
Abstract
Background
Population pharmacokinetic (popPK) models for antibiotics are used to improve dosing strategies and individualize dosing by therapeutic drug monitoring. Little is known about the differences in results of parametric versus nonparametric popPK models and their potential consequences in clinical practice. We developed both parametric and nonparametric models of imipenem using data from critically ill patients and compared their results.
Methods
Twenty-six critically ill patients treated with intravenous imipenem/cilastatin were included in this study. Median estimated glomerular filtration rate (eGFR) measured by the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation was 116 mL/min/1.73 m2 (interquartile range 104–124) at inclusion. The usual dosing regimen was 500 mg/500 mg four times daily. On average, five imipenem levels per patient (138 levels in total) were drawn as peak, intermediate, and trough levels. Imipenem concentration-time profiles were analyzed using parametric (NONMEM 7.2) and nonparametric (Pmetrics 1.5.2) popPK software.
Results
For both methods, data were best described by a model with two distribution compartments and the CKD-EPI eGFR equation unadjusted for body surface area as a covariate on the elimination rate constant (Ke). The parametric population parameter estimates were Ke 0.637 h−1 (between-subject variability [BSV]: 19.0% coefficient of variation [CV]) and central distribution volume (Vc) 29.6 L (without BSV). The nonparametric values were Ke 0.681 h−1 (34.0% CV) and Vc 31.1 L (42.6% CV).
Conclusions
Both models described imipenem popPK well; the parameter estimates were comparable and the included covariate was identical. However, estimated BSV was higher in the nonparametric model. This may have consequences for estimated exposure during dosing simulations and should be further investigated in simulation studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Parametric (NONMEM) and nonparametric (Pmetrics) population pharmacokinetic models of imipenem in critically ill patients treated with imipenem/cilastatin were developed. |
Both models have the same structure and describe imipenem concentrations well. The identical covariate results (absolute Chronic Kidney Disease Epidemiology Collaboration equation on the elimination rate constant) of the two different modelling methods strongly support the findings in this population. |
The parameter estimates of both models are comparable, except for the estimated between-subject variability, which was higher in the nonparametric model. Consequences for estimated exposure should be further investigated in simulation studies. |
1 Introduction
Because of increased antimicrobial resistance and few new antibiotics making it to market, optimization of antibiotic dosing regimens remains an important challenge to improve clinical outcomes of infections. Antimicrobial efficacy is determined by the susceptibility of a drug in vitro (usually expressed as the minimal inhibitory concentration [MIC]) and exposure to the drug in vivo, which relies on the pharmacokinetics (PK) and the dose [1]. Population pharmacokinetic (popPK) models describe the variability of exposure to a drug and are therefore used to support the optimization of dosing regimens with the objective to improve antimicrobial efficacy. During the development of new antibiotics, popPK models are recommended to support dose regimen identification and selection [2]. For already marketed antibiotics, popPK models are used in different ways to improve antibiotic dosing: individualization of dosing via therapeutic drug monitoring (TDM) software by Bayesian estimation and control; optimization of dosing regimens described in the package insert (especially for specific subpopulations); and setting clinical breakpoints [3]. Clinical breakpoints are MICs that define microorganisms as susceptible, intermediate or resistant to specific antibiotics [4].
While individual PK methods analyze the concentration-time profiles per individual subject, popPK methods analyze these profiles of a population as a whole. PopPK models describe and explain different types of PK variability, such as between-subject variability (BSV) and residual variability. PopPK modelling methods are classified as either parametric or nonparametric methods, which can each be divided in maximum likelihood or Bayesian approaches [3]. Bayesian popPK methods are used much less often than maximum likelihood popPK methods. Most published popPK models are based on parametric maximum likelihood methods (e.g. Monolix, NONMEM and Phoenix NLME), which estimate the set of PK parameters that maximize the joint likelihood of observations. Parametric methods assume that the population parameter distribution is known with population parameters to be estimated [5]. An example of nonparametric maximum likelihood software is the NPAG algorithm in the R package Pmetrics [6]. Nonparametric methods make no assumption about the shapes of the underlying parameter distributions. Another difference is that nonparametric methods use an exact likelihood function, while most parametric methods use an approximation. A disadvantage of nonparametric methods in the past was that confidence intervals about parameter estimates could not be easily determined [5, 7]. However, Pmetrics can estimate credibility intervals around median parameter estimates using a bootstrap method.
Some studies comparing parametric and nonparametric models are available in the literature. Precluding studies with currently outdated modelling software [8,9,10,11], we found eight comparison studies [12,13,14,15,16,17,18,19]. Four of these studies showed comparable parameter estimates of both models [12,13,14,15], one study showed different estimates [16], for two studies the estimates were incomparable due to a different model structure [17, 18], and in one study no parameter estimates were reported [19]. However, less similarity among methods was noticed for the BSV of parameters. The BSV is defined as the percent coefficient of variation (CV%), which is the standard deviation (SD) divided by the parameter mean. Three studies showed a higher BSV for the nonparametric model [12, 13, 16], one study showed similar BSV [14], and, for another study, the BSV of the nonparametric model was not reported [15]. Only one comparison study [18] presented goodness-of-fit (GOF) plots and visual predictive checks (VPCs) of both models. The other studies [12,13,14,15,16,17, 19] showed either GOF or VPC plots of both models, displayed the plots of only one method, or did not show any GOF or VPC plots.
Many parametric and nonparametric popPK models have been published in the literature and are often accompanied by dosing recommendations based on simulations of the model [3]. Differences in modelling results between these methods may have consequences for simulation findings and may therefore also influence dosing recommendations. Different parameter value probability distributions may influence dosing recommendations based on popPK models in TDM software.
To investigate the advantages and disadvantages of both modelling methods in practice, we developed both parametric and nonparametric popPK models using the same data, and compared the results. We used imipenem PK data from critically ill patients, given this population’s known PK variability for several antibiotics [20]. Imipenem is a carbapenem antibiotic administered in combination with cilastatin to prevent degradation by dehydropeptidase-I in the kidneys. When combined with cilastatin, approximately 70% of imipenem is recovered in the urine within 10 h and the rest is excreted as inactive metabolites via the urine [21]. The half-life is 1 h in patients with normal renal function, but is extended in patients with renal dysfunction [21]. Protein binding is reported as 10–20% [22].
Although it is known that the correlation between measured creatinine clearance (CLcr) and estimated glomerular filtration rate (eGFR) equations is weak [23], these equations are used in daily practice in many intensive care units (ICUs). In our study population, measured CLcr was unavailable, therefore we decided to test several eGFR equations during covariate model building to find the most suitable one in our population.
2 Methods
2.1 Study Population
Imipenem PK data from a previously published prospective cohort study [24] conducted between 2010 and 2013 in the ICU of the Geneva University Hospitals (Geneva, Switzerland) were used for this popPK study. The usual dosing regimen for imipenem/cilastatin was 500 mg/500 mg four times daily, administered by intermittent intravenous infusion of 30 min. Inclusion criteria were suspected or documented severe bacterial infection and age 18–60 years, while exclusion criteria were eGFR < 60 mL/min (measured by the Cockcroft–Gault [CG] equation [25]), body mass index < 18 or > 30 kg/m2, and pregnancy. The study protocol was approved by the University Hospital’s Ethics Committee (NAC 09-117). Given its observational nature, the Committee waived the requirement for informed consent from patients who were unconscious or otherwise unable to understand the study protocol.
Among the 54 critically ill patients from the Swiss study who were receiving imipenem therapy, the last 27 patients could be included because exact dosing and blood sampling times were known, in contrast to the first 27 patients, for whom levels were labelled only as trough, intermediate or peak. After excluding one subject because of missing height [26], data from the remaining 26 patients were included in the popPK study. None of these patients received probenecid, the only drug known to influence imipenem concentrations [21].
2.2 Study Procedures
Patients were included on their first or second day of imipenem therapy. Blood samples were planned on days 1, 2, 3, 4 and 6 of therapy, although in some patients not all planned samples were realized, e.g. due to discontinuation of therapy or problems with blood drawing. Imipenem TDM included peak (approximately 15–30 min after the end of the infusion), intermediate (midway between two sequential administrations, approximately 30 min) and trough (approximately 15 min before the next dose) concentrations. Creatinine was monitored daily.
Imipenem blood samples were drawn and immediately placed on ice, then transported to the laboratory for centrifugation. MOPS [3-(N-morpholino)propanesulfonic acid], a stabilizing buffer that protects imipenem from degradation [27], was added to an equivalent volume of plasma. Stabilized imipenem samples were subsequently stored at – 80 °C for a maximum of 1 month.
Imipenem plasma concentrations were analyzed by high-performance liquid chromatography (HPLC) with ultraviolet (UV) detection at 298 nm. Ceftazidime was used as an internal standard in the HPLC-UV analysis. Acetonitrile was added to the stabilized plasma for deproteinization. The calibration curve was linear from 0.5 to 80 mg/L. Limit of detection (LOD) and limit of quantification (LOQ) were 0.2 mg/L and 0.5 mg/L, respectively [24].
2.3 Parametric Population Pharmacokinetic (popPK) Analysis (NONMEM)
Parametric popPK analyses were performed using nonlinear mixed-effects modelling (NONMEM version 7.2; ICON Development Solutions, Ellicott City, MD, USA). The Intel Visual Fortran Compiler XE 14.0 (Santa Clara, CA, USA) was used. The first-order conditional estimation method with interaction (FOCE-I) was used throughout the model-building process. Tools used to evaluate and visualize the model were RStudio (version 1.1.456), R (version 3.5.1), XPose (version 4.6.1) and PsN (version 4.6.0), all with the graphical interface Pirana [28] (version 2.9.4).
General model selection criteria were decrease in objective function value (ΔOFV), GOF plots and VPCs. A decrease in the OFV of 3.84 units was considered statistically significant (p < 0.05, degrees of freedom [df] = 1) in a nested model [29]. For each VPC, a set of 1000 simulated datasets was created to compare the observed concentrations with the distribution of the simulated concentrations. A numerical predictive check (NPC) of the final model was created to compare with the NPC of the final nonparametric model.
During modelling, only lower bounds (of 0) and no upper bounds were set for each parameter [29]. One-, two-, and three-compartment distribution models were evaluated [30]. Databases with untransformed and logarithmic transformed concentrations were compared by assessing GOF plots and parameter estimates. For both databases, residual unexplained variability was tested with proportional and combined (additive and proportional) error models [31]. The proportional (exponential) error model of the final NONMEM model with log-transformed data is shown in Eq. (1). The observed concentration (OBS) consisted of the individually predicted concentration (IPRED) with added residual unexplained variability ε (epsilon, fixed to 1 in our model) weighted by an estimated error parameter:
Variability of a popPK parameter was estimated using an exponential variance model (individual popPK parameter = population popPK value × eη). Eta (η) is a random variable drawn from a normal distribution with a mean of 0 and a variance of omega (ω2) [32]. The BSV (CV%) of a population parameter is calculated by Eq. (2) [33]. The SD is subsequently calculated by multiplying the CV% with the population parameter estimate.
First, models with BSV on elimination rate constant (Ke) and BSV on V were compared by assessing ΔOFV and GOF plots. Subsequently, one-by-one addition of BSV on the other parameters was studied. A stepwise covariate model building was performed with forward addition at p < 0.05 (ΔOFV of 3.84 units, df = 1), followed by backward elimination at p < 0.001 (ΔOFV of 10.83 units, df = 1) [34]. Covariates were tested on parameters with BSV. The tested covariates are described in Sect. 2.5.
The 95% CI of each parameter in the final model was determined from a non-parametric bootstrap analysis, in which the dataset was resampled 1000 times.
2.4 Nonparametric popPK Analysis (Pmetrics)
Nonparametric popPK analysis was performed using Pmetrics version 1.5.2 (Laboratory of Applied Pharmacokinetics and Bioinformatics, Los Angeles, CA, USA) [6] in RStudio (version 1.1.456) as a wrapper for R (version 3.5.1), and the Intel Visual Fortran Compiler XE 14.0. The Non-Parametric Adaptive Grid (NPAG) program was used throughout the model-building process. The Iterative 2-Stage Bayesian (IT2B) program was used to estimate parameter ranges to pass to NPAG.
An NPAG will create a non-parametric popPK model consisting of discrete support points, each with a set of estimates for all parameters in the model plus an associated probability of that set of estimates (see Fig. 1 for an illustration of the distribution of a parameter) [6]. The sum of all probabilities is 1. There can be a maximum of 1 point for each subject in the study population [6]. Besides an overview of support points with corresponding parameter estimates, the NPAG output also contains the mean, SD and CV% of each parameter. The reported means are weighted means that are calculated by multiplying the estimate of each support point by the probability of that point and then summing up the resulting numbers. The SD is calculated from the parameter distribution. The BSV (CV%) of each parameter estimate is calculated by dividing the SD by the weighted mean.

Distribution of Ke in the NONMEM and Pmetrics popPK models. NONMEM: normal distribution (mean 0.637 h−1 and SD 0.121 h−1 [CV 19.0%]). Pmetrics: marginal distribution of 16 support points with 11 unique values for Ke (weighted mean 0.681 h−1 and SD 0.232 h−1 [CV 34.0%]). Ke elimination rate constant, popPK population pharmacokinetics, SD standard deviation, CV coefficient of variation
One-, two-, and three-compartment distribution models were evaluated. A stepwise covariate model building was performed with forward addition at p < 0.05 (decrease in − 2 times the log-likelihood [Δ− 2LL] of 3.84 units, df = 1), followed by backward elimination at p < 0.001 (Δ− 2LL of 10.83 units, df = 1) [34]. Covariates were tested on parameters selected after a graphical examination of possible covariate–parameter relationships. The tested covariates are described in Sect. 2.5.
Each observation in Pmetrics is weighted by 1/error2. Both gamma and lambda error models were tested (see Eqs. 3 and 4). The SD of an observation is based on the assay error polynomial (see Eq. 5) [6]; however, because the assay error polynomial was unavailable in our study, we estimated the error coefficients, with C0 = 0.5 × LOQ, C1 = 0.1, C2 = 0 and C3 = 0 as a starting point.
Model selection criteria were decrease in − 2LL, bias, imprecision, GOF plots and VPCs. A decrease in the − 2LL of 3.84 units was considered statistically significant (p < 0.05, df = 1) in a nested model. For each VPC, a set of 1000 simulated datasets was created to compare the observed concentrations with the distribution of the simulated concentrations. An NPC of the final model was created to compare with the NPC of the final parametric model. The raw VPC and NPC data were imported into PsN (version 4.6.0) using the Pirana interface [28] to generate plots with a similar layout as the parametric plots. VPC and NPC plots were created using XPose (version 4.6.1) within RStudio (version 1.1.456). Bias (mean weighted prediction error) and imprecision (bias-adjusted mean weighted squared prediction error) are automatically calculated by Pmetrics according to Eqs. (6) and (7), for both population and posterior predictions:
The 95% CI of each parameter in the final model was determined using a Monte Carlo simulation approach by creating 1000 samples with replacement for each support point [35], resulting in the 2.5th, 50th and 97.5th percentiles of the weighed median and the median absolute weighted deviation (MAWD). The SD was estimated by multiplying the MAWD by 1.4826 [36], while the CV% was calculated by dividing the SD by the 50th percentile of the weighted median.
2.5 Covariates
The tested covariates for both modelling approaches were total body weight (TBW), ideal body weight (IBW) [37], lean body weight (LBW) [37], CG eGFR [25], four-variable Modification of Diet in Renal Disease (MDRD) Study eGFR [38], Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) eGFR [39], and Jelliffe’s eGFR equation for patients with unstable renal function [40]. Per patient, one body weight measure was available, while a median of three creatinine samples per patient was drawn (see also Table 1). The MDRD, CKD-EPI and Jelliffe’s equations provide eGFR values adjusted for body surface area [BSA; mL/min/1.73 m2). The BSA-unadjusted (absolute) values (mL/min) were also calculated by multiplying the original eGFR by the individual BSA [41] and evaluated as covariates: MDRD-abs, CKD-EPI-abs and Jelliffe-abs. The CG eGFR equation is unadjusted for BSA (mL/min) and no adjustment was made.
All covariates were evaluated by power models with normalized covariates where the median covariate value was taken as the reference value (see Eq. 8). Because multiple creatinine samples per patient were collected, which are each used to calculate CG, MDRD and CKD-EPI eGFR values, eGFR was tested as a time-varying covariate. We also tested the possible situation of reaching a maximum value of the elimination constant Ke for high (adjusted or unadjusted) eGFR values from 150, 120 and 90. For TBW, LBW and IBW, both fixed (− 0.25 for Ke and 1 for V) and estimated values of the power exponent were evaluated [42]:
where Parind is the individual PK parameter estimate; Par is the popPK parameter estimate (for NONMEM) or weighted median value of the Bayesian posterior distribution (for Pmetrics); Covind is the individual covariate value; Covmedian is the median covariate value; power is the covariate effect; and eη is the individual variability (eη only for NONMEM). See Eqs. (9) and (10) in the Results section for further clarification of the differences between equations in the final parametric and nonparametric models.
Default covariate settings were used for each modelling approach. In NONMEM, by default, the next observation is carried backward (NOCB) until the time point of the previous covariate observation. For Pmetrics, covariates are applied at each dose event. By default, for missing covariate values, the last observation is carried forward (LOCF) until the last dose before the next covariate observation, when the last observation is linearly interpolated to the next observation.
2.6 Model Development and Comparison
Both models were developed by a medium-experienced NONMEM and Pmetrics modeler (FdV) supervised by highly experienced NONMEM (BdW) and Pmetrics (WY, MN) modelers. The development of both models occurred independently from each other according to a predefined study procedure, where the modelling workflow (e.g. model selection criteria, building of the structural model and covariate evaluation) was described. During model development, the GOF plots and VPCs were assessed in the layout of the separate programmes. During writing of the manuscript, the raw data of the GOF plots were transferred to GraphPad Prism (version 8.1.1) and the raw VPC and NPC data of Pmetrics were transferred to PsN (see Sect. 2.4) to create plots with the same layout. The R2 of nonlinear regression was calculated within GraphPad Prism by the fourth equation of Willett and Singer [43] for the GOF plots as a description of the graphical fit. R2 was not used during model selection.
3 Results
3.1 Study Population
Demographic and clinical characteristics of the 26 included patients are summarized in Table 1. None of the patients received continuous renal replacement therapy (CRRT).
3.2 Imipenem Samples
In total, 138 imipenem blood samples were collected from 26 patients and were subsequently analyzed. Fewer than 10% [30] of all concentrations (13/138, 9.4%) were below the limit of quantification (0.5 mg/L) and were excluded from the popPK analysis. The average number of levels per patient was 5 (range 1–11). Almost half of all samples (65/138, 47.1%) were drawn on the second day of therapy.
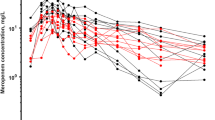
A graph of the analyzed imipenem concentrations (n = 125) plotted against the time after dose is shown in Electronic Supplementary Fig. 1. These 125 concentrations were drawn after 84 individual doses. Following 33 of these doses, two or three concentrations were taken, and after the other 51 doses, one concentration was drawn. For all PK samples, a creatinine concentration was available in the 24 h before PK sampling.
3.3 Parametric PopPK Model
The parametric popPK analysis using NONMEM showed that the data were best described by a model with two distribution compartments, BSV on Ke and CKD-EPI-abs as a covariate on Ke. The parameter estimates of the final model are displayed in Table 2. Only BSV on Ke was included (see also Fig. 1) because BSV on the central distribution volume (Vc), rate constant from the central to peripheral compartment (Kcp) and rate constant from the peripheral to central compartment (Kpc) did not significantly improve the model (ΔOFV < 3.84, and no improvement in GOF plots). Vc, Kcp and Kpc were the same for each subject (no BSV was included, therefore no CV% is shown in Table 2). Eta shrinkage was low (14%), and no large correlation (> 0.95) between the parameters was detected [29]. The GOF and VPC plots (displayed in Figs. 2a, 3a and Electronic Supplementary Fig. 2a) show good predictive performance for all concentrations, for the time range after dose and for the CKD-EPI-abs range of 18–190 mL/min (except from an outlier for the 124–141 mL/min bin). The NPC did not show points outside boundaries (data not shown). As shown in Table 2, the model-based parameter estimates were similar to the bootstrap values, indicating stability of the model.

Goodness-of-fit plots with observed against predicted concentrations of both models. a Goodness-of-fit plots of the final parametric model. The log-transformed concentrations are back-transformed for easier comparison with the untransformed concentrations in Fig. 2b. b Goodness-of-fit plots of the final nonparametric model. Solid line represents the identity (1:1) line, and the dotted line represents the regression line. Conc. concentration

VPCs of both models. a VPC of the final parametric model. The log-transformed concentrations are back-transformed for easier comparison with the untransformed concentrations in Fig. 3b. b VPC of the final nonparametric model. Circles represent observed concentrations; upper, middle and lower lines represent the 95th, 50th and 5th percentile of observations, respectively; and shaded areas represent the 95% confidence interval of the corresponding percentiles of predictions. VPCs visual predictive checks
A two-compartment model described the data better than one- or three-compartment models, according to GOF plots and an OFV increase of 22.99 for a one-compartment model and 0 for a three-compartment model. The popPK parameters of a two-compartment model with logarithmic transformed data were comparable to the same model with untransformed data. The GOF plots were improved by logarithmic transformation and therefore the following analyses were performed with transformed data. An exponential (proportional) error model (see Eq. 1 in Sect. 2.3) was preferred to a combined error model due to estimation problems with the combined model. This confirmed the findings of the untransformed data, where the proportional error model had better performance than the combined error model.
Several covariates (as described in Sect. 2.5) were tested on Ke, the only parameter with BSV. All tested eGFR covariates on Ke resulted in a significant OFV decrease (ΔOFV 24.4 until 38.3, p < 0.05) compared with the two-compartment model without covariates. However, the OFV of the model with CKD-EPI-abs as a covariate on Ke was significantly better than the other eGFR models (for example, the second best eGFR was CKD-EPI on Ke, with an OFV increase of 4.3 compared with CKD-EPI-abs; p < 0.05). Due to the observation of a maximum eta in the eta-eGFR plots, implementation of a maximum Ke value for eGFR values from 150, 120 and 90 was tested but this did not further improve the model. None of the tested measures of body weight improved the model as a covariate on Ke (ΔOFV < 3.84) during the univariate analysis.
Equation (9) describes the calculation of individual Ke (Kei) values in the final NONMEM model, using the population parameter estimates for Ke and Ke(cov) from Table 2, the individual CKD-EPI-abs value at a certain time point, and eη (individual variability). Eta (η) is drawn from normal distribution with a mean of 0 (note Eq. 9, then e0 = 1) and variance ω2 (estimated from the data as 0.0354). The corresponding BSV for Ke (CV% 19.0%) was calculated from Eq. (2):
A plot of the individual Ke against the individual CKD-EPI-abs (n = 86) is shown in Electronic Supplementary Fig. 3a. Simulated concentration-time profiles for CKD-EPI-abs of 150, 120 and 90 mL/min (n = 1000 for each eGFR) are shown in Electronic Supplementary Fig. 4a.
The median (interquartile range) of the untransformed residuals (observed minus predicted concentrations) was − 0.159 mg/L (− 0.960 to 1.267) for the population predictions and − 0.027 mg/L (− 0.649 to 0.698) for the individual predictions. Residual plots are shown in Electronic Supplementary Fig. 5a.
3.4 Nonparametric PopPK Model
The nonparametric popPK analysis using Pmetrics resulted in the same model structure as the parametric analysis: a model with two distribution compartments and CKD-EPI-abs as a covariate on the elimination constant Ke. The mean parameter estimates of the final model are displayed in Table 2. For example, the mean Kepop value is the mean of the support points weighted by population probabilities. This is illustrated in Fig. 1. Each individual has a Bayesian posterior (i.e. personal or individual) set of support points weighted by individual probabilities [6]. No large correlation (> 0.95) between the parameters was detected. The GOF and VPC plots (displayed in Figs. 2b and 3b and Electronic Supplementary Fig. 2b) show good predictive performance for all concentrations, for the time range after dose and for the CKD-EPI-abs range of 18–190 mL/min (except from an outlier for the 124–141 mL/min bin, similar to the parametric model). Also similar to the parametric model, the NPC did not show points outside boundaries (data not shown). As shown in Table 2, the model-based parameter estimates were similar to the bootstrap values, indicating stability of the model.
A two-compartment model described the data better than one- or three-compartment models, according to GOF plots and a − 2LL increase of 26.5 for a one-compartment model and 0.8 for a three-compartment model. The gamma error model was preferred to the lambda model (see Eqs. 3 and 4 in Sect. 2.4). The final values for C0 and C1 in the assay error polynomial were both 0.05, and C2 and C3 were both 0 (see Eq. 5 in Sect. 2.4). Parameter boundaries (Ke, 0–1.5; V, 1–70; Kcp and Kpc, 0–1) were set based on an IT2B run.
We evaluated several covariates (as described in Sect. 2.5) on Ke and V. All tested eGFR covariates on Ke resulted in a significant − 2LL decrease (Δ− 2LL 53.3 until 59.9; p < 0.05) compared with the two-compartment model without covariates. The − 2LL value of the four models with the largest − 2LL decrease (MDRD, MDRD-abs, Jelliffe-abs and CKD-EPI-abs) did not differ significantly from each other. The model with CKD-EPI-abs had the lowest bias and imprecision compared with the three other models. Implementation of a maximum Ke value for eGFR values from 150, 120 or 90 did not improve the model. Univariate analysis resulted in a further six significant covariates (p < 0.05): TBW, IBW and LBW on Ke (Δ− 2LL 4.0 until 8.8), and CG, Jelliffe and Jelliffe-abs on V (Δ− 2LL 4.6 until 5.5). After backward elimination at p < 0.001, none of these six covariates remained in the final model.
Equation (10) describes the calculation of individual Ke values (Kei) in the final Pmetrics model. Contrary to the parametric model with equal Ke and Ke(cov) values for each individual subject (see Eq. 9), these values are different for each individual subject in the nonparametric model. Kei,med is the weighted median value of the Bayesian posterior distribution for Ke in the ith individual, Ke(cov)i,med is the weighted median value of the Bayesian posterior distribution for Ke(cov) in the ith individual, and CKD-EPI-absi is the individual CKD-EPI-abs value at a certain time point:
A plot of the individual Ke against the individual CKD-EPI-abs (n = 86) is shown in Electronic Supplementary Fig. 3b. This plot shows a similar Kei versus CKD-EPI-abs relationship as the parametric model, although the Kei distribution is wider for the nonparametric model. A wider distribution is also shown in the simulated concentration-time profiles for CKD-EPI-abs of 150, 120 and 90 mL/min (Electronic Supplementary Fig. 4b).
The median (interquartile range) of the residuals (observed minus predicted concentrations) was − 0.045 mg/L (− 0.498 to 1.506) for the population predictions and 0.011 mg/L (− 0.295 to 0.533) for the individual predictions. Residual plots are shown in Electronic Supplementary Fig. 5b. These are similar to the parametric plots.
4 Discussion
The structure and parameter estimates of our two independently developed parametric and nonparametric popPK models of imipenem in critically ill patients treated with imipenem/cilastatin were similar; both included two distribution compartments and CKD-EPI-abs as a covariate on Ke. Body weight as a covariate was not found to be a significant covariate in either model. Both models described imipenem PK well. Two main differences between the models emerged. First, the parametric model included BSV for Ke only, while the nonparametric model included such variability on all popPK parameters. Second, the estimated BSV (defined as CV%) for Ke was higher for the nonparametric model (34.0% vs. 19.0%). The findings of similar parameter estimates but higher BSV for the nonparametric model are in line with two previously published studies comparing parametric and nonparametric models of other drugs [12, 13]. These BSV differences could be explained by the statistics behind the modelling methods. Both models have fixed (no BSV) and random (including BSV) parameters. However, while during parametric modelling the inclusion of BSV is examined for each popPK parameter, all popPK parameters for nonparametric models are, in principle, random parameters, while the residual error model is fixed. In addition, while parametric methods assume a normal or lognormal distribution of parameters, nonparametric methods make no assumption about the parameter distributions, which can cause a wider CI of the parameter estimates (e.g. see Fig. 1 for Ke, which also clearly shows the differences between the two CV% measures, and Electronic Supplementary Fig. 3). The simulated concentration-time profiles in Electronic Supplementary Fig. 4 indicate that the concentrations of the 2.5th percentile are approximately twofold lower for the nonparametric model. The consequences of this finding (e.g. higher or more frequent dosing following the nonparametric model) should be further explored by probability of target attainment calculations based on extensive Monte Carlo simulations for several dosing regimens [44].
Our finding of two distribution compartments is in accordance with other published popPK models of imipenem in critically ill patients (all with pneumoniae) [45,46,47]. However, our Vc and clearance (CL = Vc × Ke) values were higher than previously described [45,46,47]. This could be explained by a higher CLcr in our population, which could be attributed to augmented renal clearance (ARC) [48, 49]. ARC is defined as increased renal elimination of circulating solutes and drugs compared with normal baseline [23] and has been reported in approximately 30–65% of critically ill patients [48]. Our Kcp (0.2–0.4) and Kpc (0.2–0.5) values of both models were similar to the previously published parametric model [45], but remarkably different from the two nonparametric models (Kcp 3–8 and Kpc approximately 9) [46, 47]. Possibly, these differences could be explained by (unpublished) wider parameter boundaries of their nonparametric models. The parameter ranges in nonparametric software are strict boundaries wherein the optimal values are sought. To assist with optimal setting of parameter ranges in nonparametric popPK, we used the parametric IT2B module in Pmetrics to estimate the parameter ranges to pass to the nonparametric NPAG module. During parametric modelling with NONMEM, the parameter initial estimate is a starting point to search for the optimal value, and setting limits is not always necessary [29]. This was underlined by the fact that the results of our final NONMEM model with only lower boundaries were the same as a model with the same upper and lower boundaries as the final Pmetrics model.
Many types of standard GOF plots used for model evaluation are applied both in parametric and nonparametric modelling: observed versus population predicted concentrations; observed versus individually predicted concentrations; and weighted residual (WRES) plots. The observed versus predicted concentration plots of our popPK models are comparable. In Pmetrics, the standard layout of these plots includes the R2, intercept, slope, bias (mean WRES) and imprecision. In NONMEM, these measures are not automatically calculated. A residual is the difference between an observed and a predicted concentration. WRESs are used in WRES plots, as well as for the calculation of bias and imprecision. We did not show WRES plots nor calculated weighted bias and imprecision for both methods because of two reasons. First, the weighting differs between the two modelling methods. In NONMEM, the conditional WRES (CWRES) is the residual weighted by the square root of the covariance of a FOCE model [50], while in Pmetrics, WRES is the residual weighted by the squared error [35]. Second, a WRES of logarithmic transformed data (in our NONMEM model) is not the same as a WRES of untransformed data (in our Pmetrics model). We used weighted bias and imprecision in Pmetrics only to differentiate between four covariate models with a − 2LL decrease that did not differ significantly from each other. As an alternative to weighted bias, we calculated unweighted bias (the unweighted residuals of untransformed concentrations), of which the median and interquartile range were comparable for both methods.
The VPCs of both popPK models indicated a sufficient predictive performance. The means of the 95th, 50th and 5th percentiles of predictions are comparable for both models, but the 95% CIs of these percentiles differ for some bins (timeframes) of the VPCs. Most remarkably, the 95% CI of the 95th and 50th percentile of the last two bins (4.3–5.8 h after dose) is wider for the nonparametric model. This could be explained by a higher estimated BSV of Ke in the nonparametric model, leading to a wider distribution of concentrations in the elimination phase. We did not show prediction-corrected VPCs (pcVPCs) because this option has been developed for parametric methods [51] and has not yet been tested for nonparametric methods. In a pcVPC, the variability coming from binning across the independent variable, e.g. due to different doses or influential covariates, is removed [51]. The pcVPCs of the NONMEM model did not show important differences from the traditional VPCs (data not shown).
Both models include CKD-EPI-abs as a covariate on Ke. Two of the three previous mentioned published popPK models of imipenem also included renal function as a covariate, but other measures were used, i.e. 4 h CLcr in urine [45] and the CG equation [47]. Sakka et al. did not find 12 h CLcr in urine as a significant covariate [46]. Besides eGFR, other covariates were also included in the published models, i.e. body weight [45,46,47], height [46, 47], BSA [46, 47], age [46, 47], sex [47] and albumin [45]. In our covariate screening plots, sex, age and height did not show a relationship with any PK parameters. Body weight did show a relationship in the covariate plots, but inclusion as a covariate did not improve the model. Albumin data were not available. BSA was taken into account during eGFR covariate testing. We tested both BSA-unadjusted (mL/min) and BSA-adjusted (mL/min/1.73 m2) eGFR equations because the European Medicines Agency [52] and the Kidney Disease Improving Global Outcomes (KDIGO) guideline [53] recommends to base dosing on absolute instead of BSA-normalized eGFR. The KDIGO [53] also recommends using the CKD-EPI eGFR equation. However, this guideline is based on chronic kidney disease, while our study population consisted of critically ill patients with a high median CKD-EPI eGFR of 116 mL/min/1.73 m2. The correlation between measured CLcr and eGFR equations is known to be weak in critically ill patients [23, 54]. Nonetheless, the measurement of CLcr (as a surrogate for GFR) is time-consuming and is not standard practice in many ICUs. In daily practice, the eGFR is also used for dosing drugs with renal clearance, although many patients have a renal function that is not in steady state. Therefore, we decided to test several eGFR equations to find the most suitable one in our population. Minichmayr et al. performed a similar eGFR covariate analysis for meropenem in critically ill patients and found that the CG equation best described meropenem clearance [55]. We observed a maximum in the BSV–eGFR plots; however, the implementation of a maximum Ke value for eGFR values from 150, 120 and 90 did not further improve the model. As already mentioned, the correlation between measured CLcr and eGFR equations is weak, but it is also shown that this correlation varies over the eGFR range [23]. For example, in a previous study it was shown that the CKD-EPI equation performed better for measured CLcr < 120 mL/min than for CLcr > 120 mL/min [23]. We did not confirm this finding in our study (see Electronic Supplementary Fig. 2), which could be explained by the different study population.
For both modelling approaches, eGFR was implemented as a time-varying covariate using a stepwise (discontinuous) approach. The default covariate settings of both methods were slightly different (i.e. NOCB without interpolation or LOCF with linear interpolation from the last dose before the next covariate value). However, the parameter estimates of both final models, developed with the default settings, were very similar to the same models using LOCF without interpolation. This is explained by the majority (85%) of patients having reasonably stable CKD-EPI-abs eGFR values around PK sampling, according to the KDIGO definition of an eGFR drop or rise of < 25% compared with the previous value [53]. This stability statement is supported by frequent creatinine monitoring. A creatinine concentration in the 24 h before TDM was available for all PK samples. Due to the stable eGFR values, a continuous covariate approach was not necessary.
We performed our study using the most used parametric (NONMEM) and nonparametric (Pmetrics) software. For approximately a decade, a nonparametric method also exists in NONMEM, and some publications [19, 56,57,58] regarding the evaluation and optimization of this approach are available. However, this method is seldom used in clinical practice. The parametric module IT2B in Pmetrics is used to estimate parameter ranges to pass to NPAG, but underperformed compared with other parametric algorithms [12].
One of the limitations of our study is that we used eGFR equations for a critically ill population with a high frequency of augmented clearance; however, these equations are developed for more stable patients with chronic kidney disease. Nonetheless, as measured CLcr was unavailable and is also not standard practice in many ICUs, we aimed to find the eGFR equation that would best describe imipenem clearance. Another drawback is that we could not compare WRESs because the weighting is different for both methods and the residuals of untransformed data (used for the nonparametric model) are different from transformed data (used for the parametric model). Other limitations are the small number of subjects and the absence of an external validation. It would be interesting to compare the predictions of both models.
5 Conclusions
The general structure and the parameter estimates of both models were comparable. The identical covariate results (CKD-EPI-abs on Ke) of the two different modelling methods strongly support the findings in this population. The nonparametric model included BSV for all parameters, while the parametric model only included BSV on Ke. The estimated BSV of Ke was higher in the nonparametric model. The consequences of the BSV differences may affect estimated exposure during dosing simulations, and this should be further investigated in simulation studies.
References
Mouton JW, Ambrose PG, Canton R, Drusano GL, Harbarth S, MacGowan A, et al. Conserving antibiotics for the future: new ways to use old and new drugs from a pharmacokinetic and pharmacodynamic perspective. Drug Resist Updat. 2011;14(2):107–17. https://doi.org/10.1016/j.drup.2011.02.005.
European Medicines Agency. Guideline on the use of pharmacokinetics and pharmacodynamics in the development of antimicrobial medicinal products. London; 2016. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500210982.pdf.
de Velde F, Mouton JW, de Winter BCM, van Gelder T, Koch BCP. Clinical applications of population pharmacokinetic models of antibiotics: Challenges and perspectives. Pharmacol Res. 2018;134:280–8. https://doi.org/10.1016/j.phrs.2018.07.005.
Mouton JW, Brown DF, Apfalter P, Canton R, Giske CG, Ivanova M, et al. The role of pharmacokinetics/pharmacodynamics in setting clinical MIC breakpoints: the EUCAST approach. Clin Microbiol Infect. 2012;18(3):E37–45. https://doi.org/10.1111/j.1469-0691.2011.03752.x.
Racine-Poon A, Wakefield J. Statistical methods for population pharmacokinetic modelling. Stat Methods Med Res. 1998;7(1):63–84. https://doi.org/10.1177/096228029800700106.
Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit. 2012;34(4):467–76. https://doi.org/10.1097/FTD.0b013e31825c4ba6.
Tatarinova T, Neely M, Bartroff J, van Guilder M, Yamada W, Bayard D, et al. Two general methods for population pharmacokinetic modeling: non-parametric adaptive grid and non-parametric Bayesian. J Pharmacokinet Pharmacodyn. 2013;40(2):189–99. https://doi.org/10.1007/s10928-013-9302-8.
Launay-Iliadis MC, Bruno R, Cosson V, Vergniol JC, Oulid-Aissa D, Marty M, et al. Population pharmacokinetics of docetaxel during phase I studies using nonlinear mixed-effect modeling and nonparametric maximum-likelihood estimation. Cancer Chemother Pharmacol. 1995;37(1–2):47–54.
Vermes A, Mathot RA, van der Sijs IH, Dankert J, Guchelaar HJ. Population pharmacokinetics of flucytosine: comparison and validation of three models using STS, NPEM, and NONMEM. Ther Drug Monit. 2000;22(6):676–87.
Patoux A, Bleyzac N, Boddy AV, Doz F, Rubie H, Bastian G, et al. Comparison of nonlinear mixed-effect and non-parametric expectation maximisation modelling for Bayesian estimation of carboplatin clearance in children. Eur J Clin Pharmacol. 2001;57(4):297–303.
de Hoog M, Schoemaker RC, van den Anker JN, Vinks AA. NONMEM and NPEM2 population modeling: a comparison using tobramycin data in neonates. Ther Drug Monit. 2002;24(3):359–65.
Bustad A, Terziivanov D, Leary R, Port R, Schumitzky A, Jelliffe R. Parametric and nonparametric population methods: their comparative performance in analysing a clinical dataset and two Monte Carlo simulation studies. Clin Pharmacokinet. 2006;45(4):365–83. https://doi.org/10.2165/00003088-200645040-00003.
Carlsson KC, van de Schootbrugge M, Eriksen HO, Moberg ER, Karlsson MO, Hoem NO. A population pharmacokinetic model of gabapentin developed in nonparametric adaptive grid and nonlinear mixed effects modeling. Ther Drug Monit. 2009;31(1):86–94. https://doi.org/10.1097/FTD.0b013e318194767d.
Bulitta JB, Landersdorfer CB, Kinzig M, Holzgrabe U, Sorgel F. New semiphysiological absorption model to assess the pharmacodynamic profile of cefuroxime axetil using nonparametric and parametric population pharmacokinetics. Antimicrob Agents Chemother. 2009;53(8):3462–71. https://doi.org/10.1128/AAC.00054-09.
Bulitta JB, Landersdorfer CB, Huttner SJ, Drusano GL, Kinzig M, Holzgrabe U, et al. Population pharmacokinetic comparison and pharmacodynamic breakpoints of ceftazidime in cystic fibrosis patients and healthy volunteers. Antimicrob Agents Chemother. 2010;54(3):1275–82. https://doi.org/10.1128/AAC.00936-09.
Premaud A, Weber LT, Tonshoff B, Armstrong VW, Oellerich M, Urien S, et al. Population pharmacokinetics of mycophenolic acid in pediatric renal transplant patients using parametric and nonparametric approaches. Pharmacol Res. 2011;63(3):216–24. https://doi.org/10.1016/j.phrs.2010.10.017.
Woillard JB, Debord J, Benz-de-Bretagne I, Saint-Marcoux F, Turlure P, Girault S, et al. A time-dependent model describes methotrexate elimination and supports dynamic modification of MRP2/ABCC2 activity. Ther Drug Monit. 2017;39(2):145–56. https://doi.org/10.1097/FTD.0000000000000381.
Woillard JB, Lebreton V, Neely M, Turlure P, Girault S, Debord J, et al. Pharmacokinetic tools for the dose adjustment of ciclosporin in haematopoietic stem cell transplant patients. Br J Clin Pharmacol. 2014;78(4):836–46. https://doi.org/10.1111/bcp.12394.
Baverel PG, Savic RM, Wilkins JJ, Karlsson MO. Evaluation of the nonparametric estimation method in NONMEM VI: application to real data. J Pharmacokinet Pharmacodyn. 2009;36(4):297–315. https://doi.org/10.1007/s10928-009-9122-z.
Roberts JA, Lipman J. Pharmacokinetic issues for antibiotics in the critically ill patient. Crit Care Med. 2009;37(3):840–51. https://doi.org/10.1097/ccm.0b013e3181961bff(quiz 59).
Merck Sharp & Dohme BV. Summary of product characteristics Tienam 500/500 mg powder for solution for infusion. The Netherlands, Haarlem, 2015. https://www.geneesmiddeleninformatiebank.nl/smpc/h11089_smpc.pdf.
Buckley MM, Brogden RN, Barradell LB, Goa KL. Imipenem/cilastatin. A reappraisal of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy. Drugs. 1992;44(3):408–44. https://doi.org/10.2165/00003495-199244030-00008.
Baptista JP, Neves M, Rodrigues L, Teixeira L, Pinho J, Pimentel J. Accuracy of the estimation of glomerular filtration rate within a population of critically ill patients. J Nephrol. 2014;27(4):403–10. https://doi.org/10.1007/s40620-013-0036-x.
Huttner A, Von Dach E, Renzoni A, Huttner BD, Affaticati M, Pagani L, et al. Augmented renal clearance, low beta-lactam concentrations and clinical outcomes in the critically ill: an observational prospective cohort study. Int J Antimicrob Agents. 2015;45(4):385–92. https://doi.org/10.1016/j.ijantimicag.2014.12.017.
Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41.
Johansson AM, Karlsson MO. Comparison of methods for handling missing covariate data. AAPS J. 2013;15(4):1232–41. https://doi.org/10.1208/s12248-013-9526-y.
Legrand T, Chhun S, Rey E, Blanchet B, Zahar JR, Lanternier F, et al. Simultaneous determination of three carbapenem antibiotics in plasma by HPLC with ultraviolet detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;875(2):551–6. https://doi.org/10.1016/j.jchromb.2008.09.020.
Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometr Syst Pharmacol. 2013;2:e50. https://doi.org/10.1038/psp.2013.24.
Boeckmann A, Sheiner L, Beal S. NONMEM users guide—Part V. Ellicott City, MD: ICON Development Solutions; 2011. https://nonmem.iconplc.com/nonmem720/guides/v.pdf.
Byon W, Smith MK, Chan P, Tortorici MA, Riley S, Dai H, et al. Establishing best practices and guidance in population modeling: an experience with an internal population pharmacokinetic analysis guidance. CPT Pharmacometr Syst Pharmacol. 2013;2:e51. https://doi.org/10.1038/psp.2013.26.
Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development—part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometr Syst Pharmacol. 2013;2:e38. https://doi.org/10.1038/psp.2013.14.
Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development. CPT Pharmacometr Syst Pharmacol. 2012;1:e6. https://doi.org/10.1038/psp.2012.4.
Elassaiss-Schaap J, Heisterkamp SH. Variability as constant coefficient of variation: can we right two decades in error? Population Approach Group in Europe (PAGE) meeting; St Petersburg, 23–26 June 2009.
Hutmacher MM, Kowalski KG. Covariate selection in pharmacometric analyses: a review of methods. Br J Clin Pharmacol. 2015;79(1):132–47. https://doi.org/10.1111/bcp.12451.
University of Southern California Laboratory of Applied Pharmacokinetics and Bioinformatics. Pmetrics User Manual, May 2016.
Rousseeuw PJ, Croux C. Alternatives to the median absolute deviation. J Am Stat Assoc. 1993;88(424):1273–83. https://doi.org/10.1080/01621459.1993.10476408.
Alobaid AS, Hites M, Lipman J, Taccone FS, Roberts JA. Effect of obesity on the pharmacokinetics of antimicrobials in critically ill patients: a structured review. Int J Antimicrob Agents. 2016;47(4):259–68. https://doi.org/10.1016/j.ijantimicag.2016.01.009.
Levey AS, Coresh J, Greene T, Marsh J, Stevens LA, Kusek JW, et al. Expressing the Modification of Diet in Renal Disease Study equation for estimating glomerular filtration rate with standardized serum creatinine values. Clin Chem. 2007;53(4):766–72. https://doi.org/10.1373/clinchem.2006.077180.
Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604–12.
Jelliffe R. Estimation of creatinine clearance in patients with unstable renal function, without a urine specimen. Am J Nephrol. 2002;22(4):320–4.https://doi.org/10.1159/000065221
Du Bois D, Du Bois EF. A formula to estimate the approximate surface area if height and weight be known. Nutrition. 1989;5(5):303–11 (discussion 12–3).
Boxenbaum H. Interspecies pharmacokinetic scaling and the evolutionary-comparative paradigm. Drug Metab Rev. 1984;15(5–6):1071–121. https://doi.org/10.3109/03602538409033558.
Willett JB, Singer JD. Another cautionary note about R2: its use in weighted least-squares regression analysis. Am Stat. 1988;42(3):236–8.
Roberts JA, Kirkpatrick CM, Lipman J. Monte Carlo simulations: maximizing antibiotic pharmacokinetic data to optimize clinical practice for critically ill patients. J Antimicrob Chemother. 2011;66(2):227–31. https://doi.org/10.1093/jac/dkq449.
Couffignal C, Pajot O, Laouenan C, Burdet C, Foucrier A, Wolff M, et al. Population pharmacokinetics of imipenem in critically ill patients with suspected ventilator-associated pneumonia and evaluation of dosage regimens. Br J Clin Pharmacol. 2014;78(5):1022–34. https://doi.org/10.1111/bcp.12435.
Sakka SG, Glauner AK, Bulitta JB, Kinzig-Schippers M, Pfister W, Drusano GL, et al. Population pharmacokinetics and pharmacodynamics of continuous versus short-term infusion of imipenem–cilastatin in critically ill patients in a randomized, controlled trial. Antimicrob Agents Chemother. 2007;51(9):3304–10. https://doi.org/10.1128/AAC.01318-06.
Suchankova H, Lips M, Urbanek K, Neely MN, Strojil J. Is continuous infusion of imipenem always the best choice? Int J Antimicrob Agents. 2017;49(3):348–54. https://doi.org/10.1016/j.ijantimicag.2016.12.005.
Hobbs AL, Shea KM, Roberts KM, Daley MJ. Implications of augmented renal clearance on drug dosing in critically ill patients: a focus on antibiotics. Pharmacotherapy. 2015;35(11):1063–75. https://doi.org/10.1002/phar.1653.
Udy AA, Roberts JA, Boots RJ, Paterson DL, Lipman J. Augmented renal clearance: implications for antibacterial dosing in the critically ill. Clin Pharmacokinet. 2010;49(1):1–16. https://doi.org/10.2165/11318140-000000000-00000.
Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res. 2007;24(12):2187–97. https://doi.org/10.1007/s11095-007-9361-x.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–51. https://doi.org/10.1208/s12248-011-9255-z.
European Medicines Agency. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. London; 2015. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-decreased-renal-function_en.pdf.
Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney International Supplements. 2013;3(1):1–150.
Barletta JF, Mangram AJ, Byrne M, Hollingworth AK, Sucher JF, Ali-Osman FR, et al. The importance of empiric antibiotic dosing in critically ill trauma patients: are we under-dosing based on augmented renal clearance and inaccurate renal clearance estimates? J Trauma Acute Care Surg. 2016;81(6):1115–21. https://doi.org/10.1097/ta.0000000000001211.
Minichmayr IK, Roberts JA, Frey OR, Roehr AC, Kloft C, Brinkmann A. Development of a dosing nomogram for continuous-infusion meropenem in critically ill patients based on a validated population pharmacokinetic model. J Antimicrob Chemother. 2018;73(5):1330–9. https://doi.org/10.1093/jac/dkx526.
Baverel PG, Savic RM, Karlsson MO. Two bootstrapping routines for obtaining imprecision estimates for nonparametric parameter distributions in nonlinear mixed effects models. J Pharmacokinet Pharmacodyn. 2011;38(1):63–82. https://doi.org/10.1007/s10928-010-9177-x.
Savic RM, Karlsson MO. Evaluation of an extended grid method for estimation using nonparametric distributions. AAPS J. 2009;11(3):615–27. https://doi.org/10.1208/s12248-009-9138-8.
Savic RM, Kjellsson MC, Karlsson MO. Evaluation of the nonparametric estimation method in NONMEM VI. Eur J Pharm Sci. 2009;37(1):27–35. https://doi.org/10.1016/j.ejps.2008.12.014.
Acknowledgements
This paper is dedicated to the memory of Prof. Dr. Johan W. Mouton who passed away on 9 July 2019.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Funding
This work was supported by the Innovative Medicines Initiative Joint Undertaking under Grant agreement no. 115523, the resources of which are composed of a financial contribution from the European Union’s Seventh Framework Programme (FP7/2007–2013) and EFPIA companies’ in-kind contribution. The research leading to these results was conducted as part of the COMBACTE-NET consortium. For further information please refer to http://www.combacte.com/. The initial cohort study was funded by a research and development Grant awarded by the Geneva University Hospitals in 2009 (PRD 09-II-025). AH and EvD were partially supported by the EU-funded project AIDA (Grant Health-F3-2011-278348).
Conflict of interest
Femke de Velde, Brenda de Winter, Michael Neely, Walter Yamada, Elodie von Dach and Angela Huttner declare they have no conflicts of interest. Johan Mouton has received research funding from IMI, the EU, ZonMw (Dutch governmental support), Adenium, AstraZeneca, Basilea, Eumedica, Cubist, Merck & Co., Pfizer, Polyphor, Roche, Shionogi, Thermo-Fisher, Wockhardt, Astellas, Gilead and Pfizer. Birgit Koch has received research funding from ZonMw (Dutch governmental support) and Teva. Stephan Harbarth has received honoraria from Sandoz for participation in a Scientific Advisory Board. Teun van Gelder has received honoraria as a consultant/speaker from Aurinia Pharma, Vitaeris, Roche Diagnostics, Novartis, Astellas and Chiesi, along with grant support for transplant-related studies from Chiesi and Astellas.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee (Geneva University Hospital’s Ethics Committee, NAC 09-117) and with the 1964 Helsinki declaration and its later amendments.
Informed Consent
Informed consent was obtained from individual participants included in this study, unless patients were unconscious or otherwise unable to understand the study protocol. This was approved by the abovementioned Ethics Committee, given the observational nature of the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
de Velde, F., de Winter, B.C.M., Neely, M.N. et al. Population Pharmacokinetics of Imipenem in Critically Ill Patients: A Parametric and Nonparametric Model Converge on CKD-EPI Estimated Glomerular Filtration Rate as an Impactful Covariate. Clin Pharmacokinet 59, 885–898 (2020). https://doi.org/10.1007/s40262-020-00859-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00859-1