defined by the investigators, and the all-cause hospital mortality was 36%. Shields et al. reported single-center, observational experience using ceftazidime–avibactam for the treatment of CRE infection in 37 patients [13]. Of note, the patients had a variety of infectious syndromes, primarily pneumonia (12/37), but also including soft tissue infection, intra-abdominal infection, primary bacteremia, and even a single case each of subdural empyema and ventriculitis and mediastinitis. No isolates expressed VIM, IMP, NDM, or OXA-48 carbapenemases, and 78% (29/37) expressed KPC. Thirty-day survival was 76% (28/37) and clinical success was 59% (22/37). Microbiologic failures due to recurrence of infection occurred in 10 patients of which 3 isolates displayed ceftazidime–avibactam resistance [13]. Collectively, this suggests ceftazidime–avibactam is a potentially viable option with a comparable clinical response to alternative therapies. However, it raises the concern of emergence of resistance following therapy. Randomized controlled trials are needed to more definitively study the relative efficacy of ceftazidime–avibactam in comparison to other available therapies for the treatment of invasive infections caused by carbapenemase-producing Enterobacteriaceae.
2.6 Adverse Effects of Avibactam
Phase 1 data found avibactam well tolerated at multiple dose ranges [8, 17]. Overall, avibactam has had few adverse effects reported [9]. Similarly, ceftazidime–avibactam was reasonably well tolerated even when subjected to supratherapeutic doses of 3000 mg ceftazidime with 2000 mg avibactam, although 30% of volunteers experienced adverse effects, these consisted of nausea, vomiting, and headache [9]. In Phase 3 trials, adverse event rate was generally low and similar to comparator agents, and consisted most commonly of headache, nausea, or diarrhea, which rarely resulted in discontinuation of study drug [21].
3 Vaborbactam
3.1 Vaborbactam: Introduction
Boronic acids are potent inhibitors of serine proteases and have thus been of considerable interest clinically. Vaborbactam (formerly RPX7009) is the first boronic acid β lactamase inhibitor in current Phase 3 clinical trials. RPX7009 was found to exhibit potent inhibition of KPC enzymes, as well as other Ambler class A and C enzymes (Table 1). However, similar to avibactam, it lacks inhibition of class B MBLs. Based on preliminary Phase 3 treatment data, FDA submission of New Drug Application (NDA) was filed in February 2017.
3.2 Mechanism of Vaborbactam
Boronates have a high affinity for serine proteases resulting in a covalent association between the catalytic serine and the boronate moiety [26]. This is a novel mechanism as compared to current clinically available β-lactamase-inhibitors. By forming a reversible dative bond with the β-lactamase, vaborbactam acts as a competitive inhibitor and is not hydrolyzed by the β-lactamase. Vaborbactam is administered in combination with meropenem, a carbapenem approved in 1996.
3.3 Microbiologic Spectrum of Vaborbactam
In vitro testing of vaborbactam was performed by Castanheira et al. [27]. The ability of vaborbactam to augment the activity of meropenem was evaluated in vitro against 315 serine carbapenemase-producing Enterobacteriaceae isolates as identified by PCR sequencing and microarray-based assay. As expected, meropenem alone inhibited only 2.2% of the strains at the Clinical and Laboratory Standards Institute (CLSI) susceptibility breakpoint (≤1 μg/mL). However, in the presence of increasing concentrations of vaborbactam, the activity of meropenem was restored. Based on dose escalation studies, activity of meropenem was at least 64-fold greater with a fixed dosed concentration of vaborbactam at 8 μg/mL, making this concentration the target for additional study [27]. With this fixed concentration vaborbactam, activity of meropenem was restored to MIC ≤ 1 in 93.7% of isolates and ≤2 in 96.5% of isolates. Seven isolates, all K. pneumoniae, had persistent MIC values ≥16 μg/mL despite addition of vaborbactam. Of these isolates, four produced an MBL concurrently with KPC enzyme. The remaining three isolates expressed reduced levels of outer membrane protein OmpK37 and high expression of AcrAB-TolC efflux [27]. Of note, vaborbactam did not restore activity of biapenem against OXA-48-carbapenemase-producing CRE [28].
Meropenem–vaborbactam also showed favorable activity when tested against 4500 Gram-negative clinical isolates from 11 New York City hospitals [29]. When examining only multi-drug-resistant carbapenemase-producing strains of Enterobacteriaceae, inclusive of Escherichia coli, K. pneumoniae, and Enterobacter spp., 131/133 (98.5%) were inhibited by meropenem plus vaborbactam [29]. The combination had reduced activity against two KPC-producing K. pneumoniae isolates which had reduced OmpK35 and OmpK36 expression [29]. Notably, vaborbactam did not improve activity of meropenem against A. baumannii and P. aeruginosa postulated due to alternative mechanisms of drug resistance including the porin alterations and drug efflux [29]. The impact of vaborbactam on the anaerobic spectrum of carbapenems was evaluated in combination with biapenem [30]. As expected, the activity of biapenem alone against anaerobes was excellent, and the addition of vaborbactam did not significantly change the anti-anaerobic activity.
3.4 Pharmacokinetics/Pharmacodynamics of Vaborbactam
In Phase 1 studies, in 36 healthy volunteers, vaborbactam was well tolerated and had a half-life of 1.23 h, and steady-state volume of distribution of 21.0 L [26]. As such, pharmacokinetics mimicked most β-lactams which typically share a short half-life and low volume of distribution. Vaborbactam is given in clinical trials in combination with meropenem in a dosage of 2–2 g intravenous infusion every 8 h.
Wenzler et al. evaluated the plasma and epithelial lining fluid (ELF) concentrations of meropenem and vaborbactam in healthy subjects following intravenous infusion, as a potential predictor for efficacy in lower respiratory tract infections. In 26 healthy adult subjects, when administered 2–2 g meropenem–vaborbactam every 8 h as a 3-hour extended infusion, a similar time course and magnitude of meropenem and vaborbactam concentrations was observed in serum and ELF, with penetration 65 and 79% for meropenem and vaborbactam, respectively [31].
3.5 Vaborbactam: Clinical Studies
Clinical studies are summarized in Table 3. Phase 1 clinical studies by Griffith et al. showed vaborbactam was well tolerated in 36 healthy volunteers when administered via 3-h infusions at doses ranging from 250 to 1500 mg [32].
Two large multicenter Phase 3 trials were initiated in 2014 to evaluate the efficacy of meropenem–vaborbactam clinically. TANGO-1 was the first trial to complete enrollment in early 2016 [33]. TANGO-1 was a multicenter, 1:1 randomized, double-blind study comparing meropenem–vaborbactam to piperacillin–tazobactam in the treatment of cUTI in adults. Clinical success was defined as clinical cure or improvement in symptoms in addition to microbiologic eradication with follow-up urine culture reduction to less than 104 CFU/mL. In the intent-to-treat population, clinical success occurred in 188/192 patients (98.4%) in the meropenem–vaborbactam and in 171/182 patients (94.0%) in the piperacillin–tazobactam group. This FDA primary endpoint met statistical significance, with a difference of 4.5% (95% CI: 0.7–9.1%). In the microbiologic evaluable group for test-of-cure, microbiological eradication occurred in 118/178 patients (66.3%) in the meropenem–vaborbactam and 102/169 patients (60.4%) in the piperacillin–tazobactam group, a difference of 5.9%, which was not statistically significant (95% CI: −4.2–16%) [33, 34].
The TANGO-2 study (ClinicalTrials.gov Identifier: NCT02168946), currently ongoing, is a 60-site, randomized at a 2:1, experimental drug study evaluating meropenem–vaborbactam for the treatment of infections by a suspected carbapenem-resistant Enterobactericiae. Potential infectious syndromes evaluated include cUTI, HABP/VABP, cIAI, and bacteremia when compared to BAT [33]. TANGO-3 (ClinicalTrials.gov Identifier: NCT03006679) is also planned. It will compare meropenem–vaborbactam versus piperacillin–tazobactam in patients with HABP/VABP. Additionally, a new trial has begun recruitment, as a Phase 1 study to evaluate the dose, pharmacokinetics, safety and tolerability of meropenem–vaborbactam in pediatric patients (ClinicalTrials.gov Identifier: NCT02687906).
3.6 Adverse Effects of Vaborbactam
As vaborbactam remains in clinical trials there are few published data on drug tolerance. However, initial studies suggest it is likely to be well tolerated. TANGO-1 released data showed the treatment-emergent adverse event rate was 15.1 and 12.8% in the vaborbactam and piperacillin–tazobactam group, respectively. The rate of study drug discontinuation secondary to adverse effect was 2.6 and 5.1% for vaborbactam and piperacillin–tazobactam, a non-significant difference.
Wenzler et al. [31], in testing of pharmacokinetic properties of combination meropenem–vaborbactam in 26 healthy adults had one subject discontinue administration due to chest discomfort, dizziness, and dyspnea, which was considered to be potentially related to the investigational drug [31]. The remaining 25 subjects, tolerated the study drug at 2gm-2gm doses without any reportedly meaningful laboratory, vital sign, EKG, or physical examination adverse effects [31].
4 Relebactam
4.1 Relebactam: Introduction
Relebactam (formerly MK-7655) is a bridged bicyclic urea candidate molecule, which following discovery has become a clinical candidate due to its broad class A and C β-lactamase inhibition [35]. It is currently in clinical trials in co-formulated combination of the carbapenem imipenem and the renal dehydropeptidase inhibitor cilastatin.
4.2 Mechanism of Relebactam
Similar to avibactam, relebactam is a small serine-based molecule with a diazabicyclooctane core. Relebactam, in contrast, also possesses a piperidine ring. However, the predicted mechanism of action appears to be similar to that of avibactam with potent inhibition of both class A and C β-lactamases [4].
4.3 Microbiologic Spectrum of Relebactam
Relebactam has a similar activity to avibactam; it inhibits class A and class C β-lactamases including KPC enzymes (Table 1). In this way, the addition of relebactam to imipenem broadens the spectrum of that combination to include certain imipenem-resistant Enterobacteriaceae and P. aeruginosa strains. However, the addition of relebactam to imipenem did not provide added benefit against A. baumannii [36]. An early in vitro study of the potential activity of relebactam was performed by Hirsch et al. utilizing mathematical modeling in a hollow fiber infection model to assess bactericidal activity of relebactam in combination with imipenem [37]. Time-kill studies were performed on a KPC-2-producing K. pneumoniae and three P. aeruginosa isolates which exhibited OprD porin mutations and AmpC overexpression. For all four isolates, MICs were significantly reduced in the presence of relebactam at 4 mg/L. This effect was most pronounced in K. pneumoniae with a 64-fold reduction, from 128 to 2 mg/L. In the P. aeruginosa strains, although relebactam synergistic activity was seen with imipenem, the effects were far less dramatic. At least a 2-log reduction in bacterial burden was observed in all strains following initial exposure to relebactam; however, regrowth occurred in two Pseudomonas strains at 72 h [37].
Further analysis by Livermore et al. examined the ability of relebactam to restore imipenem activity in a variety of isolates via in vitro agar dilution studies. Relebactam, at a concentration of 4 mg/L, reduced the imipenem MIC for Enterobacteriaceae with KPC carbapenemases from 16–64 mg/L to 0.12–1 mg/L [38]. A minimal effect of relebactam was seen in OXA-48 producing isolates. Isolates with an initial carbapenem MIC >64 mg/L had an MIC reduction to 16 mg/L with the addition of high-dose 32 mg/L relebactam [38]. Given the similarities between avibactam and relebactam, and the fact that the addition of relebactam to a subset of OXA-48-producing K. pneumoniae does result in restoration of the activity of imipenem, further studies are needed to determine the effect of relebactam on OXA-48 [38]. In P. aeruginosa, there was an MIC reduction in OprD-deficient strains from 16–64 mg/L to 1–4 mg/L [38]. This is likely explained by inhibition of the continued AmpC function present in OprD-deficient strains [39].
In regard to anaerobic spectrum, the addition of relebactam was found to not add to the potent anaerobic spectrum of imipenem. While imipenem resistance of bacteroides is rare, with an estimated occurrence of less than 1%, a study of 451 clinical isolates of the B. fragilis showed that the addition of relebactam did not further inhibit imipenem-resistant isolates [40].
4.4 Pharmacokinetics/Pharmacodynamics of Relebactam
In murine modeling, Mavridou et al. found that the AUC (area under the curve) was the parameter best correlating to efficacy, and confirmed that peak concentration was not a significant determinant of efficacy [41].
Although the optimal dosing regimen for relebactam has not been determined, Phase 1 studies based on murine and hollow fiber modeling have suggested relebactam doses at or above 125 mg every 6 h was able to achieve an effect pharmacokinetic/pharmacodynamic target [42]. However, certain models did suggest higher concentrations of relebactam may be required for highly resistant strains of Pseudomonas [42].
4.5 Relebactam: Clinical Studies
Clinical studies are summarized in Table 4. In the introduction to their Phase 2 data, Lucasti et al. refer to Phase 1 unpublished data that relebactam is well tolerated with intravenous administration as either a single dose of up to 1150 mg or when administered at multiple doses of 625 mg every 6 h [42]. Notably, transient liver enzyme elevation did occur in several patients who received multiple administration dosing [42].
Recently, Phase 2 clinical data from a multicenter randomized controlled double-blind study has been released in the use of imipenem–cilastatin in combination with various doses of relebactam in the treatment of cIAI. Patients were randomized in a 1:1:1 fashion into groups receiving 500 mg imipenem–cilastatin every 6 h with either relebactam 250 mg, relebactam 125 mg, or placebo. Important to note, patients with APACHE >30 and those with baseline renal dysfunction were excluded. Approximately 117 subjects were assigned to each treatment group, allowing for study power of 80% to demonstrate non-inferiority of relebactam compared to control predicated upon a non-inferiority margin of −15, and an overall 90% control group clinical response rate. Clinical response rate of all three groups, relebactam 250 mg, relebactam 125 mg, and placebo group were 99.2, 98.3, 99.1%, respectively, and treatment between all three groups was considered similar [42].
Of note, in the 277 patients in the microbiologic intention-to-treat subset, 36 patients (13%) had an infection with a Gram-negative imipenem non-susceptible organism. This was inclusive of both intermediate and fully resistant organisms. Thirty-four of these patients were able to be evaluated at follow-up, with all 34 classified as having a favorable clinical response, with 14/14, 9/9, and 11/11 patients responding in the relebactam 250 mg, relebactam 125 mg, and imipenem alone groups, respectively [42]. Notably, this subset of 34 patients was responsible for 40 bacterial isolates. Subsequent testing of these 40 imipenem non-susceptible isolates resulted in 7 which had restored susceptibility to the combination of imipenem–relebactam, but the remaining 33 remained non-susceptible despite relebactam administration [42]. Interestingly, 21 of these isolates were Proteus sp., with the majority Proteus mirabilis, of which only 2 had restored activity of imipenem in the presence of relebactam. The remainder of isolates were predominantly Pseudomonas, Stenotrophomonas, and Acinetobacter spp.; all species with either intrinsic or a more diverse range of mechanisms for carbapenem resistance.
A second Phase 2 multicenter double-blinded study (Protocol 7655-003) has recently been completed for the evaluation of relebactam 250 mg and imipenem in the treatment of cUTI. Initial reported data was that a favorable microbiologic response occurred in three groups of relebactam 250 mg with imipenem 500 mg, relebactam 125 mg with imipenem 500 mg, and placebo with imipenem 500 mg in 95.5, 98.6, and 98.7%, respectively [43]. This met criteria for non-inferiority.
Based on Phase 2 data, initiation of two Phase 3 trials is being planned to compare the efficacy of imipenem–relebactam versus colistin in combination with imipenem, in particular for the treatment of imipenem-resistant bacterial infection [44]. A second Phase 3 study, is planned to evaluate imipenem–relebactam to piperacillin–tazobactam for treatment of patients with HABP/VABP [44].
4.6 Adverse Effects of Relebactam
Clinical data from Phase 1 trials have not been published, although in general data presented at American Society for Microbiology conferences, favorable tolerability has been shown. In the Phase 2 trial reported by Lucasti et al., the most common adverse events, those with an incidence >5%, were diarrhea, nausea, and vomiting, which were similar in occurrence rate between relebactam- and placebo-controlled groups [42].
5 Proposed Roles in Therapy
All three agents described in this review—in combination with a β-lactam antibiotic—may prove useful in the treatment of patients with infections caused by MDR Gram-negative bacteria such as carbapenem-resistant Enterobacteriacae and MDR P. aeruginosa. An important question that remains to be answered is how these novel β-lactamase/β-lactam combinations will perform as compared to current BAT in the treatment of carbapenem-resistant bacterial infections. In addition, whether the use of any one of these agents results in superior clinical outcomes as compared to the other novel agents is unclear. Also, the comparative threshold for resistance development after more widespread clinical use is an issue deserving of future studies. Resistance mutations to ceftazidime–avibactam have already been described. This highlights the importance of judicious use and the tenuous balance that exists with resistance selection.
Unfortunately, approval and marketing of these antibiotics is not accompanied by a timely approval of standardized methods of susceptibility testing. The lack of these methods with availability of only “research-only” methods for susceptibility testing limits the appropriate use of these important novel agents. Furthermore, a lack of CLSI breakpoints further restricts the interpretation of susceptibility testing for these new agents.
The clinical impact of differences in microbiologic spectrum will depend on the specific patient and infection type. For instance, avibactam shows in vitro activity against Enterobacteriacae strains with OXA-48 enzymes, whereas both vaborbactam and relebactam did not [4, 38]. However, the companion drug of avibactam, ceftazadime, is susceptible to efflux mechanisms to a greater extent than imipenem [38, 45]. A mutation of outer membrane porins or porin downregulation has a pronounced effect on imipenem susceptibility [46]. Meanwhile, meropenem exhibits less dramatic MIC increases with OprD mutations due to more rapid porin transit, but the combination of OprD downregulation and, in particular, expression of MexAB-OprM efflux results in resistance [45, 47].
This aspect leads to a critical point, specifically, each inhibitor’s role and spectrum of activity is closely dependent upon its companion agent. In examining differences for avibactam, vaborbactam, and relebactam, their co-formulations with ceftazidime, meropenem, and imipenem, respectively, have to be considered. These companion agents have important differences with regard to spectrum and pharmacokinetic considerations. All three combination regimens should primarily be considered as therapeutic options for drug-resistant aerobic Gram-negative rod infections. Ceftazidime has moderate anti-streptococcal, very limited anti-staphylococcal, no anti-enterococcal, and unreliable anaerobic activity. This lack of broader spectrum activity may be a positive attribute to provide less antibiotic pressure on the microbiome. Meropenem, in contrast, has excellent broad spectrum Gram-negative and anaerobic activity, with more modest anti-staphylococcal and enterococcal activity. Imipenem, has more reliable anti-enterococcal activity in conjunction with anaerobic activity, whereas the Gram-negative spectrum may be marginally less broad than meropenem. Of key consideration, Proteus, Providencia, and Morganella, based on current CLSI breakpoints, have reduced imipenem susceptibility. Therefore, these differences may have a significant impact in the scenario of a polymicrobial infection.
Another important distinction is that although extensive clinical experience exists for each of ceftazidime, meropenem, and imipenem, there is a dearth of clinical treatment experience with all of the novel inhibitors. Thus far, all Phase 3 studies for these agents have been for the indication of cUTI and cIAI. Data on pneumonia and bacteremia—the infections that carry the highest mortality risk with CRE—are limited [48]. Alveolar distribution and cerebrospinal fluid penetration have not been adequately studied in critically ill patients. Therefore, the performance of these agents, including appropriate dosing for central nervous system infections has not been clarified. Also, although data exist in healthy adult volunteers in Phase 1 studies, in critically ill patients, altered physiology, renal function, and concurrent tissue injury may drastically affect drug tissue penetrance and ultimately clinical performance. Additionally, Phase 1 and 2 studies have generally excluded patients with severe renal insufficiency.
Likewise, no sufficiently powered pathogen-specific trials with inferential statistics have been performed. This is particularly important as the majority of studies have included patients with infections caused mostly by carbapenem-susceptible Enterobacteriacae. Larger studies will be needed to study drug efficacy against pathogens with complex multi-faceted resistance mechanisms such as Pseudomonas.
None of the three novel inhibitors have activity for class B carbapenemases. In a 10-month study of bloodstream isolates at Texas MD Anderson Cancer Center, 7/11 (64%) CRE were ceftazidime–avibactam resistant, with 6 harboring an MBL phenotype [49]. This underscores the caveat that the use of these agents must be in conjunction with knowledge of patient risk factors and local epidemiology. A reliable commercial method for sensitivity testing must also be available in order to fully utilize these agents, particularly because MDR Enterobacteriaceae for which they will predominantly be employed often have multifaceted resistance mechanisms. Potentially, new microbiology techniques and the increasing availability of bacterial sequencing will allow for the identification of organisms based on their production of KPC, VIM, or NDM. As such, it will be critical to determine how to integrate diagnostic technology into real-time clinical feedback as to which patients may be candidates for novel inhibitor therapy.
Other combinations of inhibitor agents and β-lactams have also been postulated to be of clinical utility and may warrant future study. There has been a focus on the combination of ceftaroline and avibactam for cases of diabetic foot ulcer and polymicrobial wound infection [50]. This combination would potentially provide effective activity for anaerobic bacteria, drug-resistant Gram-negative bacteria, and methicillin-resistant staphylococci simultaneously [50, 51]. Ceftaroline–avibactam was studied in cUTI in a Phase 2 study that was recently completed (ClinicalTrials.gov Identifier: NCT01281462). Likewise, there is promising in vitro data utilizing the combination of aztreonam and avibactam. This combination resulted in a reduction in carbapenem MIC for CRE, and MBL-containing pathogens were reduced [4, 11, 52]. The hypothesis is that MBL-producing pathogens, often concurrently produce extended spectrum or AmpC β-lactamases, which can be inhibited by avibactam, while aztreonam intrinsically evades hydrolysis by MBLs and can thereby still exert an antimicrobial effect [4, 52].
6 Conclusion
All three agents do share common features and limited treatment experience. Limited trials have shown comparable clinical outcomes to comparators. However, randomized data from patients with carbapenem-resistant bacterial infections are not yet available. In addition, more widespread use in the future will undoubtedly lead to increasing rates of resistance development, as has already been shown for avibactam. As such, it will be the responsibility of the clinical community to ensure judicious use and to determine the optimal settings in which to employ these new agents. A renewed focus on combining antimicrobial stewardship and infection control measures in conjunction with medicinal therapy may offer the greatest benefit in prolonging the lifespan of these therapeutic options [6].
References
Öztürk H, Ozkirimli E, Özgür A. Classification of Beta-lactamases and penicillin binding proteins using ligand-centric network models. PLoS One. 2015;10(2):e0117874.
Bush K, Jacoby GA. Updated functional classification of beta-lactamases. Antimicrob Agents Chemother. 2010;54(3):969–76.
Bonomo RA. Beta-Lactamases: a focus on current challenges. Cold Spring Harb Perspect Med. 2017;7(1):a025239.
Drawz SM, Papp-Wallace KM, Bonomo RA. New β-lactamase inhibitors: a therapeutic renaissance in an MDR world. Antimicrob Agents Chemother. 2014;58(4):1835–46.
van Duin D, Doi Y. The global epidemiology of carbapenemase-producing Enterobacteriaceae. Virulence. 2016;1–10.
Wong D, Spellberg B. Leveraging antimicrobial stewardship into improving rates of carbapenem-resistant enterobacteriaceae. Virulence. 2016;1–8.
Stachyra T, Péchereau MC, Bruneau JM, Claudon M, Frère JM, Miossec C, et al. Mechanistic studies of the inactivation of TEM-1 and P99 by NXL104, a novel non-beta-lactam beta-lactamase inhibitor. Antimicrob Agents Chemother. 2010;54(12):5132–8.
Zhanel GG, Lawson CD, Adam H, Schweizer F, Zelenitsky S, Lagacé-Wiens PR, et al. Ceftazidime–avibactam: a novel cephalosporin/β-lactamase inhibitor combination. Drugs. 2013;73(2):159–77.
van Duin D, Bonomo RA. Ceftazidime/avibactam and ceftolozane/tazobactam: second-generation β-lactam/β-lactamase inhibitor combinations. Clin Infect Dis. 2016;63(2):234–41.
de Jonge BL, Karlowsky JA, Kazmierczak KM, Biedenbach DJ, Sahm DF, Nichols WW. In vitro susceptibility to ceftazidime–avibactam of carbapenem-nonsusceptible enterobacteriaceae isolates collected during the INFORM Global Surveillance Study (2012–2014). Antimicrob Agents Chemother. 2016;60(5):3163–9.
Livermore DM, Mushtaq S, Warner M, Zhang J, Maharjan S, Doumith M, et al. Activities of NXL104 combinations with ceftazidime and aztreonam against carbapenemase-producing Enterobacteriaceae. Antimicrob Agents Chemother. 2011;55(1):390–4.
Papp-Wallace KM, Winkler ML, Taracila MA, Bonomo RA. Variants of β-lactamase KPC-2 that are resistant to inhibition by avibactam. Antimicrob Agents Chemother. 2015;59(7):3710–7.
Shields RK, Potoski BA, Haidar G, Hao B, Doi Y, Chen L, et al. Clinical outcomes, drug toxicity and emergence of ceftazidime–avibactam resistance among patients treated for carbapenem-resistant Enterobacteriaceae infections. Clin Infect Dis. 2016;63(12):1615–8.
Shields RK, Chen L, Cheng S, Chavda KD, Press EG, Snyder A, et al. Emergence of ceftazidime–avibactam resistance due to plasmid-borne blaKPC-3 mutations during treatment of carbapenem-resistant Klebsiella pneumoniae infections. Antimicrob Agents Chemother. 2017;61(3). pii: e02097-16.
Sader HS, Huband MD, Castanheira M, Flamm RK. Antimicrobial susceptibility of Pseudomonas aeruginosa: results from four years (2012–2015) of the International Network for Optimal Resistance Monitoring (INFORM) Program in the United States. Antimicrob Agents Chemother. 2017;61(3). pii: e02252-16.
Citron DM, Tyrrell KL, Merriam V, Goldstein EJ. In vitro activity of ceftazidime-NXL104 against 396 strains of beta-lactamase-producing anaerobes. Antimicrob Agents Chemother. 2011;55(7):3616–20.
Merdjan H, Tarral A, Das S, Li J. Phase 1 study assessing the pharmacokinetic profile and safety of avibactam in patients with renal impairment. J Clin Pharmacol. 2017;57(2):211–8.
Nicolau DP, Siew L, Armstrong J, Li J, Edeki T, Learoyd M, et al. Phase 1 study assessing the steady-state concentration of ceftazidime and avibactam in plasma and epithelial lining fluid following two dosing regimens. J Antimicrob Chemother. 2015;70(10):2862–9.
Vazquez JA, Gonzalez Patzan LD, Stricklin D, Duttaroy DD, Kreidly Z, Lipka J, et al. Efficacy and safety of ceftazidime–avibactam versus imipenem–cilastatin in the treatment of complicated urinary tract infections, including acute pyelonephritis, in hospitalized adults: results of a prospective, investigator-blinded, randomized study. Curr Med Res Opin. 2012;28(12):1921–31.
Lucasti C, Popescu I, Ramesh MK, Lipka J, Sable C. Comparative study of the efficacy and safety of ceftazidime/avibactam plus metronidazole versus meropenem in the treatment of complicated intra-abdominal infections in hospitalized adults: results of a randomized, double-blind, Phase II trial. J Antimicrob Chemother. 2013;68(5):1183–92.
Wagenlehner FM, Sobel JD, Newell P, Armstrong J, Huang X, Stone GG, et al. Ceftazidime–avibactam versus doripenem for the treatment of complicated urinary tract infections, including acute pyelonephritis: RECAPTURE, a Phase 3 Randomized Trial Program. Clin Infect Dis. 2016;63(6):754–62.
Mazuski JE, Gasink LB, Armstrong J, Broadhurst H, Stone GG, Rank D, et al. Efficacy and safety of ceftazidime–avibactam plus metronidazole versus meropenem in the treatment of complicated intra-abdominal infection: results from a randomized, controlled, double-blind, phase 3 program. Clin Infect Dis. 2016;62(11):1380–9.
Food and Drug Administration. Briefing Package NDA 206494 ceftazidime–avibactam. 2014; pp. 1–55.
Carmeli Y, Armstrong J, Laud PJ, Newell P, Stone G, Wardman A, et al. Ceftazidime–avibactam or best available therapy in patients with ceftazidime-resistant Enterobacteriaceae and Pseudomonas aeruginosa complicated urinary tract infections or complicated intra-abdominal infections (REPRISE): a randomised, pathogen-directed, phase 3 study. Lancet Infect Dis. 2016;16(6):661–73.
King M, Heil E, Kuriakose S, Bias T, Huang V, El-Beyrouty C, et al. Outcomes with ceftazidime/avibactam in patients with carbapenem-resistant enterobacteriaceae (CRE) infections: a multi-center study. Open Forum Infect Dis. 2016;3(Supplement 1):2047.
Hecker SJ, Reddy KR, Totrov M, Hirst GC, Lomovskaya O, Griffith DC, et al. Discovery of a cyclic boronic acid β-lactamase inhibitor (RPX7009) with utility vs class A serine carbapenemases. J Med Chem. 2015;58(9):3682–92.
Castanheira M, Rhomberg PR, Flamm RK, Jones RN. Effect of the beta-lactamase inhibitor vaborbactam combined with meropenem against serine carbapenemase-producing enterobacteriaceae. Antimicrob Agents Chemother. 2016;60(9):5454–8.
Livermore DM, Mushtaq S. Activity of biapenem (RPX2003) combined with the boronate beta-lactamase inhibitor RPX7009 against carbapenem-resistant Enterobacteriaceae. J Antimicrob Chemother. 2013;68(8):1825–31.
Lapuebla A, Abdallah M, Olafisoye O, Cortes C, Urban C, Quale J, et al. Activity of meropenem combined with RPX7009, a novel β-lactamase inhibitor, against gram-negative clinical isolates in New York City. Antimicrob Agents Chemother. 2015;59(8):4856–60.
Goldstein EJ, Citron DM, Tyrrell KL, Merriam CV. In vitro activity of biapenem plus RPX7009, a carbapenem combined with a serine beta-lactamase inhibitor, against anaerobic bacteria. Antimicrob Agents Chemother. 2013;57(6):2620–30.
Wenzler E, Gotfried MH, Loutit JS, Durso S, Griffith DC, Dudley MN, et al. Meropenem-RPX7009 concentrations in plasma, epithelial lining fluid, and alveolar macrophages of healthy adult subjects. Antimicrob Agents Chemother. 2015;59(12):7232–9.
Griffith D, Loutit J, Morgan E, Durso S, Dudley M. A phase 1 study of the safety, tolerability, and pharmacokinetics of the beta-lactamase inhibitor RPX7009 in healthy adult subjects. 24th European Congress of Clinical Microbiology and Infectious Diseases. Barcelona, Spain; 2014.
Meanwell C, Loutit J, Dudley M. TANGO 1 Phase 3 trial results; conference call, PDF; 2016 June 27.
BusinessWire. The Medicines Company Announces Positive Top- Line Results for Phase 3 TANGO 1 Clinical Trial of CARBAVANCE. In: Visioli C, editor.: The Medicines Company; 2016. http://www.themedicinescompany.com/investors/news/medicines-company-announces-positive-top-line-results-phase-3-tango-1-clinical-trial. Accessed 2 Jan 2017.
Blizzard TA, Chen H, Kim S, Wu J, Bodner R, Gude C, et al. Discovery of MK-7655, a β-lactamase inhibitor for combination with Primaxin®. Bioorg Med Chem Lett. 2014;24(3):780–5.
Lapuebla A, Abdallah M, Olafisoye O, Cortes C, Urban C, Landman D, et al. Activity of imipenem with relebactam against gram-negative pathogens from New York City. Antimicrob Agents Chemother. 2015;59(8):5029–31.
Hirsch EB, Ledesma KR, Chang KT, Schwartz MS, Motyl MR, Tam VH. In vitro activity of MK-7655, a novel β-lactamase inhibitor, in combination with imipenem against carbapenem-resistant Gram-negative bacteria. Antimicrob Agents Chemother. 2012;56(7):3753–7.
Livermore DM, Warner M, Mushtaq S. Activity of MK-7655 combined with imipenem against Enterobacteriaceae and Pseudomonas aeruginosa. J Antimicrob Chemother. 2013;68(10):2286–90.
Livermore DM. Interplay of impermeability and chromosomal beta-lactamase activity in imipenem-resistant Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1992;36(9):2046–8.
Snydman DR, Jacobus NV, McDermott L. In vitro evaluation of the activity of imipenem/relebactam (imipenem/mk-7655) against 451 recent clinical isolates of bacteroides group and related species. Antimicrob Agents Chemother. 2016;60(10):6393–7.
Mavridou E, Melchers RJ, van Mil AC, Mangin E, Motyl MR, Mouton JW. Pharmacodynamics of imipenem in combination with β-lactamase inhibitor MK7655 in a murine thigh model. Antimicrob Agents Chemother. 2015;59(2):790–5.
Lucasti C, Vasile L, Sandesc D, Venskutonis D, McLeroth P, Lala M, et al. Phase 2, dose-ranging study of relebactam with imipenem/cilastatin in subjects with complicated intra-abdominal infection. Antimicrob Agents Chemother. 2016;60(10):6234–43.
Eisele P, Consalvo R. Results of phase 2 study of Merck’s Investigational beta-lactamase inhibitor relebactam in combination with imipenem/cilastatin presented at ASM microbe. June 20 2016. http://investors.merck.com/investors/financial-news/press-release-details/2016/Results-of-Phase-2-Study-of-Mercks-Investigational-Beta-Lactamase-Inhibitor-Relebactam-in-Combination-with-ImipenemCilastatin-Presented-at-ASM-Microbe/default.aspx.
Li D, McConnell I. Results of phase 2 study of Merck’s investigational beta-lactamase inhibitor relebactam. In: Presented at ICAAC/ICC 2015; 2015 http://www.mercknewsroom.com/news-release/research-and-development-news/results-phase-2-study-mercks-investigational-beta-lactama.
Köhler T, Michea-Hamzehpour M, Epp SF, Pechere JC. Carbapenem activities against Pseudomonas aeruginosa: respective contributions of OprD and efflux systems. Antimicrob Agents Chemother. 1999;43(2):424–7.
Rodríguez-Martínez JM, Poirel L, Nordmann P. Molecular epidemiology and mechanisms of carbapenem resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2009;53(11):4783–8.
Li H, Luo YF, Williams BJ, Blackwell TS, Xie CM. Structure and function of OprD protein in Pseudomonas aeruginosa: from antibiotic resistance to novel therapies. Int J Med Microbiol. 2012;302(2):63–8.
Hauck C, Cober E, Richter SS, Perez F, Salata RA, Kalayjian RC, et al. Spectrum of excess mortality due to carbapenem-resistant Klebsiella pneumoniae infections. Clin Microbiol Infect. 2016;22(6):513–9.
Aitken SL, Tarrand JJ, Deshpande LM, Tverdek FP, Jones AL, Shelburne SA, et al. High rates of nonsusceptibility to ceftazidime–avibactam and identification of new delhi metallo-β-lactamase production in enterobacteriaceae bloodstream infections at a major cancer center. Clin Infect Dis. 2016;63(7):954–8.
Goldstein EJ, Citron DM, Merriam CV, Tyrrell KL. Comparative in vitro activity of ceftaroline, ceftaroline–avibactam, and other antimicrobial agents against aerobic and anaerobic bacteria cultured from infected diabetic foot wounds. Diagn Microbiol Infect Dis. 2013;76(3):347–51.
Werth BJ, Rybak MJ. Ceftaroline plus avibactam demonstrates bactericidal activity against pathogenic anaerobic bacteria in a one-compartment in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother. 2014;58(1):559–62.
Marshall S, Hujer AM, Rojas LJ, Papp-Wallace KM, Humphries RM, Spellberg B, et al. Can ceftazidime/avibactam and aztreonam overcome beta-lactam resistance conferred by metallo-beta-lactamases in Enterobacteriaceae? Antimicrob Agents Chemother. 2017 Feb 6. pii: AAC.02243-16.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
DvD was supported by R21AI114508-1 from the National Institute of Allergy and Infectious Diseases.
Conflict of interest
No potential conflicts D.W.
Potential conflicts of interest: D.v.D. Actavis, Allergan, Achaogen, Shionogi, Tetraphase, Sanofi-Pasteur, MedImmune, Astellas, Advisory Board. Steris Inc., Research funding. Scynexis Research funding.
Additional information
The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
Rights and permissions
About this article

Cite this article
Wong, D., van Duin, D. Novel Beta-Lactamase Inhibitors: Unlocking Their Potential in Therapy. Drugs 77, 615–628 (2017). https://doi.org/10.1007/s40265-017-0725-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0725-1