
Abstract
Background and Objective
Individuals with diabetes mellitus may prefer different body regions for subcutaneous insulin administration. This trial investigated whether choice of injection region affects exposure and glucose-lowering effect of once-weekly basal insulin icodec.
Methods
In a randomised, open-label, crossover trial, 25 individuals with type 2 diabetes received single subcutaneous icodec injections (5.6 U/kg) in the thigh, abdomen or upper arm (9–13 weeks’ washout). Pharmacokinetic blood sampling occurred frequently until 35 days post-dose. Partial glucose-lowering effect was assessed 36–60 h post-dose in a glucose clamp (target 7.5 mmol/L). Steady-state pharmacokinetics following multiple once-weekly dosing were simulated using a two-compartment pharmacokinetic model.
Results
Total icodec exposure (area under the curve from zero to infinity after single dose; AUC0–∞,SD) was similar between injection in the thigh, abdomen and upper arm (estimated AUC0–∞,SD ratios [95% confidence interval]: abdomen/thigh 1.02 [0.96–1.09], p = 0.473; upper arm/thigh 1.04 [0.98–1.10], p = 0.162; abdomen/upper arm 0.98 [0.93–1.05], p = 0.610). Maximum icodec concentration (Cmax) after single dose was higher for abdomen (by 17%, p = 0.002) and upper arm (by 24%, p < 0.001) versus thigh. When simulated to steady state, smaller differences in Cmax were seen for abdomen (by 11%, p = 0.004) and upper arm (by 16%, p < 0.001) versus thigh. Geometric mean [coefficient of variation] glucose-lowering effect 36–60 h post-dose was comparable between the thigh (1961 mg/kg [51%]), abdomen (2130 mg/kg [52%]) and upper arm (2391 mg/kg [40%]).
Conclusion
Icodec can be administered subcutaneously in the thigh, abdomen or upper arm with no clinically relevant difference in exposure and with a similar glucose-lowering effect.
ClinicalTrials.gov Identifier
NCT04582448.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Total exposure of insulin icodec was shown to be independent of subcutaneous injection region (thigh, abdomen or upper arm). |
Maximum insulin icodec concentration at steady state was slightly higher after injection in the abdomen (by 11%) and upper arm (by 16%) than in the thigh. However, this is considered of limited clinical relevance since glucose-lowering effect profiles were comparable across injection regions. |
Individuals with diabetes may choose between different injection regions for subcutaneous administration of once-weekly insulin icodec with a similar glucose-lowering effect. |
1 Introduction
Several different regions of the body can be used for subcutaneous insulin injection in the treatment of diabetes mellitus: the upper arm, abdomen, thigh or buttock [1, 2]. Every individual with diabetes may have a preferred body region for subcutaneous insulin administration depending on anatomical, physiological and practical circumstances [1, 3]. However, choice of subcutaneous injection region may, at least to some extent, impact insulin pharmacokinetics and pharmacodynamics [4,5,6,7]. For insulin products in development, it is therefore highly relevant to investigate the pharmacological properties following subcutaneous injection in different body regions.
Insulin icodec (icodec) is a basal insulin in development for once-weekly dosing [8]. After subcutaneous administration and subsequent absorption into the circulation, icodec binds strongly to albumin and thereby forms an inactive icodec depot. At steady state, slow and stable release of icodec from this depot results in a half-life appropriate for once-weekly dosing [9]. In Phase 2 clinical trials in individuals with type 2 diabetes, the reductions in glycosylated haemoglobin (HbA1c) and fasting plasma glucose were similar between once-weekly icodec and once-daily insulin glargine U100 with comparable rates of level 2 and 3 hypoglycaemia [10,11,12].
The aim of the current trial was to investigate whether the choice of injection region affects the pharmacological properties of icodec. The trial compared the exposure and partial glucose-lowering effect of icodec following subcutaneous injection between the thigh, abdomen or upper arm in individuals with type 2 diabetes.
2 Methods
2.1 Trial Design and Participants
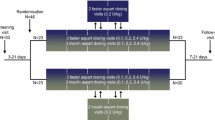
This was a single-centre (Profil, Mainz, Germany), randomised, open-label, three-period crossover trial (Fig. 1). Local health authorities (Bundesinstitut für Arzneimittel und Medizinprodukte) and an independent ethics committee (Landesärztekammer Rheinland-Pfalz) approved the trial, which was performed according to the Declaration of Helsinki and Good Clinical Practice. All participants provided written informed consent prior to any trial activities. The trial was registered at ClinicalTrials.gov (NCT04582448).

Overall trial design. Individuals participated in three 5-week periods, each consisting of single-dose subcutaneous administration of insulin icodec in the thigh, abdomen or upper arm in randomised sequence. Single dose injections were separated by 9–13 weeks of washout PK pharmacokinetic
Eligible participants were men or women aged 18–69 years, diagnosed with type 2 diabetes ≥ 180 days prior to screening, treated with basal insulin (total daily insulin dose of 0.2–1.0 U/kg) ± metformin, dipeptidyl peptidase-4 (DPP-4) inhibitors, sodium-glucose transport protein-2 inhibitors, oral combination products (of the allowed oral antidiabetic drugs) or oral/injectable glucagon-like peptide-1 receptor agonists ≥ 90 days prior to screening, with body mass index 18.5–38.0 kg/m2, HbA1c ≤ 75 mmol/mol (≤ 9.0%), and supine blood pressure of 90–159 mmHg (systolic) and 50–99 mmHg (diastolic). Individuals with highly fluctuating blood glucose levels prior to screening, previous or current cardiovascular disease, known or suspected hypersensitivity to trial product or a related product, and pregnant women were excluded from participation.
2.2 Procedures
The trial consisted of a screening visit (3–21 days before the first treatment period), 3 treatment periods of 5 weeks’ duration, and a follow-up visit (at the end of the third treatment period).
At the start of each treatment period, participants received a single dose of icodec (5.6 U/kg body weight; 700 U/mL; Novo Nordisk, Bagsværd, Denmark) injected subcutaneously into a lifted skin fold of the thigh (anterior surface), abdomen (lower abdominal wall above the inguinal area) or upper arm (the outer aspect) in randomised sequence. Single-dose injections were separated by 9–13 weeks of washout. Icodec was provided in a 3 mL PDS290 prefilled pen-injector (Novo Nordisk) and administered by trained site staff at approximately 08:00 h. Participants stayed in-house at the clinical site until 60 h after icodec administration.
Before each treatment period, participants who normally received once-daily basal insulin degludec or insulin glargine U300 discontinued their insulin treatment 48 h prior to the icodec single dose, and participants who normally received once- or twice-daily basal insulin detemir or insulin glargine U100 discontinued their insulin treatment 24 h prior to the icodec single dose. Instead, participants could use neutral protamine Hagedorn insulin for the last 24–48 h prior to the icodec administration. During the washout periods, participants received their usual basal insulin treatment guided by the investigator. Any allowed non-insulin anti-diabetic medication received at the time of entry into the trial was maintained at a stable dose and at the same frequency during the entire trial.
Blood samples for pharmacokinetic assessment were drawn pre-dose and frequently up to 840 h (35 days) after administration of each icodec single dose (Online Resource, Table S1). Total serum icodec concentrations (i.e., the sum of free, unbound icodec and icodec bound to albumin) were measured using a validated icodec specific luminescence oxygen channelling immunoassay with a minimum required dilution of 50, an analytical range from 500 to 80,000 pmol/L, and quality controls having a cumulative accuracy from − 12.0% to − 3.6% across the analytical range and an inter-batch coefficient of variation ≤ 18.2%.
The partial glucose-lowering effect of icodec was assessed between 36 and 60 h after administration of each icodec single dose in an automated glucose clamp (ClampArt; Profil, Neuss, Germany). This time interval covered the expected time of maximum glucose-lowering effect of icodec. On the day of the glucose clamp, participants received a standardised breakfast and a light lunch and then started fasting for at least 8 h prior to clamp initiation until end of the clamp procedure (except for water ad libitum). Participants stayed in a supine or semi-supine position throughout the glucose clamp. During clamp run-in, subjects received a variable intravenous infusion of regular human insulin [15 U NovoRapid® (100 U/mL; Novo Nordisk) in 49 mL saline and 1 mL of the subject’s blood] or glucose (20%) for 5 h prior to clamp initiation to obtain a plasma glucose target level of 7.5 mmol/L (135 mg/dL). From 1 hour prior to clamp initiation, the insulin infusion rate (if any) was decreased as much as possible while keeping plasma glucose at 7.5 mmol/L ±20% from − 60 to − 10 min and stable at 7.5 mmol/L ±10% during the last 10 min prior to clamp initiation. Insulin infusion (if any) was terminated at clamp initiation. From clamp initiation at 20:00 h, a variable intravenous glucose infusion was performed to ensure that plasma glucose was kept at the target level of 7.5 mmol/L throughout the clamp duration of 24 h. In case plasma glucose consistently exceeded 15.6 mmol/L, with no glucose infusion for the last 30 min, the clamp was terminated early. The quality of the conducted clamps was high and comparable across injection regions (Online Resource, Table S2) [13].
Safety assessments included adverse events, hypoglycaemic episodes classified as level 1 (plasma glucose < 3.9 mmol/L and ≥ 3.0 mmol/L), level 2 (plasma glucose < 3.0 mmol/L), and level 3 (severe cognitive impairment requiring assistance for recovery) [14], safety laboratory parameters, vital signs and physical examination.
2.3 Statistical Analyses
Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA).
The sample size calculation was based on an assumed within-participant standard deviation for the logarithmic transformed primary endpoint (area under the serum icodec concentration-time curve from zero to infinity after a single dose; AUC0–∞,SD) of 0.125 estimated from the pharmacokinetic results of previous trials with icodec (ClinicalTrials.gov identifier: NCT03723785) and insulin degludec [15]. With a sample size of 20 participants, the 95% confidence interval (CI) for the geometric mean ratio of AUC0–∞,SD between any two subcutaneous injection regions that could be obtained with at least 90% probability would be [0.91;1.10] if the observed ratio was 1.00. This was considered adequate to support the primary objective of the trial. A total of 25 individuals were randomised with the aim of achieving at least 20 completers.
The primary endpoint, AUC0–∞,SD, was calculated as the sum of two areas: first, the area under the curve from time zero to the time of last quantifiable concentration (tlast) using the linear trapezoidal technique based on observed values and actual measurement times. Second, the area from tlast to infinity under an exponential concentration-time curve with terminal rate constant, λz, and a concentration at tlast predicted from the linear part of the logarithmic transformed curve. Other pharmacokinetic endpoints after a single dose were the maximum observed concentration (Cmax,SD) and time to maximum concentration (tmax,SD).
The endpoints AUC0–∞,SD and Cmax,SD were logarithmic transformed and analysed using linear mixed models with injection region and period as fixed effects and participant as a random effect. The residual variance was depending on injection region. The pairwise differences between thigh, abdomen and upper arm were back-transformed to the original scale and presented as ratios with 95% CIs. The endpoint tmax,SD was summarised by descriptive statistics.
Glucose infusion rate (GIR) data from the glucose clamp were smoothed using the Loess smoothing technique (fixed smoothing parameter of 0.25). The partial glucose-lowering effect between 36 and 60 h after a single icodec dose (AUCGIR,36–60 h,SD) was calculated as the area under the smoothed GIR curve using the linear trapezoidal technique on interpolated points. AUCGIR,36–60 h,SD was summarised by descriptive statistics.
2.4 Pharmacokinetic Modelling
To be able to simulate the serum icodec concentration-time profiles at steady state following multiple once-weekly subcutaneous injection in the thigh, abdomen or upper arm, a pharmacokinetic model was developed based on the observed serum icodec concentrations after a single dose.
The structural part of the model was a two-compartment model with first-order absorption and first-order elimination, parameterised in terms of a bioavailability parameter (F; fixed to 1 for injection in the thigh), an absorption rate parameter (kA), two clearance parameters (CL/F and Q/F), and two volume of distribution parameters (Vc/F and Vp/F). Between-individual variability was included on CL/F, Q/F, Vc/F and Vp/F, assuming a log-normal distribution with correlation between all four parameters. Between-occasion variability was included on F and kA, assuming a log-normal distribution with no correlation. Injection region (thigh, abdomen and upper arm) was included as a covariate on F and kA. A combined (proportional + additive) error model was used to describe residual variability.
The parameters of the model were estimated with a non-linear mixed-effects approach using the first-order conditional estimation method with interaction (FOCE+I) in NONMEM version 7.3 (ICON Development Solutions, Ellicott City, MD, USA). Diagnostic plots for the model and a table with the estimated fixed effect parameters and random effect and residual error variances are provided in Online Resource, Figs. S1, S2 and Table S3. Using the post hoc parameters from the model, an individual set of values of the six model parameters was obtained for each injection region in each participant. Using these individual sets of parameter values, and assuming no change in pharmacokinetic parameters over time, simulation was performed to obtain individual-predicted steady-state serum icodec concentration-time profiles for each injection region in each participant. Specifically, once-weekly dosing of 5.6 U/kg for 10 weeks was simulated. The profile at Week 10 was considered the steady-state profile since it takes 3–4 weeks of once-weekly icodec dosing to achieve pharmacokinetic steady state [9]. Maximum concentration at steady state (Cmax,SS) and time to maximum concentration at steady state (tmax,SS) for each injection region in each participant were derived from the individual-predicted steady-state profiles. Cmax,SS was compared between injection regions by statistical analysis as described above for AUC0–∞,SD and Cmax,SD, while tmax,SS was summarised by descriptive statistics.
3 Results
3.1 Participants
A total of 52 individuals were screened and 25 were randomised and exposed to icodec (Online Resource, Fig. S3). One participant was withdrawn by the investigator during the first period (upper arm) and one participant withdrew consent after the first period (upper arm). Thus, 23 participants completed the trial. Baseline characteristics of the 25 randomised participants are shown in Table 1.
3.2 Pharmacokinetics
One-week pharmacokinetic profiles of icodec injected subcutaneously in the thigh, abdomen or upper arm after a single dose and simulated to steady state are shown in Fig. 2. Total exposure after a single dose (AUC0–∞,SD) was similar for subcutaneous injection in the thigh, abdomen and upper arm (Table 2 and Fig. 3). Because extrapolation from single dose to steady state has no effect on total exposure, total icodec exposure at steady state (AUCτ,SS) was also independent of injection region. Maximum concentration after a single dose (Cmax,SD) was higher for the abdomen and upper arm than for the thigh, while no difference was observed between the abdomen and upper arm (Table 2 and Fig. 3). At steady state, relative to single dose, smaller differences in Cmax,SS were observed for the abdomen and upper arm compared with the thigh (Table 2 and Fig. 3). Median time to maximum concentration (tmax) was comparable between injection regions after a single dose as well as when simulated to steady state (Table 2).

Mean (SEM) serum insulin icodec concentration-time profiles during a dosing interval of 1 week following subcutaneous injection of insulin icodec (5.6 U/kg) in the thigh, abdomen or upper arm. a After a single dose. b Simulated to once-weekly administration at steady state using pharmacokinetic modelling. Data are based on 23 individual pharmacokinetic profiles for thigh and upper arm and 24 individual pharmacokinetic profiles for abdomen. Error bands show standard error of the mean

Comparison of insulin icodec exposure among subcutaneous injection regions. Total exposure (AUC0–∞,SD) and maximum concentration (Cmax,SD) were derived after a single dose of insulin icodec (5.6 U/kg). Cmax,SS was derived after simulation to once-weekly administration at steady state. Results for AUC0–∞,SD also reflect total exposure at steady state (AUCτ,SS) because extrapolation to steady state has no effect on total exposure. Data are based on 23 individual pharmacokinetic profiles for thigh and upper arm and 24 individual pharmacokinetic profiles for abdomen. AUC0–∞ area under the curve from 0 to infinity, AUCτ area under the curve during a dosing interval, CI confidence interval, Cmax maximum concentration, SD single dose, SS steady state
3.3 Pharmacodynamics
Glucose-lowering effect profiles from 36 to 60 h after a single dose of icodec did not differ between subcutaneous injection in the thigh, abdomen or upper arm (Fig. 4). Accordingly, the glucose-lowering effect from 36 to 60 h (AUCGIR,36–60h,SD) was comparable across injection regions. Geometric mean AUCGIR,36–60h,SD (and coefficient of variation in percent) was 1961 mg/kg (51%) for the thigh, 2130 mg/kg (52%) for the abdomen and 2391 mg/kg (40%) for the upper arm.

Mean (SEM) glucose-lowering effect profiles from 36 to 60 h following a single dose of insulin icodec (5.6 U/kg) injected subcutaneously in the thigh, abdomen or upper arm. Data are based on 22 individual glucose-lowering effect profiles for thigh and upper arm and 23 individual glucose-lowering effect profiles for abdomen. Error bands show standard error of the mean
3.4 Safety
Icodec was well tolerated with no safety issues. During the treatment periods, 28 adverse events were reported by 12 (48%) participants. Headache (11 events) and back pain (3 events) were the most frequently reported adverse events. No injection-site reactions were reported. All adverse events were mild or moderate. No serious or severe adverse events were reported, and no adverse events led to withdrawal. A total of 27 level 1 hypoglycaemic episodes and 3 level 2 hypoglycaemic episodes were reported during the treatment periods. No severe (level 3) hypoglycaemic episodes were reported. There were no clinically significant findings in safety laboratory parameters, vital signs or physical examination.
4 Discussion
The main findings of the present trial in individuals with type 2 diabetes were that total icodec exposure was similar after subcutaneous injection of icodec in the thigh, abdomen or upper arm, that maximum icodec concentration was slightly but statistically significantly higher by up to 24% for the abdomen and upper arm versus the thigh, but these differences in maximum concentration between injection regions were not considered clinically relevant since the glucose-lowering effect of icodec was shown to be independent of injection region. Single doses of icodec injected subcutaneously in the thigh, abdomen or upper arm were safe and well tolerated, and no injection site reactions were reported.
The observation in the current trial of a greater maximum concentration of icodec after a single dose when administered in the abdomen and upper arm compared with the thigh was expected, since such a difference between injection regions has been shown in previous studies with the basal insulin products, insulin detemir and insulin degludec [15, 16]. For insulin glargine, no differences were found between the thigh, abdomen, and upper arm in the disappearance of radioactivity from the site of injection following subcutaneous injection of radio-labelled insulin glargine [17]. However, it may be questioned to what extent the indirect measure of disappearance of radioactivity from the site of injection reflects the absorption into the circulation and whether this is an appropriate method to investigate potentially modest differences in absorption between injection regions.
At steady state, which is the clinically most relevant situation, smaller differences in maximum concentration of icodec were observed for the abdomen and upper arm versus the thigh relative to the differences seen after a single dose. The explanation for this is that the icodec concentration at any time point at steady state is the result of the four to five previous once-weekly dose administrations, each of them with different relative contribution to exposure depending on the time since injection. Thus, the icodec pharmacokinetic profile, including the maximum concentration, at steady state would be less affected by small injection region-dependent differences in absorption rate from each single injection. A similar smaller difference in maximum concentration for the abdomen and upper arm versus the thigh at steady state relative to after a single dose has been observed for once-daily insulin degludec, probably also due to overlapping exposure from consecutive injections at steady state [15, 18].
Despite the slightly higher maximum concentration of icodec with subcutaneous injection in the abdomen and upper arm compared to the thigh, the glucose-lowering effect around the expected time of maximum was comparable between injection regions. This may be explained by the mode of protraction and mode of action of icodec. Thus, after absorption from the subcutaneous tissue, icodec is strongly and reversibly bound to albumin thereby forming an inactive depot of icodec in the circulation and in the interstitium [8, 9]. As with native human insulin, clearance of free icodec from the circulation occurs mainly by internalisation following binding to and activation of the insulin receptor, a process that is protracted for icodec due to its low insulin receptor binding affinity [8, 9]. It is speculated that when the absorption rate of icodec is increased, such as when icodec is injected subcutaneously in the abdomen or upper arm relative to the thigh, the inactive depot of icodec bound to albumin will act as a buffer to avoid any significant increase in the free, circulating pool of active icodec [9]. These features of icodec may help prevent any clinically relevant impact of injection region on the glucose-lowering effect of icodec. In general, since the mode of protraction of icodec occurs mainly in the circulation and at the target tissues, the glucose-lowering effect of icodec in the clinical situation is considered best reflected by its pharmacodynamic profile rather than by the pharmacokinetic profile, which reflects the total serum icodec concentrations, i.e., the sum of free, unbound icodec and icodec bound to albumin.
In individuals with type 2 diabetes on basal-only insulin therapy, Phase 2 clinical trials have shown comparable improvement in glycaemic control between once-weekly icodec and once-daily insulin glargine U100 with a similar risk of hypoglycaemia [10,11,12]. The current results suggest that individuals with diabetes initiating treatment with icodec as basal insulin can choose their preferred subcutaneous injection region among the thigh, abdomen or upper arm without any difference in the efficacy and safety profile of icodec. There are several barriers to insulin initiation in individuals with type 2 diabetes, and inconvenience and fear of pain associated with injection are among the most prominent [19, 20]. Therefore, it is reassuring for individuals with diabetes that once-weekly icodec offers not only a reduction in number of basal insulin injections by approximately 85% from 365 to 52 per year but also the option to administer basal insulin in the thigh, abdomen or upper arm according to preference.
A limitation of the current trial was that glucose-lowering effect was only assessed during part of the weekly dosing interval, since it was not feasible for participants to take part in a full-week glucose clamp. A single-dose trial design was also chosen to limit the burden on the participants, and simulation to the more clinically relevant steady-state situation was subsequently performed using a pharmacokinetic model developed based on the observed serum icodec concentrations after a single dose. A strength of the present trial was the inclusion of participants with type 2 diabetes, the population that may gain the largest advantage with a once-weekly basal insulin. With this trial population, the glucose clamp target level was set at 7.5 mmol/L to ensure that glucose infusion was required in all participants to keep the glucose concentration constant at the target. The risk of endogenous insulin secretion, which might potentially confound the pharmacodynamic results, was considered minimal due to the relatively high dose level of 5.6 U/kg icodec (corresponding to 0.8 U/kg/day) and given that the glucose clamp was conducted around the time of maximum glucose-lowering effect, where suppression of endogenous insulin secretion was also anticipated to be at its maximum. Furthermore, all participants were using basal insulin at entry into the trial, suggesting a relatively low residual insulin secretion.
5 Conclusion
The current trial shows that icodec can be administered by subcutaneous injection in the thigh, abdomen or upper arm with no clinically relevant difference in exposure and with a similar glucose-lowering effect. These results support that individuals with diabetes may choose between different injection regions for subcutaneous administration of once-weekly icodec with the same treatment effect.
References
Frid AH, Hirsch LJ, Menchior AR, et al. Worldwide Injection Technique Questionnaire study: population parameters and injection practices. Mayo Clin Proc. 2016;91:1212–23. https://doi.org/10.1016/j.mayocp.2016.06.011.
Frid AH, Kreugel G, Grassi G, et al. New insulin delivery recommendations. Mayo Clin Proc. 2016;91:1231–55. https://doi.org/10.1016/j.mayocp.2016.06.010.
De Coninck C, Frid A, Gaspar R, et al. Results and analysis of the 2008–2009 Insulin Injection Technique Questionnaire survey. J Diabetes. 2010;2:168–79. https://doi.org/10.1111/j.1753-0407.2010.00077.x.
Bantle JP, Neal L, Frankamp LM. Effects of the anatomical region used for insulin injections on glycemia in type I diabetes subjects. Diabetes Care. 1993;16:1592–7. https://doi.org/10.2337/diacare.16.12.1592.
ter Braak EW, Woodworth JR, Bianchi R, et al. Injection site effects on the pharmacokinetics and glucodynamics of insulin lispro and regular insulin. Diabetes Care. 1996;19:1437–40. https://doi.org/10.2337/diacare.19.12.1437.
Mudaliar SR, Lindberg FA, Joyce M, et al. Insulin aspart (B28 asp-insulin): a fast-acting analog of human insulin: absorption kinetics and action profile compared with regular human insulin in healthy nondiabetic subjects. Diabetes Care. 1999;22:1501–6. https://doi.org/10.2337/diacare.22.9.1501.
Hövelmann U, Heise T, Nosek L, et al. Pharmacokinetic properties of fast-acting insulin aspart administered in different subcutaneous injection regions. Clin Drug Investig. 2017;37:503–9. https://doi.org/10.1007/s40261-017-0499-y.
Kjeldsen TB, Hubálek F, Hjørringgaard CU, et al. Molecular engineering of insulin icodec, the first acylated insulin analog for once-weekly administration in humans. J Med Chem. 2021;64:8942–50. https://doi.org/10.1021/acs.jmedchem.1c00257.
Nishimura E, Pridal L, Glendorf T, et al. Molecular and pharmacological characterization of insulin icodec: a new basal insulin analog designed for once-weekly dosing. BMJ Open Diabetes Res Care. 2021;9:e002301. https://doi.org/10.1136/bmjdrc-2021-002301.
Rosenstock J, Bajaj HS, Janež A, et al. Once-weekly insulin for type 2 diabetes without previous insulin treatment. N Engl J Med. 2020;383:2107–16. https://doi.org/10.1056/NEJMoa2022474.
Bajaj HS, Bergenstal RM, Christoffersen A, et al. Switching to once-weekly insulin icodec versus once-daily insulin glargine U100 in type 2 diabetes inadequately controlled on daily basal insulin: a phase 2 randomized controlled trial. Diabetes Care. 2021;44:1586–94. https://doi.org/10.2337/dc20-2877.
Lingvay I, Buse JB, Franek E, et al. A randomized, open-label comparison of once-weekly insulin icodec titration strategies versus once-daily insulin glargine U100. Diabetes Care. 2021;44:1595–603. https://doi.org/10.2337/dc20-2878.
Benesch C, Heise T, Klein O, Heinemann L, Arnolds S. How to assess the quality of glucose clamps? Evaluation of clamps performed with ClampArt, a novel automated clamp device. J Diabetes Sci Technol. 2015;9:792–800. https://doi.org/10.1177/1932296815576957.
American Diabetes Association Professional Practice Committee. 6. Glycemic targets: standards of medical care in diabetes—2022. Diabetes Care. 2022;45(Suppl 1):S83–96. https://doi.org/10.2337/dc22-S006.
Nosek L, Coester H-V, Roepstorff C, et al. Glucose-lowering effect of insulin degludec is independent of subcutaneous injection region. Clin Drug Investig. 2014;34:673–9. https://doi.org/10.1007/s40261-014-0218-x.
Morello CM. Pharmacokinetics and pharmacodynamics of insulin analogs in special populations with type 2 diabetes mellitus. Int J Gen Med. 2011;4:827–35. https://doi.org/10.2147/IJGM.S26889.
Owens DR, Coates PA, Luzio SD, Tinbergen JP, Kurzhals R. Pharmacokinetics of 125I-labeled insulin glargine (HOE 901) in healthy men: comparison with NPH insulin and the influence of different subcutaneous injection sites. Diabetes Care. 2000;23:813–9. https://doi.org/10.2337/diacare.23.6.813.
Heise T, Korsatko S, Nosek L, et al. Steady state is reached within 2–3 days of once-daily administration of degludec, a basal insulin with an ultralong duration of action. J Diabetes. 2016;8:132–8. https://doi.org/10.1111/1753-0407.12266.
Brod M, Alolga SL, Meneghini L. Barriers to initiating insulin in type 2 diabetes patients: development of a new patient education tool to address myths, misconceptions and clinical realities. Patient. 2014;7:437–50. https://doi.org/10.1007/s40271-014-0068-x.
Ng CJ, Lai PS, Lee YK, Azmi SA, Teo CH. Barriers and facilitators to starting insulin in patients with type 2 diabetes: a systematic review. Int J Clin Pract. 2015;69:1050–70. https://doi.org/10.1111/ijcp.12691.
Acknowledgements
The authors would like to thank Carsten Roepstorff, PhD, CR Pharma Consult, Copenhagen, Denmark for providing medical writing support, which was funded by Novo Nordisk.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL. This trial was funded by Novo Nordisk.
Conflicts of interest
Leona Plum-Mörschel received travel grants and/or speaker fees from Eli Lilly, Gan & Lee Pharmaceuticals and Novo Nordisk. Solvejg Hansen is an employee of Novo Nordisk. Lizette Ravn Andersen, Niels Rode Kristensen, Lars Lang Lehrskov and Hanne Haahr are employees and shareholders of Novo Nordisk. Ulrike Hövelmann and Patricia Krawietz declare that they have no conflict of interest.
Availability of data and material
Individual participant data will be shared in data sets in a de-identified/anonymised format. Data sets from Novo Nordisk sponsored clinical research completed after 2001 for product indications approved in both the EU and US will be shared. Trial protocol and redacted Clinical Trial Report will be available according to Novo Nordisk data sharing commitments. The data will be available permanently after research completion and approval of product and product use in both EU and US. There is no end date. Data will be shared with bona fide researchers submitting a research proposal requesting access to data for use as approved by the Independent Review Board according to the Independent Review Board Charter (see novonordisk-trials.com). The data will be made available on a specialised SAS data platform.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent to participate
Informed consent was obtained from all individual participants included in the trial.
Consent for publication
Not applicable.
Code availability
Not applicable.
Author contributions
LP-M contributed with trial design, data collection, critical manuscript revision and final manuscript approval. LRA, LLL and NRK contributed with data analysis, critical manuscript revision and final manuscript approval. SH contributed with trial design, data analysis, critical manuscript revision and final manuscript approval. UH and PK contributed with data collection, critical manuscript revision and final manuscript approval. HH contributed with trial conception and design, critical manuscript revision and final manuscript approval.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Plum-Mörschel, L., Andersen, L.R., Hansen, S. et al. Pharmacokinetic and Pharmacodynamic Characteristics of Insulin Icodec After Subcutaneous Administration in the Thigh, Abdomen or Upper Arm in Individuals with Type 2 Diabetes Mellitus. Clin Drug Investig 43, 119–127 (2023). https://doi.org/10.1007/s40261-022-01243-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-022-01243-6