
Abstract
Introduction
Somapacitan is a long-acting growth hormone (GH) derivative being developed for once-weekly dosing in patients with GH deficiency (GHD). Our objective was to evaluate the impact of kidney or hepatic impairment on somapacitan exposure in adults.
Methods
In two open-label, parallel-group, single-center, 6-week trials, eligible subjects (18–75 years of age, body mass index 18.5–34.9 kg/m2, GH-naïve, without GHD) were divided into five kidney (total n = 44) or three hepatic (n = 34) function groups. Subjects with normal kidney/hepatic function were matched to those with kidney/hepatic impairment by age, sex, and body weight. Subjects received three subcutaneous somapacitan administrations (0.08 mg/kg) on days 1, 8, and 15. Blood samples were collected before each dose, at 28 time points throughout 2 weeks after the last dose, and at follow-up (3–4 weeks after the last dose). The primary endpoint was area under the somapacitan serum concentration–time curve up to 1 week after the last dose (AUC0–168 h), while secondary endpoints included AUC0–168 h of insulin-like growth factor (IGF)-I.
Results
In the kidney impairment trial, somapacitan AUC0–168 h was higher in groups with severe kidney impairment and requiring hemodialysis versus the normal kidney function group (estimated ratio and 90% confidence interval 1.75 [1.00–3.06] and 1.63 [1.01–2.61], respectively). AUC0–168 h of IGF-I was increased in the moderate impairment group (1.35 [1.09–1.66]), severe impairment group (1.40 [1.10–1.78]), and requiring hemodialysis group (1.24 [1.01–1.52]), compared with the normal function group. In the hepatic impairment trial, somapacitan AUC0–168 h was significantly higher in the moderate impairment group compared with the normal hepatic function group (4.69 [2.92–7.52]). IGF-I AUC0–168 h was lower in both hepatic impairment groups (0.85 [0.67–1.08] for the mild impairment group and 0.75 [0.60–0.95] for the moderate impairment group) compared with the normal function group. No new safety or tolerability issues were observed.
Conclusions
In summary, somapacitan exposure increased with level of kidney/hepatic impairment. Clinically, this will be taken into account when treating adults with GHD with somapacitan, as doses should be individually titrated.
Clinical Trial Registration
NCT03186495 (kidney impairment trial, registered 12 June 2017); NCT03212131 (hepatic impairment trial, registered 30 June 2017).
Plain Language Summary
Somapacitan is a long-acting growth hormone molecule for patients with growth hormone deficiency. After its administration as a subcutaneous injection, the action of somapacitan can be affected by kidney or liver disease. Thus, we conducted two trials in which the pharmacokinetic and pharmacodynamic properties of somapacitan were compared between adult subjects with different degrees of worsened kidney or liver function and their healthy counterparts. We found that subjects with severely impaired kidney function and those requiring hemodialysis had a higher somapacitan exposure in blood serum compared with subjects with normal kidney function. The concentration of insulin-like growth factor (IGF)-I, an effector molecule of growth hormone, was also increased with decreased kidney function. In subjects with moderate hepatic function impairment, somapacitan exposure was also higher than those with normal hepatic function; however, the IGF-I concentrations were lower, both at baseline and after dosing with somapacitan. Our results indicate that patients with growth hormone deficiency and kidney or liver disease may need different doses of somapacitan than people with healthy kidneys and/or liver. However, this will be taken into account because somapacitan doses will be individually titrated for each patient with growth hormone deficiency.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The exposure of somapacitan, a long-acting growth hormone, was compared between adults with kidney or hepatic function impairment and their healthy counterparts. |
Somapacitan exposure was considerably higher in subjects with severe kidney impairment and those who required hemodialysis than those with normal kidney function, and significantly higher in subjects with moderate hepatic impairment versus those with normal hepatic function. |
As the somapacitan dose is to be individually titrated, this higher observed exposure will be taken into account in patients with impaired kidney or hepatic function. |
1 Introduction
Growth hormone (GH) disorders, such as GH deficiency (GHD), are currently treated with daily subcutaneous injections of GH replacement therapy. The requirement for daily administration is burdensome for the affected patients and/or their caregivers and may, therefore, compromise adherence to treatment [1, 2].
Somapacitan is a long-acting GH derivative developed to enable once-weekly dosing in adults and children with GHD [3, 4]. Phase III development in adults with GHD has recently been completed [5] and its use in adults has been approved by the US FDA [6]. The prolonged action of somapacitan is achieved with a protraction method, which was proven to extend the half-life of insulin [7] and glucagon-like peptide-1 therapies [8, 9]. Somapacitan consists of a human GH molecule (22 kDa) with a single amino acid substitution (Leu101Cys; not involved in binding to a GH receptor) and an albumin-binding moiety (1.2 kDa), which delays elimination and prolongs half-life. In adults with GHD, the terminal half-life of somapacitan was between 2 and 3 days [10].
It has previously been suggested that recombinant GH is eliminated by the kidneys as well as the liver [11, 12]. Thus, impaired kidney or hepatic function may affect the pharmacokinetic (PK) and pharmacodynamic (PD) properties of somapacitan. We report the results of two trials with the primary objective to investigate the steady-state exposure of somapacitan in subjects with various degrees of kidney or hepatic impairment, compared with subjects with normal kidney or hepatic function. Secondary objectives included evaluation of PK/PD properties, safety, and tolerability of three once-weekly subcutaneous administrations of somapacitan in these subjects.
2 Materials and Methods
2.1 Trial Design
Both trials were open-label, multiple-dose, parallel-group, single-center trials. Trial NCT03186495 was conducted in Germany and involved subjects with normal or impaired kidney function, whereas trial NCT03212131 was conducted in Slovakia and involved subjects with normal or impaired hepatic function.
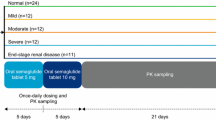
Subjects in both trials received three subcutaneous somapacitan administrations (0.08 mg/kg), one per week in the morning on days 1, 8, and 15 (Fig. 1) after an overnight fast. Subjects requiring hemodialysis continued their hemodialysis treatment throughout the trial period according to the standard clinical regimen and were administered on non-dialysis days. All somapacitan injections were administered into a skin fold parallel to the femur on the anterior aspect of the thigh by a qualified person at the clinical site. The full analysis and safety analysis sets were defined as all subjects who received at least one dose of somapacitan.

Trial design (applicable to both trials). Visits 2, 3, and 4 were in-house. PK pharmacokinetic, V visit
Trough somapacitan concentrations after the first and second somapacitan administrations were measured 10 min prior to the second and third dosing (days 8 and 15, respectively). Samples for the full profiles of somapacitan concentration versus time were collected at 15 min and 1, 2, 4, 6, 8, 12, 16, 20, 24, 26, 28, 30, 36, 42, 48, 50, 56, 64, 72, 74, 80, 86, 96, 120, 144, 168 and 336 h (i.e. from 15 min to 14 days) after the third dose, and at follow-up (21–28 days after the third dose) (Fig. 1). During the course of the trial, total blood volumes of 300 mL and 170 mL were drawn from the subjects with kidney and hepatic impairment, respectively.
2.2 Trial Participants
2.2.1 Kidney Impairment Trial
Eligible subjects were aged 18–75 years, with a body mass index (BMI) of 18.5–34.9 kg/m2, and meeting the predefined glomerular filtration rate (GFR) for any of the kidney function groups. Subjects with normal kidney function had to be considered generally healthy based on the medical history, physical examination, and the results of vital signs, electrocardiogram (ECG), and clinical laboratory tests performed during the screening visit, as judged by the investigator.
Exclusion criteria included suspected hypersensitivity to the trial product, history of GHD, being non-naïve to GH treatment, any disorder except for conditions associated with kidney impairment for subjects in the kidney impairment groups, pregnancy (or inadequate use of contraception), breastfeeding, and alcohol or drug abuse. Additionally, exclusion criteria for subjects with normal kidney function included supine blood pressure outside 90−139 mmHg (systolic) or 50−89 mmHg (diastolic), supine heart rate ≥ 90 beats/min, fasting plasma glucose (FPG) ≥ 5.6 mmol/L (100.9 mg/dL) and concomitant medication.
Subjects were enrolled in five groups in a planned ratio of 16:8:8:8:8, corresponding to normal kidney function, mild, moderate and severe impairment, and requiring hemodialysis (RH), respectively. Subjects with normal kidney function were matched to the population with kidney impairment by age, sex, and body weight. Kidney function was assessed with an exogenous filtration marker sinistrin (Inutest®; Fresenius Kabi, Austria), as recommended by the European Medicines Agency (EMA) [13]. The GFR was measured at baseline in all subjects excluding those requiring hemodialysis, and was used to classify the kidney function as normal (GFR ≥ 90 mL/min), mildly decreased (GFR 60 to < 90 mL/min), moderately decreased (GFR 30 to < 60 mL/min), and severely decreased (GFR < 30 mL/min, not requiring hemodialysis).
Estimated GFR (eGFR) was calculated for all subjects using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula, and estimated creatinine clearance (CLCr) was calculated for all subjects using the Cockcroft−Gault formula, both based on serum creatinine.
2.2.2 Hepatic Impairment Trial
Eligible subjects were aged 18–75 years, with a BMI of 18.5–39.9 kg/m2. Subjects with impaired hepatic function had a stable hepatic impairment, classified as Child–Pugh grade A or B as assessed by the investigator. Subjects with normal hepatic function (not assessed with the Child–Pugh score) were defined by their alanine aminotransferase, aspartate aminotransferase, and γ-glutamyl transpeptidase being within the lower normal limit – 100% and upper normal limit + 50%, and considered to be generally healthy (assessed as in the kidney impairment trial).
Exclusion criteria for all subjects were the same as in the kidney trial, with kidney impairment substituted with hepatic impairment. Additionally, exclusion criteria for subjects with normal hepatic function included supine blood pressure outside 90−159 mmHg (systolic) or 50−89 mmHg (diastolic), supine heart rate ≥ 90 beats/min, FPG ≥ 5.6 mmol/L (100.9 mg/dL), concomitant medication, acute hepatitis within the last 180 days, and a positive test for hepatitis B or C. For subjects with hepatic impairment, specific exclusion criteria included liver transplantation, clinical signs of acute hepatitis, esophageal variceal bleeding within the last 90 days, and biliary obstruction and/or other causes of hepatic impairment not related to parenchymal disorders and/or diseases.
Subjects were enrolled in three groups in a planned ratio of 16:9:9, corresponding to normal hepatic function, and mild and moderate hepatic impairment, respectively. Subjects with normal hepatic function were matched to the population with hepatic impairment by age, sex, race and body weight. Classification of hepatic impairment was made according to the Child–Pugh score, as recommended by the EMA and the US FDA [14, 15]. Child–Pugh Grade A (5–6 points) corresponded to mild hepatic impairment, while Child–Pugh Grade B (7–9 points) corresponded to moderate hepatic impairment. Subjects with severe hepatic impairment (Child–Pugh Grade C; 10–15 points) were not included in this trial.
2.3 Selected Pharmacokinetic/Pharmacodynamic (PK/PD) Endpoints
The primary endpoint was area under the somapacitan serum concentration–time curve (AUC) from time 0 to 168 h after the last dosing (i.e. 7 days, days 15–22). The bioanalysis of somapacitan concentrations in serum was performed using a validated somapacitan-specific luminescent oxygen channeling immunoassay (LOCI) [10, 16].
Secondary PK endpoints were derived from the somapacitan serum concentration–time curve after the last dosing on day 15 until follow-up (day 43), and included a maximum serum concentration of somapacitan (Cmax) and time to maximum serum concentration of somapacitan (tmax).
Secondary PD endpoints included AUC0–168 h of insulin-like growth factor (IGF)-I, IGF binding protein 3 (IGFBP-3), and GH-binding protein (GHBP; in the hepatic trial only). The quantification of IGF-I and IGFBP-3 in human serum was performed with an IDS-iSYS analyzer (Immunodiagnostic Systems, West Boldon, UK) as per the manufacturer’s instructions. Serum samples were incubated with two monoclonal anti-IGF-I or anti-IGFBP-3 antibodies, one biotinylated and one labeled with acridinium ester. The complexes of IGF-I or IGFBP-3 with antibodies were captured with coated magnetic particles and the luminescence, directly proportional to IGF-I or IGFBP-3 concentration, was determined [17]. The quantification of GHBP in the hepatic trial was performed using a validated enzyme-linked immunosorbent assay (ELISA). The assay was performed using a commercially available kit (Mediagnost; Reutlingen, Germany). Interassay precision was ≤ 8.7% and overall accuracy was ± 14.6%.
Safety evaluations involved incidence of adverse events (AEs), number of injection-site reactions, and occurrence of anti-somapacitan antibodies. Reports of AEs were collected and evaluated by the investigator, from signing the informed consent until the end of the trial. Injection-site reactions were evaluated by the investigator by visual and manual inspection of injection sites, at all site visits, after product administration. Blood samples for anti-somapacitan antibodies were taken prior to the first dose and at the follow-up visit.
2.4 Statistical Analyses
2.4.1 Kidney and Hepatic Impairment Trials
The sample size in both trials was not based on a formal power calculation, but was within the standard range of participants in trials investigating the PK in subjects with kidney/hepatic impairment [13, 14].
All endpoints were summarized for each of the kidney/hepatic function groups using descriptive statistics. Continuous endpoints were summarized by the geometric mean, mean, median, standard deviation, coefficient of variation and range, if applicable and relevant. Categorical endpoints were summarized by the number and percentage. AUC0–168 h of somapacitan, IGF-I, IGFBP-3, and GHBP were approximated by a linear trapezoidal technique.
Within each trial, group difference estimates (ratios) comparing kidney/hepatic impairment groups with normal function groups were obtained from an analysis of covariance (ANCOVA) with log-transformed AUC0–168 h or Cmax as dependent variables. Log weight and age were included as continuous covariates, with sex and kidney/hepatic function group as factors. Estimated differences in log-transformed values were back-transformed to original scale and presented as estimated ratio (ER) together with corresponding two-sided 90% confidence intervals (CIs). All statistical analyses and calculations of PK endpoints were carried out using SAS® software version 9.4 (SAS Institute, Cary, NC, USA).
2.4.2 Kidney Impairment Trial
Two additional analyses of the primary endpoint (somapacitan AUC0–168 h) were performed, in which the kidney function group factor was changed to log-transformed eGFR or log-transformed CLCr as the explanatory variable. Because the modeled function between eGFR or CLCr and AUC0–168 h could not, a priori, be assumed to be identical between the subjects requiring hemodialysis and those who did not, both analyses were performed without the RH group, and then repeated to include the RH group to evaluate the impact of including these data in the analysis. For these supplemental analyses, the slope estimate is presented with corresponding two-sided 95% CIs.
2.5 Ethics
Both trials were conducted in accordance with the Declaration of Helsinki [18] and International Conference on Harmonisation (ICH) Good Clinical Practice [19]. Prior to the initiation of both trials, the protocol and the consent form with the subject information sheet were reviewed and approved, according to local regulations, by appropriate health authorities, and by an independent ethics committee. Subjects were informed of the risks of the trial and that they could withdraw from the trial at any time for any reason. Consent was obtained in writing at visit 0, prior to any trial-related activities.
3 Results
3.1 Baseline Characteristics
3.1.1 Kidney Impairment Trial
Of the 94 screened subjects, 44 were enrolled in a ratio of 15:8:8:5:8 (Table 1) and administered somapacitan. The reason for not reaching the planned number of subjects (48) in a 16:8:8:8:8 ratio was premature termination of the recruitment process due to market withdrawal of Inutest® by the manufacturer. Two subjects in the normal function group withdrew their consent after the first dose and one subject from the mild impairment group was withdrawn due to non-compliance during an in-house visit. In total, 41 subjects completed the trial and all 44 subjects were included in the full analysis set and safety analysis set (Fig. 2a)

Patient flow in the a kidney and b hepatic impairment trials. FAS full analysis set, n number of subjects, SAS safety analysis set
Overall, mean age was 52 years, 43% of subjects were female, and 57% were male. The mean BMI and the mean body weight of the mild and moderate impairment groups were slightly higher compared with other kidney function groups (Table 1). Six patients were taking concomitant oral contraceptives: ethinylestradiol (n = 3), chlormadinone (n = 1), levonorgestrel (n = 1), and dienogest (n = 1). All of the six patients were in the normal kidney function group. The cause of kidney function impairment was a variety of chronic or congenital kidney disorders (e.g. various stages of chronic kidney disease, polycystic kidney disease, immunoglobulin [Ig] A, and diabetic nephropathies).
3.1.2 Hepatic Impairment Trial
After screening 41 subjects, 34 were enrolled in the planned 16:9:9 ratio and administered somapacitan. All 34 subjects completed the trial and were included in the full analysis set and safety analysis set (Fig. 2b)
The overall mean age was 57 years; 56% of subjects were female and 44% were male (Table 2). The predominant cause of hepatic disease in the mild and moderate hepatic impairment groups was hepatic cirrhosis. None of the female subjects were using concomitant oral contraceptives.
3.2 Endpoints
3.2.1 Kidney Impairment Primary Endpoint
The concentration–time profile displayed a higher geometric mean concentration of somapacitan in the groups with impaired kidney function and requiring hemodialysis, compared with the group with normal kidney function, throughout the 2-week period after the last dose (Fig. 3a).

Geometric mean curve of somapacitan full profile on the logarithmic scale in a four kidney impairment groups and the normal function group, and b mild and moderate hepatic impairment groups and the normal hepatic function group
Somapacitan AUC0–168 h was considerably higher in the severe kidney impairment and RH groups compared with the normal kidney function group (Fig. 4a). The ER [90% CI] was 1.75 [1.00–3.06] for the severe kidney impairment versus normal function group, and 1.63 [1.01–2.61] for the RH group versus the normal function group. Somapacitan AUC0–168 h in the groups with mild and moderate kidney impairment was similar to the normal function group (1.25 [0.74–2.11] and 1.27 [0.77–2.07], respectively) (Fig. 4a).

Somapacitan AUC0–168 h (ng·h/mL) after the last dose on day 15 for a four kidney impairment groups and the normal function group, and b mild and moderate hepatic impairment groups and the normal hepatic function group. Filled symbols represents the mean, center line represents the median, box represents the 25th and 75th percentiles, and whiskers represent the range. AUC0–168 h area under the plasma drug concentration-time curve, n number of subjects
In the supplemental analyses of the primary endpoint, an increase in AUC0–168 h was observed with decreasing eGFR (Fig. 5a) and CLCr (Fig. 5b). In both cases, the increase was statistically significant when the RH group was included in the analysis, and non-significant when this group was excluded. For the analysis with eGFR, the ER [95% CI] was − 0.22 [− 0.41 to − 0.03] when including the RH group, and −0.30 [− 0.63 to 0.03] when the RH group was excluded. For the association with CLCr, the ER [95% CI] was − 0.26 [− 0.48 to − 0.03] and − 0.34 [− 0.73 to 0.04] after including and excluding the RH group, respectively.

Association of AUC0–168 h with a eGFR and b CLCr in patients with kidney impairment and normal kidney function. AUC area under the plasma drug concentration–time curve, CLCr creatinine clearance, eGFR estimated glomerular filtration rate
3.2.2 Kidney Impairment Secondary PK Endpoints
The Cmax of somapacitan was similar across all kidney function groups (Fig. 6a). Compared with the normal kidney function group, the ER [90% CI] was 1.31 [0.71–2.39] for the mild impairment group, 1.40 [0.79–2.47] for the moderate impairment group, 1.47 [0.77–2.81] for the severe impairment group, and 1.34 [0.77–2.32] for the RH group.

Somapacitan Cmax in a four kidney impairment groups and the normal function group, and b mild and moderate hepatic impairment groups and the normal hepatic function group. Filled symbol represents the mean, center line represents the median, box represents the 25th and 75th percentiles, and whiskers represent the range. Cmax maximum serum concentration, n number of subjects
The median tmax varied across the different kidney function groups, but did not show a consistent pattern (8 h in the normal kidney function group, 27 h in the mild impairment group, 12 h in the moderate and severe impairment groups, and 14 h for the RH group).
3.2.3 Kidney Impairment Exploratory PD Endpoints
The AUC0–168 h of IGF-I was increased in the moderate impairment group (ER [90% CI] 1.35 [1.09–1.66]), severe impairment group (1.40 [1.10–1.78]), and RH group (1.24 [1.01–1.52]), compared with the normal function group (Fig. 7a). In the mild impairment group, AUC0–168 h of IGF-I was similar to the group with normal kidney function (1.14 [0.90–1.43]).

Mean IGF-I full profiles for a four kidney impairment groups and the normal function group, and b mild and moderate hepatic impairment groups and the normal hepatic function group. IGF-I, insulin-like growth factor-I
There was a tendency towards higher AUC0–168 h of IGFBP-3 in the kidney impairment groups compared with the group with normal kidney function (mild: 1.12 [0.99–1.27]; moderate: 1.18 [1.06–1.32]; severe: 1.36 [1.20–1.55]; and RH: 1.53 [1.37–1.70]).
3.2.4 Kidney Impairment Safety Endpoints
Twenty-four (54.5%) subjects experienced 65 AEs, of which 50 were mild and 15 were moderate. The highest proportion of patients experiencing AEs was in the severe impairment group (five patients, 100%), and the lowest was in the mild impairment group (two patients, 25%). None of the AEs were serious and all subjects recovered from the AEs. The most commonly reported AEs were headache (14 events), peripheral edema (six events), arthralgia (six events), and injection-site pain (four events). Edema occurred in one patient in each group with one event per patient, except for the moderate impairment group, where one patient experienced two events. A total of 37 AEs in 18 subjects were considered possibly or probably related to treatment. Five injection-site reactions were experienced by two subjects: one in the mild impairment group and one in the moderate impairment group; all were mild in severity.
No anti-somapacitan antibodies were observed in any of the subjects. No clinically relevant changes from baseline in biochemistry, hematology, or urinalysis parameters were observed. There were no clinically significant abnormal findings in the vital sign parameters, ECG, or body weight.
FPG and fasting serum insulin (FSI) levels were observed to be within normal ranges throughout the trial for the majority of trial subjects. Six subjects had elevated FPG, two subjects had elevated FSI, and two subjects had both parameters elevated. Most of these subjects were over 60 years of age, with a BMI ≥ 28 kg/m2.
Two subjects in the kidney impairment groups had clinically significant abnormalities in the central and peripheral nervous system, musculoskeletal system, and skin (toe amputation due to pre-existing type 2 diabetes, and swelling due to joint dislocation).
3.2.5 Hepatic Impairment Primary Endpoint
The concentration–time profile showed that, throughout the 2-week period after the last dose, the group with moderate hepatic impairment had the highest geometric mean concentration of somapacitan (Fig. 3b). The mild impairment and normal hepatic function groups had similar responses to somapacitan.
AUC0–168 h of somapacitan was significantly higher in the moderate hepatic impairment group compared with the normal function group, with exposure more than four times higher (ER [90% CI] 4.69 [2.92–7.52]) (Fig. 4b). The exposure was similar in subjects with mild hepatic impairment and normal hepatic function (1.08 [0.66–1.75]).
3.2.6 Hepatic Impairment Secondary PK Endpoints
The Cmax of somapacitan showed a similar pattern to that of AUC0–168 h across the three hepatic function groups, with nearly four times higher Cmax in the group with moderate hepatic function versus the group with normal hepatic function (ER [90% CI] 3.52 [1.97–6.31]) [Fig. 6b]. The Cmax of somapacitan was similar in subjects with mild hepatic impairment and normal hepatic function (0.95 [0.52–1.74]).
The normal hepatic function and mild impairment groups had similar median tmax (12 h in both groups), while the tmax seemed prolonged in the moderate impairment group (16 h).
3.2.7 Hepatic Impairment Exploratory PD Endpoints
The AUC0–168 h of IGF-I was lower in the hepatic impairment groups compared with the normal function group. The ER of IGF-I AUC0–168 h [90% CI] compared with the normal hepatic function group was 0.85 [0.67–1.08] for the mild hepatic impairment group and 0.75 [0.60–0.95] for the moderate hepatic impairment group (Fig. 7b).
The baseline IGF-I concentrations were lower in the mild and moderate hepatic impairment groups (mean [standard deviation] 93.7 [29.9] ng/mL and 101.1 [70.9] ng/mL, respectively), compared with the normal hepatic function group (148.1 [58.0] ng/mL).
The IGFBP-3 levels were similar between the mild and moderate impairment groups, but lower than in the normal function group. The ER of IGFBP-3 AUC0–168 h [90% CI] compared with the normal hepatic function group was 0.81 [0.68–0.97] in the mild hepatic impairment group and 0.68 [0.57–0.81] in the moderate hepatic impairment group.
The AUC0–168 h of GHBP was similar across the hepatic function groups. Compared with the normal function group, the ER [90% CI] was 0.96 [0.86–1.07] for the mild impairment group and 0.98 [0.88–1.09] for the moderate impairment group.
3.2.8 Hepatic Impairment Safety Endpoints
Nine AEs occurred in five subjects, of which six were mild and three were moderate. One patient (6.3%) in the normal hepatic function group and two patients (22.2%) in each of the mild and moderate impairment groups experienced AEs. None of the AEs were serious and all subjects recovered from the AEs. The most common AE was injection-site reaction (four events in two subjects), all of which were mild. No cases of edema were reported.
No anti-somapacitan antibodies were observed, and no clinically relevant changes were observed in individual subjects for hematology, biochemistry, lipids, and coagulation parameters across the three hepatic function groups from baseline to follow-up. No clinically relevant abnormal vital sign parameters, physical examination parameters, or ECG findings were observed.
FPG and FSI appeared stable and within normal ranges throughout the trial for the majority of subjects. Four subjects had elevated FPG, one had elevated FSI, and two subjects had both parameters elevated. Three of these four subjects had concomitant type 2 diabetes, and the fourth subject had a BMI of 35.6 kg/m2 and likely insulin resistance.
4 Discussion
Results from the two trials showed that an impairment in kidney or hepatic function was associated with an increased steady-state exposure of somapacitan, which was most pronounced in the moderate hepatic impairment group. The purpose of these trials was to determine whether kidney or hepatic impairment alters the PK of somapacitan to an extent that would create a requirement for adjusting the dosing schedule applied in phase III trials.
In the REAL 1 phase III trial, the starting dose was 1.0–2.0 mg/week (depending on age and concomitant oral estrogen), which was then individually titrated to a dose within the range of 0.1–8 mg/week [5]. The dose selected in the trials reported here (0.08 mg/kg/week) was within the dose range used in the REAL 1 phase III trial for subjects with up to 100 kg of bodyweight (8 mg/week). As the mean bodyweight was 82.1 kg and 80.6 kg in the kidney and hepatic trials, respectively, the corresponding mean doses were 6.6 and 6.4 mg/week, respectively, and the maximum doses reached 9.4 mg/week in both trials. According to the recently approved prescribing information, the maximum recommended somapacitan dose is 8 mg/week, except for patients with moderate hepatic impairment, for whom the maximum dose is reduced to 4 mg/week [20].
4.1 Kidney Impairment Trial
In the kidney impairment trial, subjects with severe kidney impairment or those requiring hemodialysis had a substantially higher AUC0–168 h compared with subjects with normal kidney function. As the hepatic function in subjects with severe kidney disease was normal, the increased somapacitan exposure suggests that clearance via kidneys also plays a role in somapacitan elimination. Somapacitan AUC0–168 h showed an increasing trend with decreasing eGFR and CLCr values, which was statistically significant when the RH group was included in the analysis. This was most likely due to the lower intersubject variability in the RH group compared with the remaining four kidney function groups.
The Cmax of somapacitan was similar across the kidney function groups, with intersubject variability observed within all groups. Median tmax varied across the different kidney function groups, with no consistent pattern.
IGF-I and IGFBP-3 levels were increased in the kidney impairment groups, most likely due to a higher somapacitan exposure compared with subjects with normal kidney function. However, this increase may not translate into higher bioactivity of IGF-I because levels of other IGFBPs (1, 2, 4, 6) are also increased in relation to kidney impairment, compromising the bioavailability of IGF-I [21, 22].
Women tend to have lower IGF-I concentrations compared with men [17]. Furthermore, in women using oral contraceptives, IGF-I levels are decreased further, thus reducing the hepatic sensitivity to GH [23, 24]. Therefore, it is possible that IGF-I levels measured in the normal kidney function group were affected by the six women taking oral contraceptives in this group, which could have led to a slight overestimation of the impact of kidney impairment in this trial.
Overall, the observed higher exposure of somapacitan and IGF-I indicate that patients with impaired kidney function may require a lower somapacitan dose relative to their counterparts with normal kidney function.
4.2 Hepatic Impairment Trial
In the hepatic impairment trial, somapacitan AUC0–168 h was significantly higher in subjects with moderate hepatic impairment compared with those with normal hepatic function. Somapacitan Cmax and tmax followed a similar pattern to that of AUC0–168 h. Both parameters were markedly higher in the moderate hepatic impairment group, and were similar between the mild impairment and normal function groups.
In the hepatic impairment groups, both IGF-I and IGFBP-3 levels were decreased relative to the subjects with normal hepatic function since baseline. However, the extent of the increase in IGF-I concentration after dosing with somapacitan in the hepatic impairment groups was similar to that in the normal hepatic function group. The decrease may be related to the resistance to GH and low levels of hepatically produced IGF-I and IGFBP-3 reported in subjects with hepatic disease [25,26,27]. This may be due to the decreased number of functional GH receptors on the liver in subjects with hepatic impairment [28]. The reduced IGF-I response may lead to the requirement of a higher somapacitan dose in patients with mild or moderate hepatic impairment compared with patients with normal hepatic function. However, the maximum recommended dose for patients with moderate hepatic impairment is reduced to 4 mg/week [20].
The levels of GHBP after somapacitan exposure were similar across the three hepatic function groups. The results from GHBP appear not to contribute to the explanation of the increased exposure of somapacitan observed in subjects with moderate hepatic impairment.
4.3 Safety
In both trials, somapacitan had a good safety profile and was well tolerated after three once-weekly subcutaneous somapacitan administrations. No new safety or tolerability issues were observed.
GH treatment can lead to a transient and reversible decrease in insulin sensitivity [29, 30]. In the present trials, increased FPG or FSI were observed in one or more subjects in each of the five kidney and three hepatic function groups. The elevated levels of FPG and FSI were mostly transient and likely related to the GH treatment, together with pre-existing concomitant diabetes mellitus and reduced insulin sensitivity.
4.4 Somapacitan Elimination
Previously, it was observed that the PK of somapacitan were approximately linear in the clinically relevant dose range (0.02–0.12 mg/kg), but non-linear at higher doses [31, 32]. This finding suggested that saturable elimination mechanisms, e.g. receptor-mediated clearance, play a key role in the elimination of somapacitan. Given the high abundance of GH receptors in the liver, high somapacitan exposure in subjects with moderate hepatic impairment further substantiates the notion of receptor-mediated clearance of somapacitan in the liver. This is in line with the previously suggested elimination of recombinant GH by receptor-mediated endocytosis followed by lysosomal degradation [11].
The observed increase in somapacitan exposure in the severe kidney impairment and RH groups may also be related to reduced receptor-mediated clearance in the kidney. This observation is consistent with the previous findings that subjects with kidney impairment had reduced kidney clearance of recombinant GH [11, 33]. However, it is likely that, since the patients with kidney impairment had normal liver function, GH was being cleared by the liver via receptor-mediated mechanisms to a normal extent, which could also translate into GH-induced IGF-I generation from the liver. This is supported by the observation that the impact of kidney disease on somapacitan clearance was less pronounced than that of hepatic impairment. The low IGF-I levels in subjects with moderate hepatic impairment, already observed at baseline, indicate a substantial degree of GH resistance. While the reason for GH resistance is unknown, it is likely that a liver with moderately impaired function expresses fewer GH receptors, or that the receptors are not functioning normally, consequently also causing reduced receptor-mediated clearance of somapacitan in subjects with moderate hepatic impairment.
A previous study on the absorption and excretion of somapacitan in healthy males has shown that urine is the major excretory route for somapacitan metabolites [34]. Therefore, it is likely that the somapacitan elimination pathway occurs primarily by metabolizing in the liver via receptor-mediated elimination, and upon entering the blood stream, the resulting metabolites are filtered and excreted by the kidney.
5 Conclusions
The exposure of somapacitan in subjects with severe kidney disease or those requiring hemodialysis, as well as those with moderate impaired hepatic function, was higher than in subjects with normal kidney or hepatic function. However, as somapacitan dose is to be individually titrated according to international guidelines based on individual clinical response, adverse effects, and serum IGF-I levels [35], the consequence of this in clinical practice is minimal. Furthermore, the results from these studies support the notion that somapacitan is degraded by receptor-mediated clearance, most pronounced in the liver, but other GH receptor-expressing organs may also contribute to the receptor-mediated clearance of somapacitan.
References
Christiansen JS, Backeljauw PF, Bidlingmaier M, Biller BM, Boguszewski MC, Casanueva FF, et al. Growth Hormone Research Society perspective on the development of long-acting growth hormone preparations. Eur J Endocrinol. 2016;174(6):C1-8.
Yuen KCJ, Miller BS, Biller BMK. The current state of long-acting growth hormone preparations for growth hormone therapy. Curr Opin Endocrinol Diabetes Obes. 2018;25(4):267–73.
Rasmussen MH, Olsen MW, Alifrangis L, Klim S, Suntum M. A reversible albumin-binding growth hormone derivative is well tolerated and possesses a potential once-weekly treatment profile. J Clin Endocrinol Metab. 2014;99(10):E1819–29.
Battelino T, Rasmussen MH, De Schepper J, Zuckerman-Levin N, Gucev Z, Savendahl L. Somapacitan, a once-weekly reversible albumin-binding GH derivative, in children with GH deficiency: a randomized dose-escalation trial. Clin Endocrinol (Oxf). 2017;87(4):350–8.
Johannsson G, Gordon MB, Rasmussen MH, Holme Hakonsson I, Karges W, Svaerke C, et al. Once-weekly somapacitan is effective and well tolerated in adults with GH deficiency: a randomized phase 3 trial. J Clin Endocrinol Metab. 2020;105(4):1–19.
ClinicalTrials.gov. Sogroya Prescribing Information. 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761156s000lbl.pdf. Accessed 12 Oct 2020.
Kurtzhals P, Havelund S, Jonassen I, Kiehr B, Ribel U, Markussen J. Albumin binding and time action of acylated insulins in various species. J Pharm Sci. 1996;85(3):304–8.
Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43(9):1664–9.
Lau J, Bloch P, Schaffer L, Pettersson I, Spetzler J, Kofoed J, et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J Med Chem. 2015;58(18):7370–80.
Rasmussen MH, Janukonyte J, Klose M, Marina D, Tanvig M, Nielsen LF, et al. Reversible albumin-binding GH possesses a potential once-weekly treatment profile in adult growth hormone deficiency. J Clin Endocrinol Metab. 2016;101(3):988–98.
Haffner D, Schaefer F, Girard J, Ritz E, Mehls O. Metabolic clearance of recombinant human growth hormone in health and chronic renal failure. J Clin Invest. 1994;93(3):1163–71.
Thorsted A, Thygesen P, Agerso H, Laursen T, Kreilgaard M. Translational mixed-effects PKPD modelling of recombinant human growth hormone—from hypophysectomized rat to patients. Br J Pharmacol. 2016;173(11):1742–55.
European Medicines Agency. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. Amsterdam: European Medicines Agency; 2015.
European Medicines Agency; Committee for Medicinal Products for Human Use. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with impaired hepatic function. Amsterdam; European Medicines Agency; 2005.
US FDA; US Department of Health and Human Services. Guidance for Industry. Pharmacokinetics in Patients with Impaired Hepatic Function: Study Design, Data Analysis, and Impact on Dosing and Labeling. Silver Spring: US FDA; 2003.
Thygesen P, Andersen HS, Behrens C, Fels JJ, Norskov-Lauritsen L, Rischel C, et al. Nonclinical pharmacokinetic and pharmacodynamic characterisation of somapacitan: a reversible non-covalent albumin-binding growth hormone. Growth Horm IGF Res. 2017;35:8–16.
Bidlingmaier M, Friedrich N, Emeny RT, Spranger J, Wolthers OD, Roswall J, et al. Reference intervals for insulin-like growth factor-1 (IGF-I) from birth to senescence: results from a multicenter study using a new automated chemiluminescence IGF-I immunoassay conforming to recent international recommendations. J Clin Endocrinol Metab. 2014;99(5):1712–21.
World Medical Association. Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–4.
International Conference on Harmonisation. ICH Harmonised Tripartite Guideline for Good Cinical Practice E6 (R2). 2016.
Novo Nordisk Inc. Sogroya, somapacitan-beco injection Prescribing Information. Novo Nordisk Inc.; 2020.
Oh Y. The insulin-like growth factor system in chronic kidney disease: Pathophysiology and therapeutic opportunities. Kidney Res Clin Pract. 2012;31(1):26–37.
Mahesh S, Kaskel F. Growth hormone axis in chronic kidney disease. Pediatr Nephrol. 2008;23(1):41–8.
Jernstrom H, Deal C, Wilkin F, Chu W, Tao Y, Majeed N, et al. Genetic and nongenetic factors associated with variation of plasma levels of insulin-like growth factor-I and insulin-like growth factor-binding protein-3 in healthy premenopausal women. Cancer Epidemiol Biomark Prev. 2001;10(4):377–84.
Blackmore KM, Wong J, Knight JA. A cross-sectional study of different patterns of oral contraceptive use among premenopausal women and circulating IGF-1: implications for disease risk. BMC Womens Health. 2011;11:15.
Donaghy A, Ross R, Wicks C, Hughes SC, Holly J, Gimson A, Williams R. Growth hormone therapy in patients with cirrhosis: a pilot study of efficacy and safety. Gastroenterology. 1997;113(5):1617–22.
de la Garza RG, Morales-Garza LA, Martin-Estal I, Castilla-Cortazar I. Insulin-like growth factor-1 deficiency and cirrhosis establishment. J Clin Med Res. 2017;9(4):233–47.
Wallace JD, Abbott-Johnson WJ, Crawford DH, Barnard R, Potter JM, Cuneo RC. GH treatment in adults with chronic liver disease: a randomized, double-blind, placebo-controlled, cross-over study. J Clin Endocrinol Metab. 2002;87(6):2751–9.
Chang TC, Lin JJ, Yu SC, Chang TJ. Absence of growth-hormone receptor in hepatocellular carcinoma and cirrhotic liver. Hepatology. 1990;11(1):123–6.
Ho KK. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: a statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur J Endocrinol. 2007;157(6):695–700.
Jorgensen JOL, Juul A. Growth hormone replacement therapy in adults: 30 years of personal clinical experience. Eur J Endocrinol. 2018;179(1):R47–56.
Battelino T RM, De Schepper J, Zuckerman-Levin N, Gucev Z, Sävendahl L. A reversible albumin-binding once-weekly growth hormone (GH) treatment (somapacitan; NNC0195-0092) induces a dose-dependent IGF-I response similar to daily GH therapy in children with growth hormone deficiency (GHD). The Endocrine Society Meeting, Boston, USA 1–4 April, 2016. Endocr Rev. 2016;Suppl 137.2.
Juul RV, Rasmussen MH, Agerso H, Overgaard RV. Pharmacokinetics and pharmacodynamics of once-weekly somapacitan in children and adults: supporting dosing rationales with a model-based analysis of three phase I trials. Clin Pharmacokinet. 2019;58(1):63–75.
Kaskel F. Chronic renal disease: a growing problem. Kidney Int. 2003;64:1141–51.
Rasmussen MH, Damholt BB, Helleberg H, Bjelke M, Suresh S, Juul RV, et al. Absorption and excretion of somapacitan in healthy male subjects. In: 100th annual meeting of the endocrine society, 17–18 Mar 2018; Chicago, IL:SUN-569.
Yuen KCJ, Biller BMK, Radovick S, Carmichael JD, Jasim S, Pantalone KM, et al. American Association of Clinical Endocrinologists and American College of Endocrinology guidelines for management of growth hormone deficiency in adults and patients transitioning from pediatric to adult care. Endocr Pract. 2019;25(11):1191–232.
Acknowledgements
Medical writing and editorial support, under the guidance of the authors, was provided by Sonia Vyskocilova and Victoria Jones of Watermeadow Medical, part of the Ashfield Group, supported by Novo Nordisk A/S.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
Both trials were conducted in accordance with the 1964 Helsinki Declaration and its later amendments and ICH Good Clinical Practice. The study was approved by the appropriate health authorities and an independent Ethics Committee (i.e. a review panel responsible for ensuring the protection of the rights, safety, and well-being of human subjects involved in a clinical investigation).
Availability of data and material
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Funding
These trials and publication fees (including open access) were sponsored by Novo Nordisk A/S.
Author contributions
All authors confirm that they meet the International Committee of Medical Journal Editors (ICJME) uniform requirements for authorship and that they have contributed to critical analysis and interpretation of the data, drafting/critically revising the article, and sharing in the final responsibility for the content of the manuscript and the decision to submit the manuscript for publication.
Conflict of interest
BBD, MDB, CB, and MHR are employed by and have shares in Novo Nordisk. SLD is employed by Novo Nordisk.
Consent to participate
Informed consent was obtained from all individual participants included in this study.
Consent for publication
Not applicable.
Code availability (Software Application or Custom Code)
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bentz Damholt, B., Dombernowsky, S.L., Dahl Bendtsen, M. et al. Effect of Kidney or Hepatic Impairment on the Pharmacokinetics and Pharmacodynamics of Somapacitan: Two Open-Label, Parallel-Group Trials. Clin Pharmacokinet 60, 1015–1027 (2021). https://doi.org/10.1007/s40262-021-00990-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-00990-7