
Abstract
Background and Objectives
Caspofungin is an echinocandin antifungal agent used as first-line therapy for the treatment of invasive candidiasis. The maintenance dose is adapted to body weight (BW) or liver function (Child-Pugh score B or C). We aimed to study the pharmacokinetics of caspofungin and assess pharmacokinetic target attainment for various dosing strategies.
Methods
Caspofungin pharmacokinetic data from 21 intensive care unit (ICU) patients was available. A population pharmacokinetic model was developed. Various dosing regimens (loading dose/maintenance dose) were simulated: licensed regimens (I) 70/50 mg (for BW <80 kg) or 70/70 mg (for BW >80 kg); and (II) 70/35 mg (for Child-Pugh score B); and adapted regimens (III) 100/50 mg (for Child-Pugh score B); (IV) 100/70 mg; and (V) 100/100 mg. Target attainment based on a preclinical pharmacokinetic target for Candida albicans was assessed for relevant minimal inhibitory concentrations (MICs).
Results
A two-compartment model best fitted the data. Clearance was 0.55 L/h and the apparent volumes of distribution in the central and peripheral compartments were 8.9 and 5.0 L, respectively. The median area under the plasma concentration–time curve from time zero to 24 h on day 14 for regimens I–V were 105, 65, 93, 130, and 186 mg·h/L, respectively. Pharmacokinetic target attainment was 100 % (MIC 0.03 µg/mL) irrespective of dosing regimen but decreased to (I) 47 %, (II) 14 %, (III) 36 %, (IV) 69 %, and (V) 94 % for MIC 0.125 µg/mL.
Conclusion
The caspofungin maintenance dose should not be reduced in non-cirrhotic ICU patients based on the Child-Pugh score if this classification is driven by hypoalbuminemia as it results in significantly lower exposure. A higher maintenance dose of 70 mg in ICU patients results in target attainment of >90 % of the ICU patients with species with an MIC of up to 0.125 µg/mL.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A population pharmacokinetic model of caspofungin in critically ill patients greatly assisted in applying simulations to derive pharmacokinetic target attainment. |
The caspofungin dose should not be reduced in non-cirrhotic intensive care unit (ICU) patients classified as Child-Pugh B or C if this classification is driven by hypoalbuminemia. |
A higher maintenance dose of caspofungin 70 mg in ICU patients results in target attainment of >90 % of the ICU patients with species with a minimal inhibitory concentration of up to 0.125 µg/mL. |
1 Introduction
Critically ill patients are at increased risk for infections and about 20 % of infections in intensive care unit (ICU) patients are caused by fungal infections such as by Candida or Aspergillus spp. [1]. Caspofungin is an echinocandin antifungal agent licensed as a first-line therapy for invasive candidiasis in patients with moderate to severe illness and/or recent exposure to azoles [2]. The efficacy of echinocandin therapy for the treatment of invasive candidiasis is higher than with other antifungal agents. An adequate response to echinocandins is generally achieved in 66–90 % of patients [3, 4]. Echinocandins work by inhibiting the synthesis of β-(1,3)-d-glucan, an important component of the fungal cell wall.
Caspofungin is administered intravenously and the dosage is based on body weight (BW). The clinical guideline of the International Infectious Disease Society recommends a loading dose of 70 mg and subsequent daily maintenance doses of 50 mg [2]. In addition, the maintenance dose is recommended to be increased to 70 mg for patients with BW >80 kg and decreased to 35 mg for patients with moderate or severe hepatic impairment, classified as Child-Pugh B or C [5, 6]. The Child-Pugh score is a marker for severity of liver function impairment that is composed of several markers of liver function such as albumin serum values and the presence of ascites [7].
Recently, we analyzed the pharmacokinetics of caspofungin in 21 ICU patients using a non-compartmental approach [8]. Total caspofungin exposure (area under the plasma concentration–time curve [AUC] from time zero to 24 h [AUC24]) did not seem to be altered in this population as compared with other populations, even though dose reductions would have been indicated in most patients based on their Child-Pugh scores [8]. It should be noted that Child-Pugh scoring was developed in patients with cirrhosis and not in ICU patients. Large inter-individual variability (IIV) in caspofungin exposures was observed among ICU patients [8].
Next to pharmacokinetic considerations, pharmacodynamic factors should be taken into account to identify the best dose strategy for caspofungin in ICU patients. Various studies have shown that AUC/minimal inhibitory concentration (MIC) best described the pharmacokinetic–pharmacodynamic relationship for caspofungin, micafungin, and anidulafungin [9–11]. Murine infection models of caspofungin explored the target AUC/MIC ratio associated with efficacy [9, 11]. An AUC/MIC ratio of 865 was associated with a 1-log kill/24 h in a neutropenic mouse model of disseminated Candida albicans [11, 12]. To date, a caspofungin pharmacokinetic–pharmacodynamic target in humans has not yet been established.
The current study analyzed the pharmacokinetics of caspofungin in ICU patients using non-linear mixed–effects modelling to obtain a better mechanistic understanding of the pharmacokinetic properties of the drug and to be able to characterize potential covariate relationships with higher statistical power. We were specifically interested in the effects of BW and Child-Pugh scores on exposures, as the current dosing of caspofungin is adapted based on these parameters. In addition, non-linear mixed–effects modelling allows simulations of different dosing regimens with corresponding exposures and assessment of target attainment for these regimens.
2 Methods
2.1 Study Design, Drug Regimen, and Population
The study was approved by the local ethics committee (Commissie Mensgebonden Onderzoek regio Arnhem-Nijmegen number 2011/346, ClinicalTrials.gov identifier NCT01533558 [38]) and informed consent was given by all participants. Inclusion criteria included admission to the ICU, caspofungin therapy for suspected or proven infection or for prophylaxis, age >18 years when starting caspofungin, started therapy a maximum of 2 days before inclusion, and management with a central venous catheter. Exclusion criteria included allergy for echinocandins or excipients, known HIV, hepatitis B or C infection, or a history of drug or alcohol abuse.
Dosing was as prescribed by the physician in attendance: a 70 mg loading dose on day 1 followed by either 50 mg maintenance daily for patients weighing ≤80 kg or a 70 mg maintenance for patients with BW >80 kg. The maintenance dose should be reduced to 35 mg for patients classified as Child-Pugh B or C. Caspofungin was administered intravenously over approximately 1 h. Patients were treated as long as clinically relevant but the duration of the study (i.e., sampling of patients) was limited to 14 days of caspofungin treatment. If patients stopped treatment before 14 days, pharmacokinetic washout samples until 3 days after cessation of therapy were taken.
Patient demographics were collected and included sex, age, race, weight, body mass index (BMI), lean body mass (LBM [13]), indication for ICU admission, indication for caspofungin, clinical characteristics, chemistry, and hematological parameters. In addition, the APACHE II score (Acute Physiology and Chronic Health Evaluation II; severity of disease classification) within 24 h of ICU admission, SOFA score (Sequential Organ Failure Assessment score, to assess patients during their stay at an ICU) and Child–Pugh class, co-medication, and (type of) renal replacement therapy were recorded.
2.2 Pharmacokinetic Sampling and Analysis
Patients were intensively sampled (2 mL) on days 3 (±1) and 7 (±1) of therapy at t = 0 (pre-dose) and 0.5, 1, 2, 4, 6, 8, 12, 16, 20, and 24 h post-dose, and additional trough samples (pre-dose) were taken on other study days. Details on the sample preparation and the assay (validated ultra-performance liquid chromatography–fluorescence method) have been described previously [8].
2.3 Pharmacokinetic Model
One-, two-, and three-compartment models were considered based on a review of the literature and on visual inspection of the data. IIV was estimated using an exponential model. The primary pharmacokinetic parameters were clearance (CL) and volume of distribution (V d). Proportional, additive, and combined additive and proportional models were evaluated for residual variability. Both a single covariance matrix on the first compartment and multiple omega blocks for the individual compartments as a full covariance matrix on all compartments were considered for the inter-individual random effects.
Model selection was initially based on the objective function value (OFV) computed as –2 log likelihood, where a decrease in OFV of ≥3.84 was considered significant (Chi-squared [χ 2], 1 degree of freedom [df], p < 0.05). In addition, standard goodness-of-fit (GOF) plots, including the observed versus the population- and individual-predicted concentrations, and conditional weighted residuals versus time after dose and population-predicted concentration were used for model evaluation. Also, the precision of the parameter estimates, eta-shrinkage, and IIV was assessed. Good candidate models were further evaluated by a visual predictive check (VPC).
After selection of the base model, various covariates were tested on CL and V d by a stepwise covariate model (scm) as implemented in Perl-speaks-NONMEM (PsN 4.2.0), using forward selection and backward elimination [14]. Only baseline values were considered. Continuous covariates included age, BW, length, LBM, BMI, body temperature, serum creatinine, ureum, albumin, liver enzymes (alanine aminotransferase, aspartate aminotransferase, γ-glutamyl transpeptidase, alkaline phosphatase, bilirubin), C-reactive protein, blood pH, and glomerular filtration rate (GFR). Categorical covariates included sex, Child-Pugh score, and abnormal/normal GFR (breakpoint >60 mL/min). All covariates were tested on both CL and apparent volume of the central compartment (V 1). Linear, exponential, and power conditions were tested for continuous covariates and linear conditions on categorical data, all using a forward inclusion criterion of p < 0.05 (OFV decrease of ≥3.84, χ 2, 1 df) and a backward exclusion criterion of p < 0.01 (OFV increase of ≥6.64, χ 2, 1 df).
Based on physiological plausibility and extensive previous evidence, BW was incorporated a priori as a covariate, both on CL (allometrically with a power exponent of 0.75) and on V d (power exponent of 1) and was standardized to a typical 70 kg patient [15–18].
To assess the predictive performance of the final model, a prediction-corrected VPC (pcVPC) was made, based on 1000 Monte-Carlo simulations. The precision of the parameter estimates of the final model was further evaluated using a non-parametric bootstrap method in which re-sampling of the dataset was performed 1000 times to produce new datasets with the same size but containing a different combination of individuals and yielding new parameter estimates and confidence intervals.
Population pharmacokinetic modelling was carried out by non-linear mixed–effect modelling using NONMEM® version 7.2 (ICON Development solutions, Ellicott City, MA, USA), PsN (version 4.2.0), and Xpose (version 4.5.3) [14, 19, 20]. The Pirana® interface was used for run interpretation [21]. The first-order conditional estimation method with interaction was used for the analysis. R (version 3.1.10) was used for exploratory graphical analysis and for evaluation of the GOF [22].
2.4 Monte-Carlo Simulations of Different Dosing Regimens
After selection of the final model, a simulation study was performed using this model to assess exposure and probability of target attainment following various alternative dosing regimens. As our ICU cohort was too small (n = 21) for a representative weight distribution for the population, the weight distribution from a hospital-based cohort of 1706 adult patients from the hematology department (2007–2014) was used to build a valid dataset for simulation purposes. Based on our clinical experience and on the literature [23, 24], it was assumed that the anthropometric characteristics in this cohort were similar to that of a typical ICU population. The cohort consisted of 61 % men (n = 672) and had a median age (range) of 59 years (18–81), median weight (range) of 76 kg (39–154) and median BMI (range) of 24.7 (13–42).
Five different caspofungin dosing regimens were simulated: two licensed regimens and three alternative regimens. These regimens were chosen at the discretion of the researcher and based on empiric rationale. The concentration–time curves for these regimens were simulated for the aforementioned patient cohort, without replications. The licensed regimens included (I) a loading dose of 70 mg followed by 50 mg maintenance in patients with BW ≤80 kg or by 70 mg maintenance in patients with BW >80 kg; and (II) 70 mg loading dose followed by 35 mg as labeled for patients with moderate or severe hepatic dysfunction [2, 5, 6]. Alternative regimens, all irrespective of BW, included (III) a 100 mg loading dose followed by 50 mg maintenance; (IV) a 100 mg loading dose followed by 70 mg maintenance; and (V) a 100 mg loading dose followed by 100 mg maintenance.
Predicted exposure to caspofungin in terms of AUC24 was assessed on days 3 and 14 of therapy. Differences in exposure both between treatment days within the same regimen and between regimens on the same day of treatment were statistically tested with the Wilcoxon signed rank and Wilcoxon rank sum test, respectively.
2.5 Pharmacokinetic Target Attainment
The human clinical pharmacokinetic–pharmacodynamic target for caspofungin in the treatment of invasive candidiasis has yet to be elucidated. Nevertheless, the preclinical target has been determined in a neutropenic murine model of disseminated Candida infection for which a minimally required AUC/MIC ratio of 865 (1-log kill/24 h) was determined for C. albicans [11]. This AUC/MIC ratio was set as preclinical pharmacokinetic target to attain the current study. Target attainment at day 14 following the different dosing regimens was assessed for a wide range of clinically relevant MIC values (0.007–1.0 µg/mL). The mode MIC for C. albicans is 0.03 µg/mL and the epidemiological cut-off value is 0.12 µg/mL [25, 26].
3 Results
3.1 Patients, Dosing, and Samples
In total, 21 ICU patients treated with caspofungin were included, yielding a total of 419 pharmacokinetic observations. Patient characteristics and their baseline values are summarized in Table 1. All patients were classified with Child-Pugh score B and all patients had hypoalbuminemia (≤34 g/L). Despite the fact that all patients should have received a reduced maintenance dose (35 mg/day) according to the label based on their Child-Pugh score, only one of 21 patients actually received this reduced dose. This patient received 35 mg/day for 3 of the total 13 treatment days. In addition, four of eight patients with BW >80 kg received a 50 mg/day maintenance dose instead of the label-indicated 70 mg/day and two of 13 patients with BW ≤80 kg received a 70 mg/day maintenance dose instead of the label-indicated 50 mg/day.
3.2 Population Pharmacokinetic Model
A two-compartment disposition model with first-order elimination from the central compartment and a combined proportional and additive residual error model fitted the data best. The addition of a third compartment led to a significant increase in OFV. IIV on CL, V 1 and volume of distribution of compartment 2 (V 2) improved the model, while the data did not support estimation of IIV on Q (intercompartmental clearance). Allowing a correlation between the IIV on CL and V 1 further improved the model (difference in OFV = 8.6). Parameter estimates of the final model are shown in Table 2. CL, V 1, Q, and V 2 were estimated to be 0.55, 8.93, 0.71, and 4.98. The IIV of CL, V 1, and V 2 were estimated to be 30.7, 25.6, and 75.8 % with eta shrinkage of 0, 6, and 16 %, respectively. Stepwise covariate modelling could not identify any additional covariates significantly affecting CL or V 1. Of note, the Child-Pugh score could not be confirmed as a covariate for CL or V 1 either.
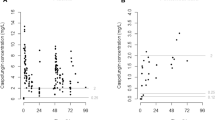
A pcVPC of the final model is shown in Fig. 1. No deviating trends were observed in the pcVPC, suggesting a good predictive performance of the model to the data. This was confirmed by the numerical predictive check, which indicated that 1.4 % (95 % CI 0–7.9) of the observed values lay below the 95 % prediction interval, and 4.1 % (95 % CI 0–7.7) fell above the 95 % prediction interval. Parameter precisions using a bootstrap re-sampling approach of the final model are listed in Table 2. Basic GOF plots are shown in Fig. 2. No major deviations were detected from the plots.

Visual predictive check for the final pharmacokinetic model of caspofungin, based on n = 1000 simulations. Prediction-corrected simulated (shaded areas) and observed (circles and lines) caspofungin concentrations versus time after dose (h). The thick red line connects the observed median values per bin. The solid blue lines connect the 5th and 95th percentiles of the observations. The blue areas are the 95 % confidence interval of the 5th and 95th percentiles. The red area indicates the confidence interval of the median

Goodness-of-fit plots for the final pharmacokinetic model of caspofungin. The solid black lines indicate the unit line or the line of identity. The thick lines (red or blue) are smooth lines showing the trend in the observations. CWRES conditional weighted residuals
3.3 Monte-Carlo Simulations of Alternative Dosing Regimens
Alternative dose regimen simulations were performed in a cohort of 1706 hospitalized patients. AUC24 values on days 3 and 14 achieved by different dosing regimens are shown in Fig. 3. Median (range) AUC24 values on day 3 for regimens I–V were 96 (39–405), 65 (22–241), 93 (33–344), 122 (44–444), and 167 (62–594) mg·h/L. Median (range) AUC24 values on day 14 for regimens I–V were 105 (39–459), 65 (22–271), 93(32–387), 130 (44–541), and 186 (64–772) mg·h/L, respectively, showing that the regimen with a 70 mg loading dose followed by 35 mg as labeled for patients with a Child-Pugh score of B (regimen II) resulted in the lowest median exposure (65 mg·h/L). The Wilcoxon signed rank test confirmed significant differences in exposure between days 3 and 14 within one regimen for all regimens (p < 0.01). There was also a significant difference in exposure (AUC24) between all five regimens at day 14 (p < 0.01, Wilcoxon rank-sum).

a AUC24 on day 3 for the whole cohort, irrespective of body weight. b AUC24 on day 14 for the whole cohort, irrespective of body weight. The horizontal line represents the AUC24 for healthy volunteers. Regimens: (I) loading dose of 70 mg followed by 50 mg maintenance in patients with body weight ≤80 kg or by 70 mg maintenance in patients with body weight >80 kg; (II) 70 mg loading dose followed by 35 mg; (III) 100 mg loading dose followed by 50 mg maintenance; (IV) 100 mg loading dose followed by 70 mg maintenance; and (V) 100 mg loading dose followed by 100 mg maintenance. AUC area under the concentration–time curve, AUC 24 AUC from time zero to 24 h
Predicted variability in exposure was considerable, with coefficient of variation percentages in AUC24 of 44–45 and 46–47 % for days 3 and 14, respectively, for all regimens.
Typical simulated plasma concentration–time profiles with 95 % confidence intervals during 2 weeks of treatment following the different dosing regimens are shown in Fig. 4. As expected, a higher loading dose resulted in higher exposure early in therapy (regimens III and IV vs. regimen I). Steady state was usually reached at day 7 (168 h).

Caspofungin concentration–time curve for a regimen I: a loading dose of 70 mg followed by 50 mg maintenance in patients with body weight ≤80 kg or by 70 mg maintenance in patients with body weight >80 kg; b regimen II: 70 mg loading dose followed by 35 mg as labeled for patients with moderate or severe hepatic dysfunction [2, 5, 6]; c regimen III: a 100 mg loading dose followed by 50 mg maintenance; d regimen IV: a 100 mg loading dose followed by 70 mg maintenance; and e regimen V: a 100 mg loading dose followed by 100 mg maintenance. The thick black lines are medians and the dotted lines are the 5 and 95 % percentiles. Conc concentration
3.4 Pharmacokinetic Target Attainment
Simulated attainment of pharmacokinetic targets for the various dose regimens is shown in Fig. 5, and Table 3 shows the probabilities of target attainment stratified by BW.

Target attainment versus MIC for all five simulated regimens based on a preclinical target AUC/MIC ratio of >865. Asterisk Indicates that the regimen is based on body weight: the maintenance dose was 50 mg for patients with body weight ≤80 kg and 70 mg for body weight >80 kg. AUC area under the concentration-time curve, MIC minimal inhibitory concentration
Both licensed regimen I (based on BW) and II (reduced based on Child-Pugh B) resulted in a pharmacokinetic target attainment of >99 % using a MIC of 0.03 µg/mL (mode).
Not surprisingly, the probability of target attainment increased with increasing doses. For example, a substantial difference in target attainment was observed between the licensed regimen II (reduced dosing based on Child-Pugh score B) and the alternative regimen V (100 mg loading + 100 mg/day maintenance): 14 versus 97 %, respectively, for an MIC of 0.125 µg/mL. From a MIC of 0.125 µg/mL, the differences in pharmacokinetic target attainment between the dosing regimens become more important. Thus, for infections with species with higher MICs, though occurring less frequently, dose will probably have an important effect on outcome.
Higher BW consistently resulted in decreased target attainment (Table 3), and this effect was statistically significant (p < 0.01). This decreased target attainment was the direct result of a relatively decreased AUC24 for higher BW patients. Certainly for the higher BW ranges combined with a Child-Pugh B score, better target attainment was observed with higher maintenance doses (regimen II vs. III) (Table 3).
Better target attainment was observed with higher loading and maintenance doses than suggested by the label, as depicted in Table 3. For example, 35 mg/day maintenance (regimen II) resulted in 59–81 % target attainment and 70–100 mg/day (regimens IV and V) resulted in >99.5 % target attainment, with an MIC of 0.06 µg/mL.
4 Discussion
This is the first study reporting a population pharmacokinetic model of caspofungin in ICU patients. The developed model was successfully used to assess the probability of pharmacokinetic target attainment for various caspofungin dose regimens. The simulations revealed insufficient target attainment with the currently licensed regimens. In non-cirrhotic ICU patients classified with Child-Pugh B, reducing the dose of caspofungin is not recommended.
Caspofungin population pharmacokinetics were comparable with those reported in earlier studies [18, 27]. In contrast to other reported studies, our data did not support a third structural peripheral pharmacokinetic compartment, possibly due to the sparseness of the data in the late elimination phase. The typical CL for caspofungin was 0.55 L/h and was slightly higher than reported in earlier studies amongst other patient groups (0.40 and 0.46 L/h) [18, 27]. V d values were also slightly higher than in earlier studies [18, 27]. Moderate IIV was observed for both CL (30.7 %) and V 1 (25.6 %) and also confirmed earlier literature [18, 27]. No additional covariates, including Child-Pugh score, could be identified that significantly affected the CL or V 1 of caspofungin in the current study. On the basis of previous results and on the well-established general physiological mechanisms supporting the standard allometric scaling of compound metabolism and disposition [28–30], BW was added a priori allometrically on CL (exponent of 0.75) and V 1 (exponent of 1). This approach is widely accepted if the drug of choice distributes widely into tissues, strengthening the assumption of BW relating to V 1 [31]. Metabolism of caspofungin involves spontaneous degradation, hydrolysis and N-acetylation, processes expected to be related to BW also [5, 31].
Conflicting results on covariates and caspofungin pharmacokinetics exist in the literature. One study could not find any covariates significantly affecting the pharmacokinetic parameters [27], whereas another study found a linear relationship between BW and CL and V 1 [18]. In a study on the pharmacokinetics of caspofungin in obese subjects, it was found that BW was allometrically related to CL (power 0.75) for subjects weighing >66.3 kg (n = 14). In addition, increased BW was associated with both lower maximum (peak) concentrations (C max) and lower AUC from time zero to 72 h (AUC72) values [16].
Despite our attempt to approach this by non-linear mixed–effect modelling, no covariate could be identified on top of a priori allometric scaling of CL and V 1 by BW. In our cohort, all ICU patients were classified as Child-Pugh B (mainly driven by their albumin status), making it impossible to distinguish between classes. To accurately identify the effect of cirrhosis (using Child-Pugh classification) on caspofungin exposure, a more heterogeneous cohort in terms of liver function should be studied. In addition, pharmacokinetics in ICU patients are infamously variable due to a mixture of factors (e.g., systemic inflammatory response, capillary leak, protein binding capacity) [32]. As such, covariates might be obscured due the overall high variability in ICU patients and our limited sample size.
Simulation of different dosing regimens showed that reducing the maintenance dose to 35 mg, which is recommended for patients with moderate to severe hepatic dysfunction (classified as Child-Pugh B or C), resulted in the lowest average exposure. The median AUC24 was 65 mg·h/L for this regimen, which is far below the 100 mg·h/L typically observed for caspofungin [8, 33]. As the Child-Pugh score is highly driven by albumin, ICU patients with hypoalbuminemia are often unfairly classified with Child-Pugh score B. In our cohort, all patients had an albumin of <28 g/L, scoring them automatically with 7 points (Child-Pugh B). Decreasing the dose in ICU patients with a Child-Pugh B score without liver cirrhosis will lead to unnecessary low exposure, risking loss of efficacy of caspofungin. As caspofungin is highly protein bound (92–97 %), hypoalbuminemia might lead to changes in caspofungin free fraction [31]. It is currently unknown whether hypoalbuminemia can influence the pharmacokinetics of caspofungin.
Due to a lack of a clinically validated pharmacokinetic–pharmacodynamic target for caspofungin, a preclinical pharmacokinetic–pharmacodynamic target was used. It is not the target attainment itself but rather the magnitude change with various dosing regimens in relation to pathogen susceptibility that is important. Despite the use of a preclinical target, our results are in line with reported response rates of caspofungin in the literature, which show that clinical and mycological cure at the end of therapy is usually 72–73 % [2, 34]. Low exposure to caspofungin might result in clinical failure; high exposure, on the other hand, does not appear to lead to safety precautions: caspofungin has an overall favorable safety profile, and therapy with 150 mg daily is well-tolerated in patients with invasive candidiasis [35–37]. To achieve an optimal probability of target attainment, we recommend the use of a caspofungin 70–100 mg maintenance dose in the setting of an infection with pathogens with an attenuated MIC (>0.125 µg/mL). Irrespective of the susceptibility profile in the invading pathogen, patients may benefit from a higher loading dose of 100 mg on day 1 to achieve early adequate exposure (see Fig. 4).
Some challenges remain for future work. First of all, data pooling is clearly needed to achieve sufficient power to detect relevant covariates specifically in the light of the heterogeneous ICU population. Clearly, a more stratified approach taking into account pathogen susceptibility needs to be validated in terms of general mycological and clinical cure. A study designed to identify the clinical breakpoint for caspofungin in invasive candidiasis is warranted. In the meantime, caspofungin dose reductions based on Child-Pugh B scores do not appear to be valid for non-cirrhotic ICU patients. Ultimately, therapeutic drug monitoring, i.e., individualized drug dosing based on the measurement and interpretation of drug concentrations recognizing the pathogen susceptibility, could be an important tool to derive an optimal exposure in the individual patient.
References
Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA. 2009;302(21):2323–9.
Pappas PG, Kauffman CA, Andes D, Benjamin DK Jr, Calandra TF, Edwards JE Jr, et al. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis. 2009;48(5):503–35.
Andes DR, Safdar N, Baddley JW, Playford G, Reboli AC, Rex JH, et al. Impact of treatment strategy on outcomes in patients with candidemia and other forms of invasive candidiasis: a patient-level quantitative review of randomized trials. Clin Infect Dis. 2012;54(8):1110–22.
Pound MW, Townsend ML, Drew RH. Echinocandin pharmacodynamics: review and clinical implications. J Antimicrob Chemother. 2010;65(6):1108–18.
European Medicines Agency. Summary of product characteristics: Cancidas 2011 [last updated 26 Oct 2014]. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000379/WC500021033.pdf. Accessed 27 Mar 2015.
US Food and Drug Administration. Cancidas label information 2014 [last updated 26 Oct 2014]. http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021227s033lbl.pdf. Accessed 27 Mar 2015.
Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60(8):646–9.
Muilwijk EW, Schouten JA, van Leeuwen HJ, van Zanten AR, de Lange DW, Colbers A, et al. Pharmacokinetics of caspofungin in ICU patients. J Antimicrob Chemother. 2014;69(12):3294–9.
Andes DR, Diekema DJ, Pfaller MA, Marchillo K, Bohrmueller J. In vivo pharmacodynamic target investigation for micafungin against Candida albicans and C. glabrata in a neutropenic murine candidiasis model. Antimicrob Agents Chemother. 2008;52(10):3497–503.
Louie A, Deziel M, Liu W, Drusano MF, Gumbo T, Drusano GL. Pharmacodynamics of caspofungin in a murine model of systemic candidiasis: importance of persistence of caspofungin in tissues to understanding drug activity. Antimicrob Agents Chemother. 2005;49(12):5058–68.
Andes D, Diekema DJ, Pfaller MA, Bohrmuller J, Marchillo K, Lepak A. In vivo comparison of the pharmacodynamic targets for echinocandin drugs against Candida species. Antimicrob Agents Chemother. 2010;54(6):2497–506.
Wiederhold NP, Kontoyiannis DP, Chi J, Prince RA, Tam VH, Lewis RE. Pharmacodynamics of caspofungin in a murine model of invasive pulmonary aspergillosis: evidence of concentration-dependent activity. J Infect Dis. 2004;190(8):1464–71.
Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean bodyweight. Clin Pharmacokinet. 2005;44(10):1051–65.
Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit–a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–57.
Hall RG, Swancutt MA, Gumbo T. Fractal geometry and the pharmacometrics of micafungin in overweight, obese, and extremely obese people. Antimicrob Agents Chemother. 2011;55(11):5107–12.
Hall RG 2nd, Swancutt MA, Meek C, Leff R, Gumbo T. Weight drives caspofungin pharmacokinetic variability in overweight and obese people: fractal power signatures beyond two-thirds or three-fourths. Antimicrob Agents Chemother. 2013;57(5):2259–64.
Hope WW, Seibel NL, Schwartz CL, Arrieta A, Flynn P, Shad A, et al. Population pharmacokinetics of micafungin in pediatric patients and implications for antifungal dosing. Antimicrob Agents Chemother. 2007;51(10):3714–9.
Wurthwein G, Cornely OA, Trame MN, Vehreschild JJ, Vehreschild MJ, Farowski F, et al. Population pharmacokinetics of escalating doses of caspofungin in a phase II study of patients with invasive aspergillosis. Antimicrob Agents Chemother. 2013;57(4):1664–71.
Beal SL, Sheiner LB, Boeckmann A. NONMEM user’s guides (1989–2006). Ellicott City: Icon Development Solutions; 2006.
Jonsson EN, Karlsson MO. Xpose–an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58(1):51–64.
Keizer RJ, van Benten M, Beijnen JH, Schellens JH, Huitema AD. Pirana and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101(1):72–9.
R Development Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2013.
Pickkers P, de Keizer N, Dusseljee J, Weerheijm D, van der Hoeven JG, Peek N. Body mass index is associated with hospital mortality in critically ill patients: an observational cohort study. Crit Care Med. 2013;41(8):1878–83.
Bruggemann RJ, Van Der Velden WJ, Knibbe CA, Colbers A, Hol S, Burger DM, et al. A rationale for reduced-frequency dosing of anidulafungin for antifungal prophylaxis in immunocompromised patients. J Antimicrob Chemother. 2015;70(4):1166–74.
Pfaller MA, Boyken L, Hollis RJ, Kroeger J, Messer SA, Tendolkar S, et al. Wild-type MIC distributions and epidemiological cutoff values for the echinocandins and Candida spp. J Clin Microbiol. 2010;48(1):52–6.
Pfaller MA, Diekema DJ, Andes D, Arendrup MC, Brown SD, Lockhart SR, et al. Clinical breakpoints for the echinocandins and Candida revisited: integration of molecular, clinical, and microbiological data to arrive at species-specific interpretive criteria. Drug Resist Updat. 2011;14(3):164–76.
Wurthwein G, Young C, Lanvers-Kaminsky C, Hempel G, Trame MN, Schwerdtfeger R, et al. Population pharmacokinetics of liposomal amphotericin B and caspofungin in allogeneic hematopoietic stem cell recipients. Antimicrob Agents Chemother. 2012;56(1):536–43.
Anderson BJ, Holford NH. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet. 2009;24(1):25–36.
Holford N, Heo YA, Anderson B. A pharmacokinetic standard for babies and adults. J Pharm Sci. 2013;102(9):2941–52.
West GB, Brown JH, Enquist BJ. A general model for the origin of allometric scaling laws in biology. Science. 1997;276(5309):122–6.
European Medicines Agency (EMA). Cancidas: EPAR scientific discussion 2004. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000379/WC500057186.pdf. Accessed 27 Mar 2015.
Blot SI, Pea F, Lipman J. The effect of pathophysiology on pharmacokinetics in the critically ill patient–concepts appraised by the example of antimicrobial agents. Adv Drug Deliv Rev. 2014;20(77):3–11.
Mistry GC, Migoya E, Deutsch PJ, Winchell G, Hesney M, Li S, et al. Single- and multiple-dose administration of caspofungin in patients with hepatic insufficiency: implications for safety and dosing recommendations. J Clin Pharmacol. 2007;47(8):951–61.
Mora-Duarte J, Betts R, Rotstein C, Colombo AL, Thompson-Moya L, Smietana J, et al. Comparison of caspofungin and amphotericin B for invasive candidiasis. N Engl J Med. 2002;347(25):2020–9.
Betts RF, Nucci M, Talwar D, Gareca M, Queiroz-Telles F, Bedimo RJ, et al. A Multicenter, double-blind trial of a high-dose caspofungin treatment regimen versus a standard caspofungin treatment regimen for adult patients with invasive candidiasis. Clin Infect Dis. 2009;48(12):1676–84.
Safdar A, Rodriguez G, Rolston KV, O’Brien S, Khouri IF, Shpall EJ, et al. High-dose caspofungin combination antifungal therapy in patients with hematologic malignancies and hematopoietic stem cell transplantation. Bone Marrow Transplant. 2007;39(3):157–64.
Cornely OA, Vehreschild JJ, Vehreschild MJ, Wurthwein G, Arenz D, Schwartz S, et al. Phase II dose escalation study of caspofungin for invasive Aspergillosis. Antimicrob Agents Chemother. 2011;55(12):5798–803.
Radboud University. Pharmacokinetics of caspofungin (Cancidas®) given intravenously as therapy to patients with an invasive fungal infection in the intensive care unit—a search for co-variates (CASCADE) [ClinicalTrials.gov identifier NCT01533558]. US National Institutes of Health, ClinicalTrials.gov. https://www.clinicaltrials.gov. Accessed 7 Nov 2015.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study has been approved by the appropriate ethics committee and has therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All participants gave their informed consent prior to their inclusion in the study.
Funding declaration
The current study was funded by the Radboud University Medical Center.
Individual funding declarations
LC. Martial has no conflict of interest. R.J.M. Brüggemann received grands from Merck, Pfizer, and Gilead; all payments have been made to the Radboud university medical center. J.A. Schouten has no conflict of interest. H.J. van Leeuwen has no conflict of interest. A.R. van Zanten has received payment for lectures and chairing scientific meetings from MSD. D.W. de Lange has no conflict of interest. E.W. Muilwijk has no conflict of interest. P.E. Verweij has received grands from Gilead Sciences, Pfizer, Astellas, Basilea, MSD, and F2G, has received consulting fees from Gilead Sciences and MDS, and has received payment fees from Gilead Sciences, MSD, and Biorad. D.M. Burger has no conflicts of interest. R.E. Aarnoutse has no conflict of interest. P. Pickkers has no conflict of interest. T.P.C Dorlo has no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Martial, L.C., Brüggemann, R.J.M., Schouten, J.A. et al. Dose Reduction of Caspofungin in Intensive Care Unit Patients with Child Pugh B Will Result in Suboptimal Exposure. Clin Pharmacokinet 55, 723–733 (2016). https://doi.org/10.1007/s40262-015-0347-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-015-0347-2