
Abstract
Finerenone, a selective and nonsteroidal antagonist of the mineralocorticoid receptor, has received regulatory approval with the indication of cardiorenal protection in patients with chronic kidney disease associated with type 2 diabetes. It is rapidly and completely absorbed and undergoes first-pass metabolism in the gut wall and liver resulting in a bioavailability of 43.5%. Finerenone can be taken with or without food. The pharmacokinetics of finerenone are linear and its half-life is 2 to 3 h in the dose range of up to 20 mg. Cytochrome P450 (CYP) 3A4 (90%) and CYP2C8 (10%) are involved in the extensive biotransformation of finerenone to pharmacologically inactive metabolites, which are excreted via both renal (80%) and biliary (20%) routes. Moderate or severe renal impairment, or moderate hepatic impairment result in area-under-the-curve increases of finerenone (< 40%), which do not require a dose adjustment per se, as the starting dose is based on estimated glomerular filtration rate (eGFR) and titrated according to serum potassium levels and eGFR decline. No relevant effects of age, sex, body size or ethnicity on systemic finerenone exposure were identified. Modulators of CYP3A4 activity were found to affect finerenone exposure, consistent with its classification as a sensitive CYP3A4 substrate. Serum potassium should be monitored during drug initiation or dosage adjustment of either a moderate or weak CYP3A4 inhibitor or finerenone, and the dose of finerenone should be adjusted as appropriate. Its use with strong inhibitors is contraindicated and strong or moderate inducers of CYP3A4 should be avoided. Finerenone has no potential to affect relevant CYP enzymes and drug transporters.
Plain Language Summary
Finerenone is a drug that is used to treat patients with chronic kidney disease and type 2 diabetes. Many of these patients take several medicines to treat other conditions. This review summarizes several studies showing the suitability of finerenone for these patients. Taken as a daily tablet, the dose circulates in the body before being quickly removed. The age, sex, body weight, and ethnicity of a patient do not affect dosing. As finerenone can cause an increase of serum potassium levels, potassium levels and kidney function should be measured before a patient starts treatment. The starting dose will depend on a patient’s kidney function, with the dose changed according to potassium levels and changes in kidney function. A protein called cytochrome P450 3A4 (CYP3A4) is key to removing finerenone from the body. Anyone taking medicines that strongly inhibit CYP3A4 should not take finerenone. Serum potassium levels should be measured before starting finerenone or changing the dose of either finerenone or ‘moderate’ or ‘weak’ CYP3A4 inhibitors, with the dose of finerenone adjusted as appropriate. Finerenone should not be taken alongside drugs that result in ‘moderate’ or ‘strong’ increases in CYP3A4 activity. In patients with moderate hepatic impairment, potassium should be monitored and finerenone doses be adjusted as appropriate. Finerenone is not expected to affect other drugs. Finerenone slows decline in kidney function, a treatment effect associated with reducing urine albumin. Potassium level-guided starting dose and dose changes support finerenone being effectively used and well tolerated in patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Finerenone is an approved medication used to treat patients with chronic kidney disease associated with type 2 diabetes. |
The pharmacokinetic profile of finerenone was extensively characterized in healthy volunteers and in patients. The relationship of pharmacokinetics and pharmacodynamics was assessed in patients. |
Recommendations on the initiation and dosing of finerenone in patients are supported by multiple studies and analyses, which are reported in this review. |
1 Introduction
Aldosterone is synthesized in the adrenal cortex and its synthesis and release are stimulated by low sodium or high potassium levels in the plasma or directly via angiotensin II [1]. The physiological role of aldosterone as a key regulator of electrolyte homeostasis and control of blood pressure is to promote the retention of sodium and the loss of magnesium and potassium. Aldosterone acts via the specific nuclear mineralocorticoid receptor (MR), which is mainly expressed in the heart, kidneys, and blood vessels [1]. The MR belongs to the superfamily of nuclear hormone receptors, which translocate from the cytosol to the nucleus upon ligand binding [1, 2].
Chronically elevated aldosterone release and pathophysiological MR activation contribute to various deleterious molecular mechanisms in the development of cardiorenal diseases such as chronic kidney disease (CKD) and heart failure (HF). The available steroidal MR antagonists (MRAs) spironolactone and eplerenone have been shown to be effective in reducing cardiovascular mortality and morbidity in patients with chronic HF and reduced left ventricular ejection fraction (HFrEF). However, they remain underutilized due to the associated risks of inducing hyperkalemia and worsening kidney function [1].
Finerenone (Kerendia®, Firialta®, previous nomenclature BAY 94-8862) is a compound with a nonsteroidal structure (see Fig. 1) that exhibits higher selectivity for the MR than all other steroid hormone receptors and a high binding affinity (half-maximal inhibitory concentration 18 nM). Unlike spironolactone and eplerenone, finerenone binds to the MR as a “bulky” antagonist to inhibit the transcriptional cofactor recruitment implicated in the expression of hypertrophic, proinflammatory, and profibrotic genes. With regard to MR inhibition, finerenone is more potent than eplerenone, at least as potent as spironolactone, and more selective compared with spironolactone or eplerenone [3].

The structure of finerenone
In preclinical models of kidney injury, finerenone was associated with kidney-protective effects mediated through downregulation of renal proinflammatory/profibrotic genes in response to kidney injury, which were paralleled by a dose-dependent improvement in albuminuria. In animal models of cardiac fibrosis, finerenone led to regression of cardiac hypertrophy, accompanied by a reduction in albuminuria and other markers of inflammation and fibrosis. These beneficial effects were observed at dosages not inducing significant hemodynamic effects, suggesting that the protection of the heart and kidney with finerenone is independent of such effects. Preclinical studies with finerenone also provided preliminary evidence of cardiorenal protection with a minimal impact on serum potassium levels [4].
The Phase I program comprised 28 studies in healthy subjects and subjects with renal or hepatic impairment [5,6,7]. The Phase II clinical trial program to investigate the safety and efficacy of finerenone included > 2000 patients with HFrEF, CKD, and/or type 2 diabetes (T2D) or patients with CKD and T2D [8,9,10]. In the double-blind, placebo-controlled Phase IIb study, ARTS-DN, in 823 randomized patients with T2D and persistent albuminuria, finerenone reduced urinary albumin to creatinine ratio (UACR) in a dose-dependent fashion [8]. The Phase III clinical trial program of finerenone in patients with CKD and T2D comprised two double-blind, randomized, placebo-controlled studies, FIDELIO-DKD and FIGARO-DKD [11,12,13,14]. In the FIDELIO-DKD trial, finerenone compared to placebo reduced the occurrence of the primary composite kidney endpoint, defined as the time to onset of kidney failure, sustained ≥ 40% decline in estimated glomerular filtration rate (eGFR) from baseline or death from renal causes by 18% (hazard ratio [HR]: 0.82; 95% confidence interval [CI]: 0.73–0.93) [11]. In the FIGARO-DKD trial, finerenone compared to placebo lowered the occurrence of the primary composite cardiovascular outcome, defined as the time to cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, or hospitalization for HF by 13% (HR: 0.87; 95% CI 0.76–0.98) [12]. The frequency of hyperkalemia leading to discontinuation of the trial regimen was 2.3% versus 0.9% in FIDELIO-DKD and 1.2% versus 0.4% in FIGARO-DKD under finerenone versus placebo, respectively, while the frequency of other adverse events was similar between the finerenone and placebo groups [11, 12].
Finerenone has received regulatory approval with the indication of cardiorenal protection in adults with CKD associated with T2D [4].
In this review, we focus on the pharmacokinetic (PK) profile of finerenone (Sect. 3), how it is affected by intrinsic (Sect. 4) and extrinsic factors (Sect. 5.1), and summarize how PKs correlate with pharmacodynamic (PD) effects of the drug (Sect. 6). This is complemented by a summary of studies investigating the effects of finerenone on the PKs of other drugs (Sect. 5.2). In addition to dedicated clinical Phase I studies followed by noncompartmental analysis of PK data, the PKs of finerenone were also evaluated with model-informed approaches, i.e., population PK (popPK) and physiologically based PK (PBPK) analyses. PopPK evaluations were conducted based on Phase I, II, and III data and mainly supported the characterization of the PKs in the target population, i.e., patients with CKD and T2D, compared to healthy volunteers as well as the characterization of intrinsic factors and will be discussed as part of Sect. 4. Physiologically based PK evaluations complemented the assessment of finerenone as the victim in drug-drug interactions (DDIs) and will be described as part of Sect. 5.1.2.
2 Physiochemical Properties
Finerenone is an enantiomerically pure dihydronaphthyridine derivative with a log D value of 2.40, which is relatively more polar compared to the steroidal MRAs spironolactone and eplerenone (log D of 3.13 and 3.03, respectively) [1]. Owing to its basic nature, the solubility of finerenone decreases continuously with increasing pH, rendering finerenone a low-solubility compound with an equilibrium solubility of 0.024 mg/mL at pH 6.8 [5] according to the criteria of the Biopharmaceutical Classification System (BCS) [15]. Considering this, together with its high apparent permeability in Caco-2 cells [5], finerenone can be classified as a compound belonging to class II of the BCS [6].
3 Pharmacokinetic Properties
3.1 Absorption and the Effect of Food
In humans, finerenone is completely absorbed after oral administration, as indicated by the complete recovery of radioactivity in excreta almost entirely in the form of metabolites with only negligible amounts of unchanged drug in a mass-balance study (Fig. 2) [16]. Absorption in the fasted state is rapid, with time to peak drug concentration (tmax) typically in the range of 0.5 to 1.25 hours (h) [5] after administration of a finerenone tablet releasing micronized drug substance. Its absolute bioavailability as a 5-mg tablet, determined in comparison to a 1-mg tablet as an intravenous infusion, was equal to 43.5%, resulting from first-pass metabolism in the gut wall and liver. Estimated extraction ratios of 0.425 in the gut wall (EG) and 0.244 in the liver (EH) indicate a quantitatively more important contribution of first-pass metabolism in the gut wall to total bioavailability compared to the liver [17].

Summary of finerenone absorption, excretion profile, distribution, and clearance. AE,fec amount of drug excreted in feces, AE,ur amount of drug excreted in urine, CLsys systemic clearance, CLR renal clearance, CYP2C8 cytochrome P450 2C8, CYP3A4 cytochrome P450 3A4, fm fractions metabolized, h hour, Vss steady state volume of distribution
The absorption of finerenone in solution shows profound differences depending on the nature of the solvent. Taken as an oral aqueous solution in the fasted state, finerenone was rapidly absorbed with a median tmax of 0.5 h, and both area-under-the-curve (AUC) and peak drug concentration (Cmax) were comparable to the tablet formulation [16]. This contrasts with data obtained from a study using a solution of finerenone in macrogol (polyethylene glycol [PEG]) 400, which indicate a reduction in oral bioavailability of about 50% compared to the tablet formulation, although the absorption process was also rapid (tmax of 0.5 h as a 10 mg PEG solution). The detrimental effect of PEG 400 on bioavailability is believed to be due to shortened small intestinal transit time caused by osmotic effects with increased fluid volume and enhanced peristalsis in the small intestinal lumen [18]. This can be a confounding factor for investigational compounds formulated as PEG solutions in the context of early clinical trials.
Intake in the fed condition (after a high-fat, high-calorie meal) showed the expected retarding effects on the rate of absorption, resulting in a Cmax decrease of 19% (20-mg tablet) or 32% (10-mg tablet), and prolonged tmax (2.5 h vs 0.75 h, respectively), compared to administration in the fasted condition. Minor increases in finerenone AUC of 21% (20-mg tablet) or 10% (10-mg tablet) after intake in the fed state may reflect alterations in first-pass clearance or food-induced changes in fraction unbound, given the complete absorption of the drug in the fasted state [18, 19].
Dose proportionality in finerenone AUC and Cmax was demonstrated in two studies, between the 1.25-, 2.5-, 5.0-, 7.5- and 10-mg tablets, and between the 10-mg and 20-mg tablets [19]. Both studies together confirmed linear PKs between 1.25 mg and the highest intended therapeutic dose of 20 mg. A third exploratory study indicated dose-proportionality between 10-mg and 80-mg (8 × 10 mg) tablets, i.e., up to a dose fourfold higher than the highest therapeutic dose [18].
3.2 Distribution
The ratio of finerenone concentrations in human blood to plasma was equal to 0.935 [5]. The major binding protein of finerenone in human plasma was albumin, with α1-acidic glycoprotein, low density lipoprotein, and γ-globulins being of less relevance [5, 6]. In vitro, 91.7% of finerenone in human plasma was bound to proteins corresponding to a fraction unbound (fu) of 8.3% [5]. The fu determined ex vivo was slightly higher (10.6% in the healthy controls of a study in renal impairment, and 11.5% in the control subjects of a study in hepatic impairment). Protein binding of finerenone was insensitive to the disease states mild, moderate, or severe renal impairment and mild or moderate hepatic impairment [20, 21] (see Sects. 4.5 and 4.6). The volume of distribution at steady-state of finerenone was 52.6 L [17].
An interesting aspect of distribution was investigated when finerenone PKs after oral administration were compared between venous plasma and capillary plasma sampled simultaneously in the finger pad. Capillary concentrations in the absorption phase were up to 71% higher than venous concentrations, while the difference disappeared during the distribution and elimination phase. This is believed to reflect the arterio-venous difference in drug concentration before reaching a distribution equilibrium. Finger-prick-based blood is a mixture of arterial, venous, and capillary blood, with a greater ratio of arterial to venous blood due to the greater pressure in arterioles and the arterial limb of capillaries [22].
Preclinical studies with radioactively labeled finerenone in rats showed no irreversible binding or retention. Radioactivity concentrations in the brain were very low [6]. The distribution of finerenone into cardiac and kidney tissues of healthy rats was balanced, as demonstrated using quantitative whole-body autoradiography with carbon-14–labeled finerenone [3].
3.3 Metabolism and Elimination
Two main biotransformation pathways were identified for finerenone: (i) oxidation of the dihydronaphthyridine to the naphthyridine derivative M-1, followed by hydroxylation (M-2) and oxidation to a carboxylic acid (M-3) as a major clearance pathway; and (ii) a presumably intermediate epoxidation with subsequent hydrolysis to M-4 and M-5. The metabolites of finerenone have no on-target pharmacological activity [16].
Finerenone is a pure S-enantiomer and there is no racemization, as only this isomer was found in human plasma. The predominant human metabolites M-1, M-2, and M-3 exhibit axial chirality with a clear preponderance of one atropisomer (> 80%) observed for each metabolite in plasma [6].
CYP3A4 and CYP2C8 were relevant contributors to the metabolic clearance of finerenone. Based on in vitro studies, and corresponding clinical DDI studies with inhibitors of the respective CYP enzymes, fractions metabolized of 0.88–0.89 (CYP3A4) and 0.1 (CYP2C8) were estimated [16]. CYP3A4 expressed in the gut wall contributes to the first-pass conversion of finerenone to its metabolite M-1 via oxidation of the dihydronaphthyridine to a naphthyridine moiety [17].
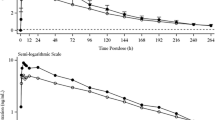
Following oral administration of finerenone, 79.6% and 21.2% of the dose was recovered in urine and feces, respectively, i.e., recovery in the excreta was complete (101%). Excretion of unchanged finerenone (0.825% in urine and 0.184% in feces) represented about 1% of the administered dose (see Fig. 2) [16].
The renal clearance of finerenone (0.310–0.473 L/h in healthy subjects) is indicative of glomerular filtration as mechanism [17, 18, 20, 21]. The blood clearance determined in healthy subjects after intravenous administration was 24.8 L/h, which classifies finerenone as a low-clearance compound with regard to hepatic clearance [17]. Terminal elimination half-life (t1/2) of finerenone was within 2 to 3 h in the dose range up to 20 mg.
A small deviation from strict time-linearity was observed in healthy subjects in an approximate 10% increase in AUC after once-daily (OD) administration, which is believed to be due to CYP3A4 auto-inhibition [6]. Once a steady-state of this effect was achieved, the PKs of finerenone became time-linear, as confirmed in patients after OD administration for up to 3 months in Phase II studies [23]. As eGFR is a covariate for finerenone PKs and patients’ eGFR was slowly deteriorating even under finerenone treatment, this disease progression led to an additional time-nonlinearity in the long term, as was seen in FIDELIO-DKD, where time-varying eGFR described the data better than baseline eGFR [24].
Low intra-individual variability with percent coefficients of variation (CVs) of 12% for oral finerenone clearance CL/F, and 22% for Cmax of finerenone was observed, accompanied by low-to-moderate inter-individual variability (CV < 36% for both CL/F and Cmax) [18].
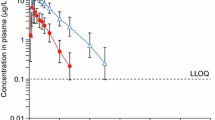
Plasma concentration-time data observed in FIDELIO-DKD along with the popPK model-predicted profiles are shown in Fig. 3 in a prediction-corrected visual predictive check based on model parameters shown in Table 3 [24].

Adapted from van den Berg, Ruppert et al [24] under the terms of the CC-BY-NC license (http://creativecommons.org/licenses/by-nc/4.0/)
Prediction-corrected visual predictive check [52] of the PK model for finerenone based on FIDELIO-DKD data. Black/gray dots: prediction-corrected observations; red line: observed median; red dashed lines: 2.5th (as long as above the lower limit of quantification) and 97.5th percentiles of observations; red area: 95% variability-based prediction interval of the simulated medians; gray area: 95% prediction interval; blue symbols: observations below the lower limit of quantification. CI confidence interval, h hour, LLOQ, lower limit of quantification. Model parameters are provided in Table 3.
4 Pharmacokinetics in Special Populations/Intrinsic Factors
An overview of intrinsic factors is provided in Fig. 4 and Table 1 and summarized as follows.

Impact of intrinsic factors on finerenone exposure based on dedicated Phase I studies and model-informed approaches. AUC area under the curve, CI confidence interval, CKD chronic kidney disease, CLCR creatinine clearance, Cmax peak drug concentration, popPK population pharmacokinetic, T2D type 2 diabetes
4.1 Age
Elderly subjects (aged ≥ 65 years) showed a decrease (−25%) in CL/F paralleled by increases in AUC and Cmax of 34% and 51%, respectively, compared to young subjects (aged ≤ 45 years), based on the total population of male and female subjects in a specific Phase I study ([5], see also Tables 1 and 2 of the Electronic Supplementary Material). This trend was confirmed in the popPK analysis of FIDELIO-DKD [24].
4.2 Sex
Based on the Phase I study in young and elderly male and female subjects, the mean ratios (females/males) of finerenone AUC and Cmax were contained within the range 80–125% [5], indicating no relevant effect of sex. Also in popPK analyses of FIDELIO-DKD, sex was not a significant covariate [24], and slight increases in exposure in females compared to males, in line with findings from elderly Phase I subjects, may be explained by differing body size.
4.3 Body Size
In an integrated Phase I popPK analysis, body weight was a covariate on volume but not on clearance, and consequently, Cmax decreased with increasing body weight, while AUC was not affected [23]. Similar effects were observed in popPK analyses of Phase II and III data [23, 24]. In FIDELIO-DKD, body height was identified as an additional covariate, however, on clearance and not volume. The known correlation between the two size descriptors and potential additional covariates is considered in Fig. 4 showing post hoc parameter estimates for FIDELIO-DKD subjects with low (< 5th percentile) and high (≥ 95th percentile) body weight relative to the main (5–95th percentile) population investigated. In this multivariate analysis, Cmax decreased with increasing body weight, as did AUC to a smaller extent. Similar to body-weight effects, finerenone exposure was also slightly decreased for patients with a high body mass index (≥ 30 compared to < 30 kg/m2).
4.4 Ethnicity
An integrated analysis of Phase I studies indicated a tendency in Asian subjects towards reduced finerenone CL/F (−25%) compared to Caucasian subjects. Integral and peak exposure of finerenone after single-dose administration (AUC divided by dose and Cmax divided by dose) was about 30% higher in Asian subjects (see also Table 2 of the Electronic Supplementary Material). This difference was greatly reduced by normalization to body weight, reflecting the large contribution of lower body weight in Asian subjects [6].
While Korean ethnicity was statistically significant in popPK analyses of FIDELIO-DKD, forward-simulations of effects in racial and ethnic subgroups were contained within typical equivalence ranges (80–125%) and are thus considered clinically not significant [24].
4.5 Hepatic Impairment
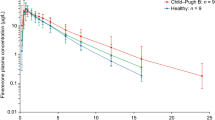
In subjects with moderate hepatic impairment (Child-Pugh B, score 7–9) compared to healthy subjects, finerenone CL/F was reduced by about 28%, accompanied by increases in AUC (+ 38%), fu in plasma (+ 12%), unbound AUC (+ 55%) and t1/2 (3.18 h vs 2.29 h). Finerenone Cmax was unchanged. The minor increase in fu in plasma from 11.5% (healthy participants) to 12.9% (Child-Pugh B) was due to a decrease in serum concentrations of albumin, the main binding protein of finerenone, from 3.96 g/dL (healthy) to 3.07 g/dL (Child-Pugh B). No clear effects on AUC, Cmax, or protein binding of finerenone were seen in subjects with mild hepatic impairment (Child-Pugh A, score 5–6). Cirrhotic patients with severe hepatic impairment (Child-Pugh C) were not studied [21].
Hepatic function based on criteria analogous to the Child-Pugh score was not a significant covariate in the popPK analysis of FIDELIO-DKD [24].
4.6 Renal Impairment
Finerenone CL/F was decreased (−25%, excluding an outlier) in subjects with moderate renal impairment (creatinine clearance [CLCR] 30 to < 60 mL/min), compared with control subjects with CLCR ≥ 90 mL/min. The effect was similar (CL/F −27%) in subjects with severe impairment (CLCR < 30 mL/min). This resulted in AUC increases by 34% and 36% in subjects with moderate and severe impairment, compared to control subjects [6]. The changes in finerenone clearance were paralleled by slightly prolonged t1/2 (2.6–2.8 h vs 2.3 h) but there was no clear effect on the absorption phase and Cmax. Mild renal impairment did not affect AUC or Cmax. An evaluation of this study based on different categories [25] of mild or moderate impairment (CLCR 50–80 or 30–< 50 mL/min, respectively) and normal renal function (CLCR > 80 mL/min) showed similar effects of moderate or severe impairment on AUC (+ 61% and + 45%), compared to healthy subjects [20]. Moreover, this analysis demonstrated an fu of finerenone in plasma independent of renal function (10.3%–10.7% across categories) and minimal renal excretion of the drug (amount excreted decreasing from 0.81% of the dose [healthy subjects] to 0.38% [subjects with severe impairment]). For a drug with such a minor contribution of glomerular filtration (see Sect. 3.3) to overall elimination, the observed increase in AUC in subjects with moderate or severe renal impairment, although small, is unexpected, suggesting effects on nonrenal routes of elimination.
Renal impairment had the expected effects on plasma exposure of the pharmacologically inactive metabolites M-1, M-2, and M-3 (AUC ratio “subjects with severe impairment vs healthy subjects” of 1.68, 1.91, and 5.75, respectively), which show relevant renal elimination in contrast to the parent drug finerenone (see Sect. 3.3) [20].
Estimated glomerular filtration rate as a marker of renal function was also consistently identified as a covariate for finerenone exposure in popPK analysis with comparable effect sizes to those observed in Phase I [23, 24].
4.7 Patients with CKD and T2D
Integral exposure (+ 50%) and, to a minor extent, peak exposure (+ 9%) were increased in a typical simulated FIDELIO-DKD popPK patient compared to a typical simulated Phase I subject with a CLCR ≥ 50 mL/min. Patients from FIDELIO-DKD were older, had impaired renal function, and a somewhat higher body weight compared to healthy volunteers from Phase I studies. Thus, the higher exposure in patients can be mainly attributed to the correlated intrinsic factors of decreased renal function and increased age.
5 Drug–Drug Interactions
5.1 Finerenone as a Substrate
The influence of extrinsic factors is illustrated in Fig. 5 and food effects are discussed in Sect. 3.1, the effect of comedications affecting gastric pH and metabolic DDIs are discussed in Sect. 5.1.1 based on Phase I studies, and in Sect. 5.1.2 based on model-informed approaches.

Effect of extrinsic factors on finerenone exposure; Phase I studies presented as geometric LS mean and 90% CI for ratio “finerenone + extrinsic factor/finerenone alone,” model-informed approaches based on an exploratory Phase IIa popPK covariate effect (amiodarone), and PBPK simulations (other substances) as geometric mean ratios for “finerenone + extrinsic factor/finerenone alone” and range based on sensitivity scenarios assuming ± 5% altered hepatic fm via CYP3A4 and CYP2C8 compared to the reference model with ~90% fm(CYP3A4) versus 10% fm(CYP2C8). AUC area under the curve, BID twice daily, CI confidence interval, Cmax peak drug concentration, CYP cytochrome P450, fm fractions metabolized, LS least squares, OD once daily, TID three times daily, PBPK physiologically based pharmacokinetic, popPK population pharmacokinetic
5.1.1 Phase I Studies
Comedications increasing gastric pH were investigated in Phase I studies, as the absorption of finerenone could theoretically be altered given its decreased aqueous solubility with increasing pH. The proton pump inhibitor omeprazole (40 mg on the day of finerenone administration, and 40 mg OD on the previous 4 days) had no effect on finerenone AUC or Cmax. Algeldrate (aluminum hydroxide/magnesium hydroxide 70 mVal suspension 10 mL; Maalox®) did not influence finerenone AUC and decreased Cmax by about 19% [19].
Finerenone was investigated as a substrate/victim in DDIs focusing on CYP3A4 and 2C8 as the only enzymes of relevance for its metabolism. Studies were conducted with index inhibitors [26] of these CYP isoforms, with erythromycin and verapamil as moderate CYP3A4 inhibitors and gemfibrozil as a strong CYP2C8 inhibitor [17]. Erythromycin (500 mg three times a day) reduced finerenone CL/F (−71.3%), resulting in increased AUC (+ 248%), Cmax (+ 88.2%), and t1/2 (from 1.6 to 2.5 h) compared to finerenone alone. Verapamil (240 mg OD) decreased finerenone CL/F (−63.0%), leading to AUC and Cmax increases by 170% and 122%, respectively, and a prolonged t1/2 (from 1.9 to 3.2 h). Changes observed in the concentration versus time profiles of finerenone metabolites M-1, M-2, and M-3 in plasma in the presence of erythromycin or verapamil confirmed the relevance of CYP3A4 for this pathway. Gemfibrozil (600 mg twice daily) reduced finerenone CL/F (−9.1%), resulting in AUC and Cmax increases by 10.1% (90% CI within 80%–125%) and 15.7%, respectively, without effect on t1/2. The effects of erythromycin and verapamil observed in vivo were consistent with results of a static prediction algorithm including the fraction metabolized by CYP3A4 (0.88–0.89 based on in vitro data) and the reported in vivo potencies of CYP3A4 inhibitors [27, 28]. Similarly, the effect of gemfibrozil confirms the fraction metabolized via the CYP2C8 pathway to be approximately 0.10 [17].
5.1.2 Model-Informed Approaches
A PBPK model of finerenone was developed based on physiochemical, in vitro, and clinical Phase I PK data. Informed by preclinical data and clinical Phase I DDI studies, a hepatic fraction metabolized by CYP3A4 of approximately 90% was assumed (reference scenario). As sensitivity scenarios, alternative finerenone models with hepatic fractions metabolized by CYP3A4 constituting 85% or 95% of total hepatic metabolic clearance were implemented as well and used to generate prediction intervals [29].
Model development was conducted in the open-source PBPK platform PK-Sim, qualified for the application of assessing substances as an object of CYP3A4-mediated DDIs [30,31,32,33,34]. The finerenone model was coupled with precipitant/perpetrator models of interest included in the platform without additional adaptions to the models. The coupled models were simulated in 1000 virtual Phase I-like individuals to evaluate the DDI [29].
First, to further validate the finerenone model, especially regarding the contribution of CYP3A4 metabolism to total clearance, DDIs with erythromycin and verapamil were simulated and compared to data observed in dedicated clinical Phase I studies. The simulated versus observed geometric mean ratios for finerenone were 3.46 versus 3.48 (AUCR) and 2.00 versus 1.88 (CmaxR) with erythromycin, and 2.91 versus 2.70 (AUCR) and 1.86 versus 2.22 (CmaxR) with verapamil [29] (corresponding to simulated decreases in CL/F of −71.1% and −65.6%, respectively).
Then, the validated model was coupled to other available precipitant models to predict clinically untested DDIs with various CYP3A4 modulators. An AUCR and CmaxR of 6.31 and 2.37, respectively, were predicted with itraconazole, and of 5.28 and 2.25, respectively, with clarithromycin, in line with the general classification of these drugs as strong inhibitors [35]. This corresponded to simulated CL/F decreases of −84.2% and −81.1%, respectively. An AUCR of 1.59 and a CmaxR of 1.40 was predicted with cimetidine, and of 1.57 and 1.38 with fluvoxamine, corresponding to simulated CL/F decreases of −37.1% and −36.3%, respectively. While the PBPK evaluations with the sensitive CYP3A4 substrate finerenone [36] support a “weak” classification for both cimetidine and fluvoxamine, the latter is labeled “weak to moderate” in Fig. 5 and Table 2, reflecting the FDA’s update of the fluvoxamine classification from a “weak” to a “moderate” inhibitor in 2019. With respect to CYP3A4 induction, an AUCR of 0.19 and a CmaxR of 0.32 were predicted with efavirenz (600 mg OD), and of 0.07 and 0.14 with rifampicin, which are considered moderate and strong inducers, respectively (see Fig. 5 and Table 2) [29]. This would indicate increases in CL/F by > 400% and > 1000%, respectively.
Predictions pertinent to strong inhibitors and moderate/strong inducers can be considered extrapolations, which are seen as more uncertain than interpolation from stronger to weaker modulation classes. The overall well-qualified models and robust predictions, with the reference scenario (hepatic fm [CYP3A4] = 90%) being closer to observed data than investigated sensitivity scenarios (hepatic fm [CYP3A4] = 85% or 95%), led to the inclusion of the predictions in relevant labels, e.g., US prescribing information [37]. This was complementary to the information based on dedicated Phase I studies and provided a quantitative basis to guide clinical use of finerenone with concomitant CYP3A4 modulators [29].
Apart from PBPK assessments, the use of the weak CYP3A4 inhibitor amiodarone resulted in a 17% lower apparent clearance after forward inclusion corresponding to a 21% AUC increase in an explorative popPK analysis of Phase IIa data [5, 10, 23]. In FIDELIO-DKD, CYP3A4 inhibitor use was identified as a covariate; however, with smaller effects than expected based on the above Phase I and PBPK studies. The discrepancy likely reflects the less controlled and documented study and analysis design as far as the identification of comedication effects in Phase III studies is concerned [24].
Interestingly, in the FIDELIO-DKD popPK analysis, longer-term concomitant use of SGLT-2 inhibitors was identified as a covariate on finerenone CL/F and F, slightly decreasing finerenone exposure. SGLT-2 inhibitor use may improve the general renal pathophysiology in CKD patients and result in decreased levels of uremic toxins and increased CYP3A4 activity [24, 38]. However, additional data are needed to confirm and better understand this incidental finding.
5.2 Finerenone as Precipitant
5.2.1 Effect on CYP Enzymes
In vitro studies had indicated a potential for finerenone or some of its metabolites to affect the activity of CYP3A4, both via inhibition (reversible and irreversible) and induction. In addition, a degree of CYP2C8 inhibition was observed in vitro, for which clinical relevance at the exposure level of 20 mg finerenone could not be excluded. Subsequently, studies were performed with the index substrates midazolam (CYP3A4) and repaglinide (CYP2C8) [26]. Although the primary rationale of a DDI study conducted with warfarin was to assess the PDs of this drug with narrow therapeutic range [39], S-warfarin also served as an index substrate of CYP2C9 [26] (see Fig. 6).

Effect of finerenone on substrate PKs presented as mean and 90% CI for ratio “substrate + finerenone/substrate alone” in dedicated Phase I studies. AUC area under the curve, BCRP breast cancer resistance protein, CI confidence interval, Cmax peak drug concentration, CYP cytochrome P450, h hour, OATP organic anion transporting polypeptide, OD once daily, P-gp permeability glycoprotein, PK pharmacokinetic
Finerenone at the steady-state of its highest approved dose (20 mg OD) had no relevant effect on CYP3A4, i.e., the mean Cmax and AUC of midazolam were increased by 9% and 11%, respectively, and the 90% CI of the AUC ratio was included in the conventional equivalence interval of 80–125% [39]. This observed mean increase in midazolam AUC is comparable to the increase in finerenone AUC following repeated administration (about 10%, see Sect. 3.3), which reflects the common characteristic of midazolam and finerenone being sensitive CYP3A4 substrates. A single dose of 20 mg finerenone administered concomitantly with or 3 h before repaglinide had no relevant effect on repaglinide clearance, considering the demonstrated equivalence in AUC (and Cmax) of the CYP2C8 substrate (90% CIs of repaglinide AUC [and Cmax] ratios within 80%–125%) [39]. In a third study, finerenone (20 mg OD over 6 days) had no effect on AUC and Cmax of R- or S-warfarin, with point estimates and 90% CI of treatment ratios contained within 80%–125%. This confirms the lack of effect of finerenone on CYP2C9, the metabolizing enzyme of S-warfarin. Warfarin also had no effect on the plasma exposure of finerenone, as expected. Finerenone AUC and Cmax ratios (finerenone + warfarin / finerenone point estimates [exploratory 90% CI]) were 0.967 (0.939; 0.997) and 0.933 (0.792; 1.10), respectively [5]. With respect to the PD focus of the study, finerenone did not affect the AUC (0–96 h) of prothrombin time, nor of factor II and factor X activity. For the AUC (0–96 h) of factor VII activity, the upper 90% CI limit of the ratio (1.12) fell above the predefined upper equivalence limit of 1.11, owing to an outlier value in one subject [39].
5.2.2 Effect on Transport Proteins
In vitro studies have indicated that the permeability glycoprotein (P-gp) and breast cancer resistance protein (BCRP) are inhibited by finerenone [5, 7]. Moreover, finerenone and/or some of its metabolites showed a potential to inhibit the organic anion transporting polypeptide (OATP) isoforms 1B1 and 1B3 [5, 7]. A risk for finerenone to inhibit BCRP and OATP in vivo could not be completely ruled out based on prediction algorithms, while a risk for clinically relevant P-gp inhibition was not evident based on predictive algorithms and predefined threshold values [5]. Studies were performed with finerenone to investigate effects on index substrates of the above drug transporters (see Fig. 6).
Digoxin, an index substrate of P-gp [26], and drug with a narrow therapeutic range, demonstrated equivalence in trough concentration within a dosing interval after multiple dosing (Ctrough,md) on three subsequent days, and in AUC within the dosing interval after multiple dosing (AUCT,md), and Cmax within a dosing interval after multiple dose administration (Cmax,md) (ratios “digoxin + finerenone/digoxin alone” and 90% CIs within 80%–125%), when administered alone or together with finerenone 20 mg OD, with both drugs having reached steady-state. Finerenone AUCτ,md and Cmax,md were also unchanged in the absence or presence of digoxin, as expected [5] (see also Table 3 of the Electronic Supplementary Material).
The AUC or Cmax of rosuvastatin were not altered in a clinically relevant manner when administered on a background of 40 mg finerenone OD at steady-state, simultaneously with finerenone, or separated by 4 h, compared to administration of rosuvastatin alone. Rosuvastatin is an index substrate of both OATPs and BCRP [26]. In addition, finerenone had no effect on the systemic exposure of the endogenous OATP substrates coproporphyrin I and III [7].
The studies show that finerenone does not affect the activity of the drug-transporters P-gp, BCRP, or OATPs 1B1 and 1B3 in vivo.
6 Pharmacokinetic/Pharmacodynamic
The finerenone dose–exposure–response relationship was modeled in PK/PD and time-to-event analyses based on ARTS-DN and FIDELIO-DKD data for serum potassium, UACR, eGFR, and renal outcome as well as explored for additional safety markers including hemodynamics in FIDELIO-DKD [23, 24, 40, 41].
Pharmacokinetic/pharmacodynamic models were generally indirect response models reflecting the hormonal mode of action of aldosterone. While finerenone has a short PK half-life of 2 to 3 h, the model-predicted times to reach the full (99%) steady-state drug effect on serum potassium, UACR, and eGFR were 20, 138, and 85 days, respectively, indicating a time-scale separation between dosing of finerenone and relevant PD effects, which likely contributes to its safety in a titration setting. Overall, the finerenone dose–exposure–response relationships evaluated indicate shallow relations that start to saturate around the labeled doses of 10 mg and 20 mg finerenone OD. Nevertheless, numerically higher effects are predicted with 20 mg compared to 10 mg finerenone for both efficacy and safety markers and the therapeutic window is considered to extend below and above the labeled doses. For example, statistically significant UACR reduction was observed in ARTS-DN already at a dose of 7.5 mg finerenone OD [8], while neither higher doses (40 mg multiple doses or 80 mg single dose) in Phase I nor the combination of 10 mg or 20 mg finerenone with moderate CYP3A4 inhibitors in Phase III patients indicated safety concerns [5, 18, 41].
Hyperkalemia is the only identified risk based on the clinical development program including FIDELIO-DKD. Hyperkalemia is strongly influenced by baseline serum potassium values and fluctuations due to food intake, for example. Based on the physiological role of aldosterone and MR in electrolyte homeostasis, increasing serum potassium is a result of the mode of action of MRAs [1]. Serum potassium was modeled based on data from ARTS-DN as well as FIDELIO-DKD [23, 41]. Implemented and subsequently labeled eGFR- and serum potassium-based dose titration, which is not evaluable in “classical” dose-response analysis, was evaluated based on data from FIDELIO-DKD. The analyses provide support for patients who were being up-titrated to, and maintained on, the more effective dose of 20 mg, if tolerated, while guiding patients to the lower dose or interruption if they are more susceptible to hyperkalemia. Interestingly, the observed data show even higher serum potassium levels and rates of hyperkalemia in patients on the lower dose of 10 mg compared to 20 mg. This apparent paradox is explained by the PK/PD analysis that reveals an underlying relationship with monotonously increasing effects for higher doses in a simulated fixed-dose setting, however, this is inverted by titration [41, 42].
The dose–exposure–response analyses of kidney outcomes in FIDELIO-DKD indicated saturation towards 20 mg finerenone OD. Relevant components of the endpoint are directly based on relative eGFR decline or absolute eGFR levels. Interestingly, eGFR was evaluated in ARTS-DN mainly as a safety marker, where finerenone was administered for only 90 days, and PK/PD models indicated a decline saturating with dose and reaching a plateau during treatment. FIDELIO-DKD provided relevant additional insight into the kidney-protective effects of finerenone. The initial decline observed in ARTS-DN can also be observed in FIDELIO-DKD and, importantly, was followed by a sustained slowing of the chronic eGFR decline in patients receiving finerenone during a median follow-up of 2.6 years [11, 40]. With increasing time, the latter effect on eGFR trajectory provides benefits in preserving kidney function that outweigh the initial decline. In addition to MRAs, such a biphasic PD has been described for renin–angiotensin system inhibitors and sodium-glucose co-transporter-2 (SGLT-2) inhibitors, and the initial decline has been postulated to reflect reduced intraglomerular pressure. A larger initial decline was associated with better long-term preservation of kidney function [43,44,45,46]. The PK/PD analysis also shows that the initial decline is reversible upon finerenone discontinuation. The complex eGFR dynamics hint to potential challenges in directly relating finerenone exposure to outcome.
The analysis of eGFR in FIDELIO-DKD indicates a chronic eGFR decline, as well as a treatment effect of finerenone slowing this decline. This beneficial treatment effect can be explained by effects on UACR and is fully described by modeled UACR, supporting UACR as a bridging biomarker for finerenone. Generally, the UACR PK/PD model shows that finerenone leads to an exposure-dependent, sustained UACR reduction. The eGFR and especially UACR models also allow assessments of the effects of comedication with SGLT-2 inhibitors and more conclusively support the independent and additive benefits of finerenone and SGLT-2 inhibitors [40].
7 Discussion
The PK/PD relationship is discussed in the previous Section and the PKs and effects of intrinsic and extrinsic factors were evaluated in light of the PK/PD relationships and dose-titration guidance [11, 23, 37, 40, 41]. Generally, finerenone exposure is correlated with safety and efficacy markers but the relationships are considered shallow. Especially and importantly, the observed serum potassium relationship is inversed in the investigated and approved titration regimen, i.e., higher finerenone dose and exposures are associated with lower relative potassium concentrations [41].
The complete and rapid absorption of finerenone after oral administration as a film-coated tablet reflects the high permeability observed in Caco-2 cells in vitro. Although classified as a drug in BCS class II due to its low solubility at pH 6.8, the equivalent bioavailability of tablet and aqueous solution (at 10 mg) together with the demonstrated dose-linearity of the tablet across the investigated dose range show that solubility is not limiting for its absorption. The finding that food has no relevant effect on the bioavailability of the finerenone tablet is reflected in the posology in clinical Phase II and III trials as well as the approved finerenone label, which allow flexible administration regardless of meals [5, 16, 18, 19, 37].
In view of a hepatic extraction ratio < 0.3, the observed absolute bioavailability of finerenone of 43.5% can be explained by a large contribution of CYP3A4-associated first-pass metabolism in the gut wall, quantitatively more relevant compared to the liver [17]. Similar first-pass properties were also shown by other drugs containing a dihydropyridine structure moiety [47, 48].
The fu of finerenone in plasma was hardly affected by disease states associated with reduced concentrations of the main binding protein albumin, e.g., renal or hepatic impairment. With about 90% of finerenone in plasma bound to proteins, dialysis is not expected to have a relevant effect on its PKs. The distribution volume of finerenone (52.6 L), similar to the volume of total body water, is indicative of limited distribution into tissues [17, 20, 21].
The effect of renal dysfunction on finerenone PKs was investigated in a dedicated Phase I study, and in patients with CKD and T2D based on a popPK approach. Given the minimal renal excretion of finerenone (< 1% of the dose), the observed increase in AUC by 34%–36% in subjects with moderate or severe renal impairment was likely due to effects on the expression or activity of intestinal and hepatic CYP enzymes mediated by the accumulation of uremic toxins [49, 50]. In the target population of CKD patients, the starting dose of finerenone depends on the patient’s renal function and is guided by eGFR with a threshold of 60 mL/min/1.73 m2 to distinguish between 10 mg (≥ 25–< 60) and 20 mg (≥ 60) doses. Both the starting dose recommendation and overall eGFR cut-off of 25 mL/min/1.73 m2 were not based on the minor effect of renal impairment on finerenone PKs but reflected the importance of renal function for potassium serum concentration as a relevant safety marker. Current assessments of serum potassium are also utilized to guide potential dose adjustments after initiating treatment with finerenone [37, 41].
Effects of hepatic impairment on finerenone exposure were also small [21]. The minor effect in subjects of the Child-Pugh B category (increase in AUC by 38%) should be interpreted in connection with the low hepatic extraction of finerenone, and its quantitatively more important first-pass clearance in the gut wall. The small and not relevant increase in fu (+ 12%) in this group is in accordance with its plasma protein binding < 90% (based on ex vivo measurements) or slightly above this threshold (based on in vitro data, see Sect. 3.2). The threshold of 90% for the fraction bound to plasma proteins was proposed to assign a high- and low-risk category for relevant changes with hepatic impairment [51]. The effect of moderate hepatic impairment Child-Pugh B class (“no dosage adjustment”, “consider additional serum potassium monitoring”) is reflected in the drug’s label, as well as the lack of effect seen in the Child-Pugh A class (“no dosage adjustment”) and the absence of data on patients in Child-Pugh class C with severe impairment (“avoid use”) [37].
Other intrinsic factors such as age, sex, body size, and ethnicity were investigated in a dedicated Phase I study, a pooled analysis of various Phase I studies, and/or popPK analyses of patients. Phase I and popPK approaches were consistent in identifying a trend for increased finerenone AUC and Cmax with age. Investigations of various descriptors of body size showed the expected decrease in Cmax with increasing body weight or body mass index that can be related to the volume of distribution. In addition, there was some evidence, although less clear, for a decrease in AUC with increasing body size (height, weight, or body mass index) that may be related to the size of relevant clearance organs. The effect of body size is likely to be the underlying cause of other small apparent covariate effects. For example, the finerenone exposure tended to be higher in females, but overall, the effect of sex was not significant. The difference in finerenone AUC and Cmax between Asian and Caucasian Phase I subjects was greatly diminished by normalization to body weight. Effects of racial and ethnic subgroups were not clinically significant in popPK analyses of Phase III studies. This conclusion also pertains to the effects of age, sex, and body size (Table 1 and Fig. 4).
With respect to extrinsic factors, modulators of CYP3A4 activity showed the expected effects on finerenone as a sensitive substrate. Various approaches, i.e., dedicated Phase I studies, static prediction algorithms, a popPK evaluation in patients, and PBPK models [5, 17, 23, 29], were utilized and provided generally consistent results, also with regard to observed versus predicted effects. Effect sizes (finerenone AUC increases or decreases in combination with investigated precipitant drugs) were generally in accordance with their assigned categories as weak, moderate, or strong inhibitors and moderate or strong inducers of CYP3A4. However, individual examples also highlight the inherent limitations of these categorizations and/or the employed methodological approaches, with fluvoxamine (a moderate CYP3A4 inhibitor) having a “weak” effect, or efavirenz (a moderate inducer) having a “strong” effect on finerenone [26]. As per the approved label, the concomitant use of strong inhibitors of CYP3A4 is contraindicated and the concomitant use of strong or moderate inducers should be avoided with finerenone [37]. Modulators of CYP3A4 activity are the only relevant extrinsic factor and no relevant changes were identified in additional Phase I studies investigating effects of a strong CYP2C8 inhibitor (gemfibrozil) on apparent clearance [17], and effects of altered gastric pH (with omeprazole or antacid) or food on absorption/bioavailability [19].
In vitro studies have suggested that finerenone can modulate the activities of certain CYP enzymes (3A4, 2C8, and 2C9 to a lesser degree) and drug transporters (P-gp, BCRP, and OATP). The clinical relevance of all potentially relevant in vitro findings was subsequently assessed in Phase I studies employing specific substrates [7, 39]. None of the investigated substrates demonstrated relevant changes in exposure when administered with finerenone. Finerenone has no potential to affect relevant CYP enzymes and drug transporters.
8 Conclusion
The PK and PD properties of finerenone have been comprehensively assessed in healthy subjects and patients with CKD associated with T2D. Finerenone is a drug with low inter- and intraindividual variability in clearance and volume of distribution. The PK/PD models describe the indirect and time-delayed nature of the exposure–response relationship for relevant clinical markers of safety and efficacy. Intrinsic demographic parameters or the investigated stages of kidney or liver impairment had no relevant effects on systemic finerenone exposure, which would require dose adjustment. Finerenone does not present a risk of influencing the PKs of other drugs in a relevant fashion. The influence of modulators of CYP3A4, the main metabolizing enzyme, on the PKs of finerenone is adequately reflected in the prescribing information. In conjunction with a potassium-based dose titration regimen, this supports the safe and efficacious use of finerenone in the target population of the drug.
References
Kolkhof P, Jaisser F, Kim SY, Filippatos G, Nowack C, Pitt B. Steroidal and novel non-steroidal mineralocorticoid receptor antagonists in heart failure and cardiorenal diseases: comparison at bench and bedside. Handb Exp Pharmacol. 2017;243:271–305. https://doi.org/10.1007/164_2016_76.
Arriza JL, Weinberger C, Cerelli G, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237(4812):268–75. https://doi.org/10.1126/science.3037703.
Kolkhof P, Baerfacker L. 30 years of the mineralocorticoid receptor: mineralocorticoid receptor antagonists: 60 years of research and development. J Endocrinol. 2017;234(1):T125–40. https://doi.org/10.1530/JOE-16-0600.
Georgianos PI, Agarwal R. The non-steroidal MRA finerenone in cardiorenal medicine: a state-of-the-art review of the literature. Am J Hypertens. 2022;2:135–43. https://doi.org/10.1093/ajh/hpac124.
FDA. Integrated review Kerendia (finerenone). 2021. www.accessdata.fda.gov/drugsatfda_docs/nda/2021/215341Orig1s000IntegratedR.pdf. Accessed February 12, 2023.
EMA. Assessment report Kerendia (finerenone), EMA/78746/2022. 2021. www.ema.europa.eu/en/documents/assessment-report/kerendia-epar-public-assessment-report_en.pdf. Accessed February 12, 2023.
Heinig R, Fricke R, Wertz S, Nagelschmitz J, Loewen S. Results from drug–drug interaction studies in vitro and in vivo investigating the inhibitory effect of finerenone on the drug transporters BCRP, OATP1B1, and OATP1B3. Eur J Drug Metab Pharmacokinet. 2022;2:803–15. https://doi.org/10.1007/s13318-022-00794-5.
Bakris GL, Agarwal R, Chan JC, et al. Effect of finerenone on albuminuria in patients with diabetic nephropathy: a randomized clinical trial. JAMA. 2015;314(9):884–94. https://doi.org/10.1001/jama.2015.10081.
Filippatos G, Anker SD, Bohm M, et al. A randomized controlled study of finerenone vs. eplerenone in patients with worsening chronic heart failure and diabetes mellitus and/or chronic kidney disease. Eur Heart J. 2016;37(27):2105–14. https://doi.org/10.1093/eurheartj/ehw132.
Pitt B, Kober L, Ponikowski P, et al. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: a randomized, double-blind trial. Eur Heart J. 2013;34(31):2453–63. https://doi.org/10.1093/eurheartj/eht187.
Bakris GL, Agarwal R, Anker SD, et al. Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. N Engl J Med. 2020;383(23):2219–29. https://doi.org/10.1056/NEJMoa2025845.
Pitt B, Filippatos G, Agarwal R, et al. Cardiovascular events with finerenone in kidney disease and type 2 diabetes. N Engl J Med. 2021;385:2252–63. https://doi.org/10.1056/NEJMoa2110956.
Ruilope LM, Agarwal R, Anker SD, et al. Design and baseline characteristics of the finerenone in reducing cardiovascular mortality and morbidity in diabetic kidney disease trial. Am J Nephrol. 2019;50(5):345–56. https://doi.org/10.1159/000503712.
Bakris GL, Agarwal R, Anker SD, et al. Design and baseline characteristics of the finerenone in reducing kidney failure and disease progression in diabetic kidney disease trial. Am J Nephrol. 2019;50(5):333–44. https://doi.org/10.1159/000503713.
FDA. M9 biopharmaceutics classification system-based biowaivers: guidance for industry. 2021. www.fda.gov/media/148472/download. Accessed March 7, 2023.
Gerisch M, Heinig R, Engelen A, et al. Biotransformation of finerenone, a novel nonsteroidal mineralocorticoid receptor antagonist, in dogs, rats, and humans, in vivo and in vitro. Drug Metab Dispos. 2018;46(11):1546–55. https://doi.org/10.1124/dmd.118.083337.
Heinig R, Gerisch M, Engelen A, Nagelschmitz J, Loewen S. Pharmacokinetics of the novel, selective, non-steroidal mineralocorticoid receptor antagonist finerenone in healthy volunteers: results from an absolute bioavailability study and drug–drug interaction studies in vitro and in vivo. Eur J Drug Metab Pharmacokinet. 2018;43(6):715–27. https://doi.org/10.1007/s13318-018-0483-9.
Lentini S, Heinig R, Kimmeskamp-Kirschbaum N, Wensing G. Pharmacokinetics, safety and tolerability of the novel, selective mineralocorticoid receptor antagonist finerenone-results from first-in-man and relative bioavailability studies. Fundam Clin Pharmacol. 2016;30(2):172–84. https://doi.org/10.1111/fcp.12170.
Heinig R, Nagelschmitz J, Loewen S. Results from phase I studies investigating the dose linearity of finerenone tablets and the influence of food or pH-modifying comedications on its pharmacokinetics in healthy male volunteers. Eur J Drug Metab Pharmacokinet. 2022;47(4):10. https://doi.org/10.1007/s13318-022-00770-z.
Heinig R, Kimmeskamp-Kirschbaum N, Halabi A, Lentini S. Pharmacokinetics of the novel nonsteroidal mineralocorticoid receptor antagonist finerenone (BAY 94-8862) in individuals with renal impairment. Clin Pharmacol Drug Dev. 2016;5(6):488–501. https://doi.org/10.1002/cpdd.263.
Heinig R, Lambelet M, Nagelschmitz J, Alatrach A, Halabi A. Pharmacokinetics of the novel nonsteroidal mineralocorticoid receptor antagonist finerenone (BAY 94–8862) in individuals with mild or moderate hepatic impairment. Eur J Drug Metab Pharmacokinet. 2019;44(5):619–28. https://doi.org/10.1007/s13318-019-00547-x.
Rohde G, Loewen S, Heinig R. Determination of finerenone-a novel, selective, nonsteroidal mineralocorticoid receptor antagonist-in human plasma by high-performance liquid chromatography-tandem mass spectrometry and its application to a pharmacokinetic study in venous and capillary human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2021;1172: 122643. https://doi.org/10.1016/j.jchromb.2021.122643.
Snelder N, Heinig R, Drenth HJ, et al. Population pharmacokinetic and exposure-response analysis of finerenone: onsights based on phase IIb data and simulations to support dose selection for pivotal trials in type 2 diabetes with chronic kidney disease. Clin Pharmacokinet. 2020;59(3):359–70. https://doi.org/10.1007/s40262-019-00820-x.
van den Berg P, Ruppert M, Mesic E, et al. Finerenone dose-exposure-response for the primary kidney outcome in FIDELIO-DKD phase III: population pharmacokinetic and time-to-event analysis. Clin Pharmacokinet. 2022;61(3):439–50. https://doi.org/10.1007/s40262-021-01082-2.
EMA. Note for guidance on the evaluation of the pharmacokinetics of medicinal products in patients with impaired renal function CHMP/EWP/225/02. 2004. www.ema.europa.eu/en/documents/scientific-guideline/note-guidance-evaluation-pharmacokinetics-medical-products-patients-impaired-renal-function_en.pdf. Accessed March 8, 2023.
FDA. Drug development and drug interactions: table of substrates, inhibitors and inducers. 2020. www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers#inVivo. Accessed April 10, 2023.
Ohno Y, Hisaka A, Suzuki H. General framework for the quantitative prediction of CYP3A4-mediated oral drug interactions based on the AUC increase by coadministration of standard drugs. Clin Pharmacokinet. 2007;46(8):681–96. https://doi.org/10.2165/00003088-200746080-00005.
Loue C, Tod M. Reliability and extension of quantitative prediction of CYP3A4-mediated drug interactions based on clinical data. AAPS J. 2014;16(6):1309–20. https://doi.org/10.1208/s12248-014-9663-y.
Wendl T, Frechen S, Gerisch M, Heinig R, Eissing T. Physiologically-based pharmacokinetic modeling to predict CYP3A4-mediated drug-drug interactions of finerenone. CPT Pharmacometrics Syst Pharmacol. 2021;2:199–211. https://doi.org/10.1002/psp4.12746.
Britz H, Hanke N, Volz AK, et al. Physiologically-based pharmacokinetic models for CYP1A2 drug–drug interaction prediction: a modeling network of fluvoxamine, theophylline, caffeine, rifampicin, and midazolam. CPT Pharmacometr Syst Pharmacol. 2019;8(5):296–307. https://doi.org/10.1002/psp4.12397.
Frechen S, Solodenko J, Wendl T, et al. A generic framework for the physiologically-based pharmacokinetic platform qualification of PK-Sim and its application to predicting CYP3A4-mediated drug–drug interactions. CPT Pharmacometr Syst Pharmacol. 2021. https://doi.org/10.1002/psp4.12636.
Hanke N, Frechen S, Moj D, et al. PBPK models for CYP3A4 and P-gp DDI prediction: a modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacometr Syst Pharmacol. 2018;7(10):647–59. https://doi.org/10.1002/psp4.12343.
Turk D, Hanke N, Wolf S, et al. Physiologically based pharmacokinetic models for prediction of complex CYP2C8 and OATP1B1 (SLCO1B1) drug-drug-gene interactions: a modeling network of gemfibrozil, repaglinide, pioglitazone, rifampicin, clarithromycin and itraconazole. Clin Pharmacokinet. 2019;58(12):1595–607. https://doi.org/10.1007/s40262-019-00777-x.
OSP. Open systems pharmacology | CYP3A4 DDI qualification report v9.1.1. 2021. https://github.com/Open-Systems-Pharmacology/OSP-Qualification-Reports/blob/v9.1.1/DDI_Qualification_CYP3A4/report.md. Accessed February 25, 2023.
FDA. Drug development and drug interactions: table of substrates, inhibitors and inducers. 2022. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers. Accessed February 12, 2023.
University of Washington. Drug interaction database. 2022. https://didb.druginteractionsolutions.org. Accessed February 25, 2023.
FDA. Kerendia (finerenone) US prescribing information. 2021. www.accessdata.fda.gov/drugsatfda_docs/label/2021/215341s000lbl.pdf. Accessed February 12, 2023.
Ali BH, Al Salam S, Al Suleimani Y, et al. Effects of the SGLT-2 inhibitor canagliflozin on adenine-induced chronic kidney disease in rats. Cell Physiol Biochem. 2019;52(1):27–39. https://doi.org/10.33594/000000003.
Heinig R, Gerisch M, Bairlein M, Nagelschmitz J, Loewen S. Results from drug–drug interaction studies in vitro and in vivo investigating the effect of finerenone on the pharmacokinetics of comedications. Eur J Drug Metab Pharmacokinet. 2020;45(4):433–44. https://doi.org/10.1007/s13318-020-00610-y.
Goulooze SC, Heerspink HJL, van Noort M, et al. Dose–exposure–response analysis of the nonsteroidal mineralocorticoid receptor antagonist finerenone on UACR and eGFR: an analysis from FIDELIO-DKD. Clin Pharmacokinet. 2022;61(7):1013–25. https://doi.org/10.1007/s40262-022-01124-3.
Goulooze SC, Snelder N, Seelmann A, et al. Finerenone dose–exposure–serum potassium response analysis of FIDELIO-DKD phase III: the role of dosing, titration, and inclusion criteria. Clin Pharmacokinet. 2022;61(3):451–62. https://doi.org/10.1007/s40262-021-01083-1.
Schnider TW, Minto CF, Filipovic M. The drug titration paradox: correlation of more drug with less effect in clinical data. Clin Pharmacol Ther. 2021;2:401–8. https://doi.org/10.1002/cpt.2162.
Holtkamp FA, de Zeeuw D, Thomas MC, et al. An acute fall in estimated glomerular filtration rate during treatment with losartan predicts a slower decrease in long-term renal function. Kidney Int. 2011;80(3):282–7. https://doi.org/10.1038/ki.2011.79.
Weir MR. Acute fall in glomerular filtration rate with renin-angiotensin system inhibition: a biomeasure of therapeutic success? Kidney Int. 2011;80(3):235–7. https://doi.org/10.1038/ki.2011.132.
Kobayashi H, Abe M, Nakamura Y, et al. Association between acute fall in estimated glomerular filtration rate after treatment for primary aldosteronism and long-term decline in renal function. Hypertension. 2019;74(3):630–8. https://doi.org/10.1161/HYPERTENSIONAHA.119.13131.
Adamson C, Docherty KF, Heerspink HJL, et al. Initial decline (dip) in estimated glomerular filtration rate after initiation of dapagliflozin in patients with heart failure and reduced ejection fraction: insights from DAPA-HF. Circulation. 2022;2:438–49. https://doi.org/10.1161/circulationaha.121.058910.
Holtbecker N, Fromm MF, Kroemer HK, Ohnhaus EE, Heidemann H. The nifedipine-rifampin interaction. Evidence for induction of gut wall metabolism. Drug Metab Dispos. 1996;24(10):1121–3.
Heinig R. Clinical pharmacokinetics of nisoldipine coat-core. Clin Pharmacokinet. 1998;35(3):191–208. https://doi.org/10.2165/00003088-199835030-00003.
Lalande L, Charpiat B, Leboucher G, Tod M. Consequences of renal failure on non-renal clearance of drugs. Clin Pharmacokinet. 2014;53(6):521–32. https://doi.org/10.1007/s40262-014-0146-1.
EMA. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function EMA/CHMP/83874/2014. 2015. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-decreased-renal-function_en.pdf. Accessed March 27, 2023.
Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64(12):1147–61. https://doi.org/10.1007/s00228-008-0553-z.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–51. https://doi.org/10.1208/s12248-011-9255-z.
Acknowledgments
We thank Stephanie Loewen (Chrestos Concept GmbH & Co. KG) for her valuable contributions in providing the Forest plots included in this publication as well as Dr. Sebastiaan Camiel Goulooze (Leiden Experts on Advanced Pharmacokinetics and Pharmacodynamics, LAP&P) and Dr. Thomas Wendl (Bayer AG) for proofreading presented model-informed results.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was funded by Bayer AG, Wuppertal, Germany. Editorial support, including fact checking, referencing, figure preparation, formatting, proofreading, and submission was provided by Moamen Hammad, PhD, Ian Norton, PhD, and Melissa Ward, BA, of Scion, London, UK, supported by Bayer AG, Wuppertal, Germany according to Good Publication Practice guidelines (https://www.acpjournals.org/doi/10.7326/M22-1460).
Conflict of Interest
RH and TE are employees of Bayer AG. In addition, RH and TE have stock in Bayer AG.
Ethics Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Consent to Participate
Informed consent was obtained from all individual participants included in the studies.
Consent for Publication
Not applicable.
Availability of Data and Material
Availability of the data underlying this publication will be determined later according to Bayer’s commitment to the EFPIA/PhRMA principles for responsible clinical trial data sharing. This pertains to scope, time point, and process of data access. As such, Bayer commits to sharing, upon request from qualified scientific and medical researchers, participant-level clinical trial data, study-level clinical trial data, and protocols from clinical trials in participants for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 1, 2014. Interested researchers can use www.clinicalstudydatarequest.com/ to request access to anonymized participant-level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information is provided in the study sponsor’s section of the portal. Data access will be granted to anonymized participant-level data, protocols, and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that participant privacy is safeguarded.
Code Availability
Not applicable.
Authors’ Contributions
All authors drafted the article and revised it critically for important intellectual content and provided final approval of the published version.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Heinig, R., Eissing, T. The Pharmacokinetics of the Nonsteroidal Mineralocorticoid Receptor Antagonist Finerenone. Clin Pharmacokinet 62, 1673–1693 (2023). https://doi.org/10.1007/s40262-023-01312-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01312-9