
Abstract
Background and Objectives
Sifalimumab is a fully human immunoglobulin G1κ monoclonal antibody that binds to and neutralizes a majority of the subtypes of human interferon-α. Sifalimumab is being evaluated as a treatment for systemic lupus erythematosus (SLE). The primary objectives of this analysis were (a) to develop a population pharmacokinetic model for sifalimumab in SLE; (b) to identify and quantitate the impact of patient/disease characteristics on pharmacokinetic variability; and (c) to evaluate fixed versus body weight (WT)-based dosing regimens.
Methods
Sifalimumab serum concentration-time data were collected from a phase Ib study (MI-CP152) designed to evaluate the safety and tolerability of sifalimumab in adult patients with SLE. Sifalimumab was administered every 14 days as a 30- to 60-minute intravenous infusion with escalating doses of 0.3, 1.0, 3.0, and 10 mg/kg and serum concentrations were collected over 350 days. A total of 120 patients provided evaluable pharmacokinetic data with a total of 2,370 serum concentrations. Sifalimumab serum concentrations were determined using a validated colorimetric enzyme-linked immunosorbent assay (ELISA) with a lower limit of quantitation of 1.25 μg/mL. Population pharmacokinetic modeling of sifalimumab was performed using a non-linear mixed effects modeling approach with NONMEM VII software. Impact of patient demographics, clinical indices, and biomarkers on pharmacokinetic parameters were explored using a stepwise forward selection and backward elimination approach. The appropriateness of the final model was tested using visual predictive check (VPC). The impact of body WT-based and fixed dosing of sifalimumab was evaluated using a simulation approach. The final population model was utilized for phase IIb dosing projections.
Results
Sifalimumab pharmacokinetics were best described using a two-compartment linear model with first order elimination. Following intravenous dosing, the typical clearance (CL) and central volume of distribution (V 1) were estimated to be 176 mL/day and 2.9 L, respectively. The estimates (coefficient of variation) of between-subject variability for CL and V 1 were 28 and 31 %, respectively. Patient baseline body WT, interferon gene signature from 21 genes, steroid use, and sifalimumab dose were identified as significant covariates for CL, whereas only baseline body WT was a significant covariate for V 1 and peripheral volume of distribution (V 2). Although the above-mentioned covariates were statistically significant, they did not explain variability in pharmacokinetic parameters to any relevant extent (<7 %). Thus, no dosing adjustments are necessary. VPC confirmed good predictability of the final population pharmacokinetic model. Simulation results demonstrate that both fixed and body WT-based dosing regimens yield similar median steady state concentrations and overall variability. Fixed sifalimumab doses of 200, 600, and 1,200 mg monthly (with a loading dose at Day 14) were selected for a phase IIb clinical trial.
Conclusion
A two-compartment population pharmacokinetic model adequately described sifalimumab pharmacokinetics. The estimated typical pharmacokinetic parameters were similar to other monoclonal antibodies without target mediated elimination. Although the population pharmacokinetic analysis identified some statistically significant covariates, they explained <7 % between-subject variability in pharmacokinetic parameters indicating that these covariates are not clinically relevant. The population pharmacokinetic analysis also demonstrated the feasibility of switching to fixed doses in phase IIb clinical trials of sifalimumab.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Background
Systemic lupus erythematosus (SLE) is a prototypic systemic autoimmune disease [1] characterized by acute and chronic inflammation of various organs [2]. The disease includes constitutional symptoms and signs; and musculoskeletal, mucocutaneous, renal, pulmonary, cardiac, hematologic, and neuropsychiatric manifestations. The prevalence of SLE in the United States is approximately 40 to 50 cases per 100,000 people [3]. SLE may occur at any age and in men or women, but predominantly affects women of childbearing years [4, 5]. Overall female-to-male ratio is about 9:1 or 10:1. About 20 % of patients develop lupus before 16 years of age, 65 % between 16 and 65 years, and 15 % after the age of 65 years.
SLE can be a serious and life-threatening illness with a substantial unmet medical need [6]. Belimumab (Benlysta®) is the only recently approved biologic for SLE treatment. Patients with moderate-to-severe disease are treated with corticosteroids [2, 7] and other agents such as azathioprine, cyclophosphamide, or mycophenolate mofetil.
Interferons (IFNs) are classified as type I or type II [8]. Type I IFNs include IFN-α (13 subtypes) and IFNs β, ω, and κ. Evidence suggests that type I IFNs have a role in SLE [9–13]. Type I IFNs stimulate the maturation of plasmacytoid dendritic cells and the generation and function of natural killer cells, T cells, and B cells. IFN-α activity is elevated in the sera of SLE patients [11, 14]. In murine models of SLE, type I IFN receptor deficiency appears to protect against lupus [15]. IFN levels are strongly associated with SLE disease activity [11, 14]. SLE patients with high anti-dsDNA antibody titers, lupus nephritis, and progressive rashes have high serum levels of type I IFN [14]. In addition, patients with acute skin involvement tend to have elevated IFN in blood and skin [11]. Treatment with IFN-α has been associated with the development of SLE autoantibodies and clinical features of the disease [16, 17]. Increased expression of genes induced by type I IFNs (known as the gene expression signature) is prominent in peripheral blood mononuclear cells in approximately 60 % of SLE patients and is associated with greater disease activity [9, 10, 12, 18–20]. Skin biopsies from both the affected and unaffected skin of patients with SLE also show increased type I IFN gene signature [21, 22]. These data support the hypothesis that inhibiting type I IFN may reduce disease activity in SLE.
Sifalimumab is a fully human immunoglobulin (Ig) G1κ monoclonal antibody (mAb) that binds to and neutralizes a majority of the subtypes of human IFN-α. Sifalimumab is being currently evaluated for the treatment of SLE. Single intravenous dose pharmacokinetics of sifalimumab have been evaluated in a phase I study (MI-CP126; NCT00299819) following 0.3, 1, 3, 10, and 30 mg/kg in mild to moderate SLE patients and multiple intravenous dose pharmacokinetics of sifalimumab have been evaluated in a phase Ib study (MI-CP152; NCT00482989). Pharmacokinetic parameters can be affected by various patient demographics, concomitant medication, and disease status. Therefore this population pharmacokinetic analysis was conducted to (a) develop a population pharmacokinetic model for sifalimumab in SLE; (b) to quantitate the ability of various patient demographics and disease characteristics to explain pharmacokinetic variability; and (c) compare sifalimumab exposures after fixed and body weight (WT)-based dosing.
2 Methods
2.1 Study Design and Patient Population
Sifalimumab serum concentration-time data were obtained from a multicenter, randomized, double-blind, placebo-controlled, dose-escalation phase Ib (MI-CP152) study (with an open-label extension) designed to evaluate the safety and tolerability of multiple intravenous doses of sifalimumab in adult patients with SLE. The study was conducted in compliance with the Declaration of Helsinki and the Guidelines for Good Clinical Practice. Written informed consent was obtained from all patients. Approximately 30 to 40 international sites from North and South America participated in this study.
In this phase Ib study (MI-CP152), a total of 161 SLE patients were randomized in a 3:1 ratio to receive either sifalimumab or placebo; of which 121 patients received sifalimumab. Sifalimumab was administered as a 30- to 60-minute intravenous infusion with escalating doses of 0.3, 1.0, 3.0, and 10 mg/kg every 2 weeks for a total of 14 doses. Individual patients remained on the randomized dose throughout the study. Venous blood samples for measuring serum concentrations of sifalimumab were collected at pre-dose, end of infusion, and 2 hours after completion of infusion (after first and last dose on Day 182); every 2 weeks prior to drug administration (from dose 2 through 13); and on Days 185, 189, 196, 210, 238, 266, 294, 322, and 350 post last dose.
2.2 Assay Methodology
Sifalimumab serum concentrations were determined using a validated colorimetric enzyme-linked immunosorbent assay (ELISA). In the assay, microtiter plates were coated with 0.5 μg/mL goat anti-sifalimumab idiotype antibody, blocked with casein buffer and washed. Calibration standards (0.3–160 μg/mL) and control samples were prepared by diluting sifalimumab reference standard into human serum. Following incubation, plates were washed and goat anti-human IgG conjugated with horseradish peroxidase was added for binding of captured sifalimumab. Plates were washed and pre-warmed 2,2′-azino-bis-(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) peroxidase substrate was added. The reaction was stopped by addition of 1 % sodium dodecyl sulfate (SDS) stop solution. Plates were measured at a 405 nm wavelength using a Molecular Devices SpectraMax microplate reader and the results were analyzed using SOFTmax® PRO software. The color intensity of the reaction was proportional to the amount of sifalimumab present in the sample. The assay lower limit of quantitation was determined to be 1.25 μg/mL and the upper limit of quantitation was determined to be 40 μg/mL.
2.3 Population Pharmacokinetic Analysis
A non-linear mixed-effect modeling approach [23–28] was used to analyze sifalimumab pharmacokinetic data. The population pharmacokinetic modeling was performed using NONMEM Version VII software (ICON Development Solutions, Ellicott City, MD, USA), G-Fortran (http://gcc.gnu.org/fortran/) and Perl-speaks-NONMEM (PSN) [29, 30] (http://psn.sourceforge.net/). Data management and graphical analyses were performed using S-plus 8.1 (TIBCO Software Inc., Somerville, MA, USA), Xpose 4.0 (University of Uppsala, Uppsala, Sweden) and R 2.7.1 (http://cran.r-project.org/) software. The various steps involved in the modeling processes are described below.
A series of structural models were evaluated based on Akaike information criteria (AIC) value, objective function values (OFV), precision, plausibility of parameter estimates, and goodness-of-fit plots. The between-subject variability in pharmacokinetic parameters was assumed to follow a log-normal distribution and was modeled using exponential functions, while the residual variability was evaluated using heteroscedastic (proportional), or combined proportional and additive models.
After the structural model was identified, covariate model-building was carried out to assess the effect of patient/disease characteristics on sifalimumab pharmacokinetic parameters. Various patient/disease characteristics including age, gender, ethnicity, region, baseline body WT, baseline steroid use (BSTEROID), baseline SLE disease activity index (BSLEDAI) score, baseline gene signature from 21 genes (BGENE21), baseline gene signature from 4 genes (BGENE4), sifalimumab dose and immunogenicity (IM) were evaluated. BGENE21 was the baseline gene signature (or type I IFN-inducible transcript) from selected 21 IFN genes whose expression can be utilized as a marker of type I IFN inducible pathway activation in SLE. BGENE4 was the baseline gene signature from 4 IFN genes and was a subset of BEGENE21. Missing individual covariate values were imputed with population median value of that particular covariate. A preliminary assessment of covariate influence was conducted using generalized additive modeling (GAM) approach as implemented in Xpose [31]. Based on GAM results and mechanistic understanding of sifalimumab disposition, the relevant covariates for each parameter were further tested using NONMEM for their significance. The model building was carried out using a stepwise forward addition (p < 0.05; reduction in OFV [ΔOFV] >3.84) followed by backward elimination (p < 0.01; ΔOFV >6.63) approach. The impact of potential covariates was first tested individually and the most significant covariate was included in the model. In the next steps, the remaining covariates were investigated. This process was repeated until all significant covariates were included in the model to form the full model (forward selection). Then the covariates were removed from the full model one by one. Covariates were retained in the final model provided the covariates were reasonable based on the pharmacology of sifalimumab and if their removal resulted in a statistically significant increase (>6.63) in OFV (backward elimination). The first-order conditional estimation method with interaction option was used throughout the modeling process. The subroutines ADVAN3 and TRANS4 in the NONMEM library were used for modeling.
The relationship between continuous covariates and pharmacokinetic parameters was modeled using non-linear power functions (Eq. 1) with the covariate normalized to the population median for the data set. The categorical covariates were modeled using fractional change functions (Eq. 2).
where the θs are the parameters to be estimated. In Eq. 1, θ 1 represents the typical value of pharmacokinetic parameter (P) in an individual with the median value for the covariate and θ 2 represents the coefficient for particular covariate effect. In Eq. 2, θ 1 represents the typical value of pharmacokinetics parameter for an individual with covariate value = 0 and θ 2 represents the fractional change in pharmacokinetics parameter for an individual with covariate value = 1 relative to covariate value = 0. A multiplicative covariate regression approach was implemented during covariate model building.
Improvement in the model at each step was assessed based on reduction in OFV, improvement in goodness-of-fit diagnostics, reduction in between and within-subject variability, and improvement in parameter precision.
The performance of the final population pharmacokinetic model was evaluated using visual predictive check (VPC) [32], a technique whereby model appropriateness is tested by means of comparing prediction intervals of the observed data to simulation data using the final population model. The impact of body WT-based and fixed dosing of sifalimumab was evaluated by comparing predicted median steady state serum concentrations (C ss), 5th and 95th percentiles using the final population model. A population of 1,000 SLE patients was simulated using covariate distribution from study MI-CP152. The final population model was utilized for predicting pharmacokinetic exposure following various fixed intravenous doses of sifalimumab to support phase IIb dosing.
3 Results
3.1 Data
A total of 120 patients provided evaluable pharmacokinetic data for the population analysis. The population pharmacokinetic data consisted of 2,370 sifalimumab serum concentrations following escalating doses of 0.3, 1.0, 3.0, and 10 mg/kg sifalimumab. On average, approximately 20 samples were available per patient, over a collection time of up to 1 year. One patient from the 10 mg/kg cohort was excluded from the analysis due to very low observed serum concentrations compared with the average concentrations in the 10 mg/kg cohort. The reasons for this low exposure are unclear. Table 1 provides a summary of important patient demographics and other baseline characteristics included in the pharmacokinetic analysis. The patient population was predominantly from North America (71 %) and female (95 %), with a median age of 43 years and median body WT of 73 kg. A total of eight subjects did not have BGENE4 information available; hence a population median value was imputed for these subjects.
3.2 Population Pharmacokinetic Modeling
A two-compartment model best described the sifalimumab concentration-time data and was considered as the base model. The pharmacokinetic model was parameterized using clearance (CL), central volume of distribution (V 1), peripheral volume of distribution (V 2) and the inter-compartmental clearance (Q). The between-subject variability parameters (η s) were included for all the pharmacokinetic parameters using exponential functions, whereas the residual error was modeled using a proportional model. Inclusion of the covariance terms \( \eta_{{{\text{CL}} }} - \eta_{{V_{1} }} \) and \( \eta_{{V_{1} }} - \eta_{{V_{2} }} \) improved the fit significantly. The mean (% between-subject variability) estimates from the base model were as follows: CL = 0.2 L/day (35 %); V 1 = 2.88 L (33 %); V 2 = 2.12 L (60 %); and Q = 0.175 L/day (75 %). All model parameters were estimated with good precision.
The covariate model evaluated the influence of age, sex, ethnicity, region, body WT, BSTEROID, BSLEDAI, BGENE21, BGENE4, and IM. In the final model, BGENE21, body WT, BSTEROID, and sifalimumab dose were identified as statistically significant covariates for the sifalimumab CL. Body WT was also found to be a statistically significant covariate for V 1 and V 2.
The final model functions for typical value of CL, V 1, V 2 and Q are presented as follows (Eqs. 3–6).
where θ 1 is the CL of a typical/standard patient with body WT = 75 kg, BGENE21 = 32, dose = 1 mg/kg, and BSTEROID = 0. θ 2 and θ 3 represent the V 1 and V 2 of a typical/standard subject with body WT = 75 kg, respectively. θ 5–θ 10 are the exponents of covariate effect on respective pharmacokinetic parameters.
The final population pharmacokinetic parameters are presented in Table 2. The estimated values of CL, V 1, V 2, and Q for a typical/standard patient were about 176 mL/day, 2.9 L, 2.12 L and 171 mL/day, respectively. The estimates (coefficient of variation) of between-subject variability associated with CL, V 1, V 2, and Q were 28, 31, 58, and 71 %, respectively. The η-shrinkage was estimated to be 4 % (CL), 12 % (V 1), 16 % (V 2), and 39 % (Q). The ε-shrinkage was 6 %. The condition number was 382. All pharmacokinetic parameters were estimated with good precision, as reflected by relative standard errors. The performance of the final model fit is represented by goodness-of-fit plots as shown in Fig. 1. Figure 1a, b show good agreement between observed and model predicted [population predicted (PRED)/individual predicted (IPRED)] sifalimumab serum concentrations. The plots of conditional weighted residual (CWRES) versus the PRED concentrations (Fig. 1c) or time (Fig. 1d) do not show any obvious pattern.

Final model goodness-of-fit plots for sifalimumab serum concentrations. The thin black line (diagonal and horizontal) and thick black line represent line of unity and loess fit, respectively
The coefficients of body WT effect on CL (θ 5), V 1 (θ 9), and V 2 (θ 10) were 0.481, 0.489, and 0.646, respectively, indicating a less than proportional increase in pharmacokinetic parameters with body WT. Addition of body WT reduced between-subject variability by 3, 2, and 3 % on CL, V 1, and V 2, respectively. In addition to body WT, sifalimumab CL was also impacted by changes in BGENE21, BSTEROID, and sifalimumab dose. The coefficients for the effect of BGENE21 (θ 6), BSTEROID (θ 8), and sifalimumab dose (θ 7) on CL were estimated to be 0.0558, 0.195, and 0.0542, respectively. Inclusion of BGENE21, BSTEROID, and sifalimumab dose as significant covariates on CL explained about only 1.5, 1, and 2 % of between-subject variability in CL. The CL of sifalimumab changed from 135 to 221 mL/day over the body WT range of 43–120 kg. Over the range of BGENE21 scores (0.63–87), sifalimumab CL changed from 141 to 186 mL/day. Sifalimumab CL changed from 165 to 199 mL/day over the dose range of 0.3–10 mg/kg. Patients who received steroid therapy had 19.5 % higher sifalimumab CL compared with subjects with no steroid administration. Over the body WT range of 43–120 kg, V 1 and V 2 changed from 2.2 to 3.6 and 1.5 to 2.9 L, respectively. Overall, inclusion of all the previously-mentioned covariates in the final population model explained only about 7 % between-subject variability and key pharmacokinetic parameters changed <30 %. The evaluation of immunogenicity as a covariate demonstrated no impact of anti-drug antibody (ADA) on sifalimumab pharmacokinetics.
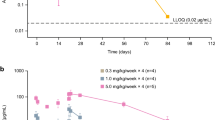
VPC results demonstrated good predictability of the final population pharmacokinetic model. As shown in Fig. 2, the observed median (solid red line), 5th and 95th data percentiles (dashed red lines) were adequately captured by the corresponding simulation-based prediction intervals (solid and dashed black lines) and 95 % confidence intervals (green and beige shaded areas).

Visual predictive check for sifalimumab serum concentrations. Observed median (solid red line), 5th and 9th percentiles (dashed red lines) and corresponding simulation-based prediction intervals (solid and dashed black lines) and 95 % confidence intervals (green and beige shaded areas)
3.2.1 Comparison of Fixed (mg) Versus Body Weight-Based (mg/kg) Dosing
The impact of fixed versus body WT-based dosing was evaluated by comparing 200 mg (fixed) with 3 mg/kg (body WT-based) every 14-day dosing of sifalimumab in a simulated SLE population of 1,000 patients. The fixed dose of 200 mg was selected to approximate 3 mg/kg (based on 75 kg body WT). The body WT distribution (43–120 kg) from MI-CP152 was used for the simulations. The final population pharmacokinetic model was used to predict 5th, median, and 95th percentile concentration-time profiles. Simulation results demonstrate that both fixed and body WT-based dosing regimens yield similar median C ss and overall variability as shown in Fig. 3.

Similarity of predicted pharmacokinetic profiles (median, 5th and 95th percentiles) following fixed (200 mg every 14 days) and body weight-based (3 mg/kg every 14 days) dosing of sifalimumab. Q14D every 14 days, WT body weight
3.2.2 Predicted Serum Concentrations for Phase IIb Clinical Trial (MI-CP1067; NCT01283139)
The final population pharmacokinetic model was used to predict concentration–time profiles following 200, 600, and 1,200 mg monthly (with an additional dose at Day 14) dose of sifalimumab in a simulated SLE population of 1,000 patients. The predicted concentration-time profiles (median, 5th, and 95th percentiles) are shown in Fig. 4. The expected steady state pharmacokinetic exposure values following 200, 600, and 1,200 mg monthly (with an additional dose at Day 14) dose of sifalimumab are presented in Table 3.

Predicted serum concentrations following 200, 600, and 1,200 mg monthly intravenous dosing of sifalimumab (with single loading dose at Day 14)
4 Discussion
To improve the understanding of sifalimumab pharmacokinetics, we developed a population pharmacokinetic model to describe serum concentration-time data following various doses of sifalimumab using a nonlinear mixed-effects modeling approach. A further objective was to identify patient/disease characteristics that influence sifalimumab pharmacokinetic parameters. Systematic understanding of the effect of patient/disease covariates would allow more rational insight on the impact of different covariates on pharmacokinetics and the potential for dose individualization for further development of sifalimumab.
The elimination and degradation of mAbs occur primarily via two mechanisms: (a) non-specific protein catabolism by the reticuloendothelial system (RES) and (b) specific binding to the target leading to complex internalization (saturable target-mediated clearance) [33, 34]. It is anticipated that sifalimumab, a mAb that binds to IFN-α, a soluble target, will exhibit linear pharmacokinetics [35]. In this population analysis, a two-compartment model without target-mediated elimination adequately described sifalimumab concentration-time data. The estimate of CL was about 176 mL/day for a typical individual, which is similar to the phase I study (MI-CP126) and other mAbs [36–42]. Based on population pharmacokinetic modeling, sifalimumab distributes into a central compartment volume (V 1) of 2.9 L for a typical patient, which is approximately equal to the plasma volume. The V 2 of 2.1 L suggests an apparently limited distribution outside the vascular space, which is consistent with behavior of endogenous IgG immunoglobulins and other therapeutic mAbs [36–43]. Modest between-subject variability estimates of about 28 % (CL) and 31 % (V 1) were also in line with literature-reported values for other mAbs [39–41]. Slightly higher between-subject variability was estimated for V 2 (58 %) and Q (71 %), which could be a result of a non-uniform collection of primarily trough blood samples. The estimated η-shrinkage and ε-shrinkage was <20 % for key parameters (except for Q) representing the informativeness of model diagnostics such as Empirical Bayes Estimates (EBEs), IPRED, and CWRES. The condition number of 382 implies that the model was stable and was not overparameterized (condition number <1,000).
Based on the covariate relationships, higher sifalimumab CL was estimated for patients with higher BGENE21, body WT, sifalimumab dose, and steroid use. Both V 1 and V 2 also increased with increase in body WT. The correlation of body WT with CL and volume of distributions has been shown for both small and large molecules [39, 40, 42, 44, 45]. The coefficient/exponent of body WT on CL, V 1, and V 2 were 0.481, 0.489, and 0.646, respectively, resulting in a modest impact of body WT on these parameters. Addition of body WT resulted in a minor reduction in between-subject variability of about 3, 2, and 3 % in CL, V 1, and V 2, respectively. Over a wide range of body WT (43–120 kg), relative to the typical individual, only a <30 % change in key pharmacokinetic parameters was estimated.
Patients with higher BGENE21 had slightly higher clearance of sifalimumab. The elevated baseline type I IFN gene signatures are an indicator of increased IFN-α levels [18–20]. However, the impact of this covariate was not substantial since only ~1.5 % of between-subject variability was explained and CL changed by <20 % over the range of BGENE21 scores. During covariate model building, BGENE4 was also found to be a significant covariate for sifalimumab CL. This was expected because BGENE4 was a subset of BGENE21 and there was a high degree of correlation between BGENE4 and BGENE21. After accounting for the impact of BGENE21 in the model, BGENE4 did not significantly influence the CL of sifalimumab.
A slight increase in CL was estimated with increasing sifalimumab doses. The reasons for this increase in CL are unclear at this point. The dose-dependent pharmacokinetics were not expected and could be a data artifact since a single-dose phase I study (MI-CP126) demonstrated linear and dose proportional pharmacokinetics over the dose range of 0.3–30 mg/kg. However, the impact of this covariate was not clinically relevant because only about 2 % of between-subject variability in CL was explained and the difference in CL between doses was within variability.
Patients who received steroid therapy had 19.5 % higher sifalimumab CL compared with subjects with no steroid administration. Corticosteroids (e.g. methylprednisolone) were used throughout the study to control the disease activity. High disease activity in these patients might be linked to higher type I IFN-α expression (or gene signature) [11, 14] and higher CL. However, addition of this covariate accounted for only 1 % reduction in between-subject variability in CL. The low titers of ADA did not impact pharmacokinetics of sifalimumab.
Although the previously-mentioned covariates were identified as statistically significant, they did not substantially explain between-subject variability (<7 %) in pharmacokinetic parameters. This suggests that a decrease or increase in sifalimumab exposure over the range of these covariates is not clinically relevant. Hence, no dosing modifications are necessary to administer sifalimumab.
Other possible sources of variability might be related to the elimination pathway of mAbs. mAbs are primarily eliminated by RES through the interaction with neonatal Fc receptors (FcRns). FcRns are expressed on phagocytic cells of the RES and protect IgG from rapid clearance. Between-subject variability in expression profiles and distribution of FcRns could account for some of the variability in pharmacokinetic profiles. However, information on these variables are lacking in the literature.
This study (MI-CP152) was designed to evaluate safety and tolerability of sifalimumab in a SLE population following multiple intravenous administrations over the dose range of 0.3 to 10 mg/kg. Measurements of clinical/disease activity were included only as exploratory end points. Sifalimumab demonstrated an acceptable safety and tolerability profile following multiple intravenous doses over 1 year. No statistically significant differences in clinical activity (SLEDAI and British Isles Lupus Assessment Group [BILAG] score) between sifalimumab and placebo were observed. The heterogeneous baseline characteristics, ascending dose design, and small number of patients in each group made comparisons between groups difficult and the study did not yield a clear estimate of target exposures. The results of safety, tolerability, and preliminary clinical activity of sifalimumab from the MI-CP152 study were published recently [46].
Body size/WT-based dosing has been a common practice for large molecules including mAbs, therapeutic proteins, and peptides. It is generally assumed that body-size correction would reduce variability in pharmacokinetic parameters across a wide range of patient body WTs. Body WT-based dosing may lead to over-correction of body size for low body WT patients and under-correction of body size for patients with high body WT. A fixed dosing approach is preferred by the prescribing community due to ease of use and reduced dosing errors. In addition, patient convenience and adherence can be greatly improved. In the current population analysis, comparison of fixed versus body WT-based dosing demonstrates that serum pharmacokinetic exposures and associated between-subject variability following both fixed and body WT-based dosing were almost identical. Similar findings have been reported by others [47–49]. Wang and colleagues investigated 12 mAbs and found that fixed and body size-based dosing perform similarly, with fixed dosing being better for 7 of 12 antibodies [48]. In addition, they investigated 18 therapeutic proteins and peptides and showed that fixed dosing performed better for 12 of 18 in terms of reducing the inter-individual variability in pharmacokinetic/pharmacodynamic parameters [49]. Our findings support the fixed dosing regimen for future clinical studies of sifalimumab.
Given no major safety concerns, no clear target exposure, and expectation of similar pharmacokinetic exposure, we considered it feasible to switch to fixed dosing regimens for evaluation in a phase IIb study. A wide range of doses including 200, 600, and 1,200 mg sifalimumab was selected for the ongoing phase IIb study to evaluate the clinical activity of sifalimumab. Population pharmacokinetic/pharmacodynamic and exposure-response modeling of clinical end point data from the ongoing phase IIb study are planned in order to assess dose–response and guide phase III dose selection for future pivotal studies of sifalimumab.
5 Conclusion
A two-compartment population pharmacokinetic model adequately described the sifalimumab concentration-time data in SLE patients. The estimated typical pharmacokinetic parameters were similar to other therapeutic mAbs without target-mediated elimination. Although the population analysis identified some statistically significant covariates, they are not anticipated to be clinically relevant because only <7 % between-subject variability in key pharmacokinetic parameters was explained. The population pharmacokinetic analysis also demonstrated the feasibility of switching to fixed doses of sifalimumab in phase IIb clinical trials.
References
American College of Rheumatology Ad Hoc Committee on Systemic Lupus Erythematosus Guidelines. Guidelines for referral and management of systemic lupus erythematosus in adults. Arthritis Rheum. 1999;42:1785–96.
Petri M. Long-term outcomes in lupus. Am J Managed Care. 2001;7:S480-5.
Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778–99.
Cooper GS, Dooley MA, Treadwell EL, et al. Hormonal, environmental, and infectious risk factors for developing systemic lupus erythematosus. Arthritis Rheum. 1998;41(10):1714–24.
Lahita RG. The role of sex hormones in systemic lupus erythematosus. Curr Opin Rheumatol. 1999;11(5):352–6.
Wallace D. Advances in the management of systemic lupus erythematosus. Bull Rheum Dis. 2004;51(11):1–7.
Urowitz MB, Bookman AA, Koehler BE, et al. The bimodal mortality pattern of systemic lupus erythematosus. Am J Med. 1976;60(2):221–5.
Theofilopoulos AN, Baccala R, Beutler B, et al. Type I interferons (α/β) in immunity and autoimmunity. Immunol Rev. 2005;204:9–26.
Crow M. Interferon-α: a new target for therapy in systemic lupus erythematosus? Arthritis Rheum. 2003;48:2396–401.
Kirou KA, Lee C, George S, et al. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005;52(5):1491–503.
Dall’era MC, Cardarelli PM, Preston BT, et al. Type I interferon correlates with clinical and serologic manifestations of systemic lupus erythematosus. Ann Rheum Dis. 2005;64:1692–7.
Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci. 2003;100(5):2610–5.
Baechler EC, Gregersen PK, Behrens TW. The emerging role of interferon systemic lupus erythematosus. Curr Opin Immunol. 2004;16(6):801–7.
Bengtsson AA, Sturfelt G, Truedsson L, et al. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus. 2000;9:664–71.
Santiago-Raber M-L, Baccala R, Haraldsson KM, et al. Type I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med. 2003;197:777–88.
Niewold TB, Swedler WI. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin Rheumatol. 2005;24(2):178–81.
Ioannou Y, Isenberg DA. Current evidence for the induction of autoimmune rheumatic manifestations by cytokine therapy. Arthritis Rheum. 2000;43(7):1431–42.
Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–23.
Kirou KA, Lee C, George S, et al. Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum. 2004;50(12):3958–67.
Feng X, Wu H, Grossman JM, et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54(9):2951–62.
Blomberg S, Eloranta ML, Cederblad B, et al. Presence of cutaneous interferon-alpha producing cells in patients with systemic lupus erythematosus. Lupus. 2001;10(7):484–90.
Farkas L, Beiske K, Lund-Johansen F, et al. Plasmacytoid dendritic cells (natural interferon-alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. 2001;159(1):237–43.
Wang Y. Derivation of various NONMEM estimation methods. J Pharmacokinet Pharmacodyn. 2007;34(5):575–93.
Ette EI, Williams PJ. Population pharmacokinetics I: background, concepts, and models. Ann Pharmacother. 2004;38(10):1702–6.
Bonate PL. Pharmacokinetic-pharmacodynamic modeling and simulation. New York: Springer; 2006.
Boeckmann AJ, Scheiner LB, Beal SL. NONMEM user guide: part V. San Francisco: NONMEM Project Group; 1994.
Mandema JW, Verotta D, Sheiner LB. Building population pharmacokinetic–pharmacodynamic models. I. Models for covariate effects. J Pharmacokinet Biopharm. 1992;20(5):511–28.
Sheiner LB, Beal SL. Evaluation of methods for estimating population pharmacokinetic parameters. III. Monoexponential model: routine clinical pharmacokinetic data. J Pharmacokinet Biopharm. 1983;11(3):303–19.
Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit–a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–57.
Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN)—a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75(2):85–94.
Jonsson EN, Karlsson MO. Xpose–an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58(1):51–64.
Holford N. The visual predictive check: superiority to standard diagnostic (Rorschach) plots. PAGE. 2005;14:738.
Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006;11(1–2):81–8.
Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–58.
Mould DR, Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies: concepts and lessons for drug development. BioDrugs. 2010;24(1):23–39.
Tabrizi M, Wang B, Lu H, et al. Population pharmacokinetic evaluation of a fully human IgG monoclonal antibody in patients with inflammatory diseases. Inflamm Allergy Drug Targets. 2010;9(4):229–37.
Singh D, Kane B, Molfino NA, et al. A phase 1 study evaluating the pharmacokinetics, safety and tolerability of repeat dosing with a human IL-13 antibody (CAT-354) in subjects with asthma. BMC Pulm Med. 2010;10(1):3.
Oh CK, Faggioni R, Jin F, et al. An open-label, single-dose bioavailability study of the pharmacokinetics of CAT-354 after subcutaneous and intravenous administration in healthy males. Br J Clin Pharmacol. 2010;69(6):645–55.
Lu JF, Bruno R, Eppler S, et al. Clinical pharmacokinetics of bevacizumab in patients with solid tumors. Cancer Chemother Pharmacol. 2008;62(5):779–86.
Ng CM, Bruno R, Combs D, et al. Population pharmacokinetics of rituximab (anti-CD20 monoclonal antibody) in rheumatoid arthritis patients during a phase II clinical trial. J Clin Pharmacol. 2005;45(7):792–801.
Dirks NL, Nolting A, Kovar A, et al. Population pharmacokinetics of cetuximab in patients with squamous cell carcinoma of the head and neck. J Clin Pharmacol. 2008;48(3):267–78.
Mould DR, Holford NH, Schellens JH, et al. Population pharmacokinetic and adverse event analysis of topotecan in patients with solid tumors. Clin Pharmacol Ther. 2002;71(5):334–48.
White B, Leon F, White W, et al. Two first-in-human, open-label, phase I dose-escalation safety trials of MEDI-528, a monoclonal antibody against interleukin-9, in healthy adult volunteers. Clin Therap. 2009;31(4):728–40.
Narwal R, Akhlaghi F, Asberg A, et al. Development of a population pharmacokinetic model for atorvastatin acid and its lactone metabolite. Clin Pharmacokinet. 2010;49(10):693–702.
Holford NH. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996;30(5):329–32.
Petri M, Wallace DJ, Spindler A, et al. Sifalimumab, a human anti-interferon-α monoclonal antibody, in systemic lupus erythematosus. Arthritis Rheum. 2013;65(4):1011–21.
Ng CM, Lum BL, Gimenez V, et al. Rationale for fixed dosing of pertuzumab in cancer patients based on population pharmacokinetic analysis. Pharm Res. 2006;23(6):1275–84.
Wang DD, Zhang S, Zhao H, et al. Fixed dosing versus body size-based dosing of monoclonal antibodies in adult clinical trials. J Clin Pharmacol. 2009;49(9):1012–24.
Zhang S, Shi R, Li C, et al. Fixed dosing versus body size-based dosing of therapeutic peptides and proteins in adults. J Clin Pharmacol. 2012;52(1):18–28.
Acknowledgments
This study was sponsored by MedImmune, LLC, which was involved in the study design; data collection, analysis, and interpretation; writing of the manuscript; and the decision to submit the manuscript for publication. Rajesh Narwal, Lorin K. Roskos, and Gabriel J. Robbie are employees of MedImmune and own stock in AstraZeneca. All authors have pending patent application PCT/US12/39098, published as WO 2012/162367, which is directed, in part, to subject matter described in this publication.
Author information
Authors and Affiliations
Corresponding author
Additional information
ClinicalTrials.gov Identifier: NCT00482989.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Narwal, R., Roskos, L.K. & Robbie, G.J. Population Pharmacokinetics of Sifalimumab, an Investigational Anti-Interferon-α Monoclonal Antibody, in Systemic Lupus Erythematosus. Clin Pharmacokinet 52, 1017–1027 (2013). https://doi.org/10.1007/s40262-013-0085-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-013-0085-2