
Abstract
Background and Objective
Insulin degludec is a basal insulin with a slow and distinct absorption mechanism resulting in an ultra-long, flat, and stable pharmacokinetic profile in patients with diabetes mellitus. The aim of this study was to examine the effect of hepatic impairment on the single-dose pharmacokinetics of insulin degludec.
Methods
Twenty-four subjects, allocated to one of four groups (n = 6 per group) based on level of hepatic impairment (normal hepatic function, Child–Pugh grade A, B, or C), were administered a single subcutaneous dose of 0.4 U/kg insulin degludec. Blood samples up to 120 h post-dose and fractionated urine samples were collected to measure pharmacokinetic parameters.
Results
No difference was observed in pharmacokinetic parameters [area under the 120-h serum insulin degludec concentration–time curve (AUC120 h), maximum insulin degludec concentration (C max), and apparent clearance (CL/F)] for subjects with impaired versus normal hepatic function after a single dose of insulin degludec. The geometric mean [coefficient of variation (CV) %] AUC120 h values were 89,092 (16), 83,327 (15), 88,944 (23), and 79,846 (19) pmol·h/L for normal hepatic function and mild, moderate, and severe hepatic impairment, respectively. Simulated steady-state insulin degludec pharmacokinetic profiles showed an even distribution of exposure across a 24-h dosing interval regardless of hepatic function status.
Conclusions
The ultra-long pharmacokinetic properties of insulin degludec were preserved in subjects with hepatic impairment and there were no statistically significant differences in absorption or clearance compared with subjects with normal hepatic function.
Similar content being viewed by others

1 Introduction
Insulin has unsurpassed efficacy in the treatment of diabetes mellitus [1], but is often underutilized, primarily due to the risk or fear of hypoglycemia [2] and the requirement for strict dosing regimens [3]. Underutilization of insulin in patients who require such therapy impedes proper glycemic control [4], thereby increasing the risk of long-term diabetes-related complications [5].
Insulin degludec is a new-generation basal insulin. Upon subcutaneous injection, insulin degludec forms multi-hexamers, resulting in a soluble depot in the subcutaneous tissue from which monomers gradually separate [6, 7]. This mechanism provides slow and continuous absorption, leading to a flat, ultra-long pharmacokinetic profile and four times less variable glucose-lowering effect compared with insulin glargine [8, 9]. The duration of action, which extends beyond 42 h, allows for the possibility of more flexible once-daily dose timing. This permits patients to alter the timing of their insulin dose when needed due to changes in daily activities without compromising glycemic control [10]. A pre-planned meta-analysis of phase 3a insulin degludec trials showed that similar improvements in glycosylated hemoglobin (HbA1c) can be achieved with fewer hypoglycemic episodes (most notably nocturnal episodes) with insulin degludec than with insulin glargine across a broad spectrum of patients with diabetes [11].
Tailored, patient-focused treatment is important in the management of diabetes [1], and a range of conditions that may affect insulin pharmacokinetics and pharmacodynamics have been identified [12], including hepatic dysfunction [13]. Insulin requirements can be lower in patients with hepatic dysfunction due to reduced clearance of insulin combined with reduced hepatic gluconeogenesis [14]. A statement to this effect is made in the summary of product characteristics for insulin glargine, although no specific data are available [15]. For insulin detemir, no clinically relevant difference in pharmacokinetics for subjects with renal or hepatic impairment has been reported, but as these populations have not been studied extensively, it is advised that plasma glucose is monitored closely [16]. The pharmacokinetics of shorter-acting insulins have not been associated with any significant changes as a result of hepatic dysfunction. Single-dose pharmacokinetic studies involving insulin aspart found that hepatic impairment and renal impairment had no significant impact on insulin aspart pharmacokinetics [17]. Hepatic dysfunction did not appear to affect the glucodynamic response to insulin lispro [18]. Considering the limited evidence available, and that hepatic disease may be associated with increased insulin resistance, specifically in cases of increased hepatic fat content [19], it is important to investigate the effect of varying levels of hepatic insufficiency on insulin pharmacokinetics.
The aim of this study was to evaluate whether the pharmacokinetic properties and safety profile of insulin degludec are comparable after a single dose in subjects with mild (Child–Pugh grade A), moderate (Child–Pugh grade B), and severe (Child–Pugh grade C) hepatic impairment compared with subjects with normal hepatic function.
2 Subjects and Methods
2.1 Study Populations
Inclusion criteria included men and women aged 18–75 years with a body mass index of ≤40 kg/m2 and either normal hepatic function or stable hepatic impairment classified as mild, moderate, or severe (Child–Pugh grades A, B, and C, respectively) as assessed by the investigator (subjects with diabetes were eligible for participation within the hepatic impairment groups) [20].
Exclusion criteria for all subjects included known or suspected allergy to the trial product or related products, clinically significant abnormality in hematology, biochemistry or urinalysis (when accounting for the underlying disease), clinically significant renal disease (creatinine clearance <60 mL/min as calculated by the Cockcroft–Gault formula at screening), or liver transplantation. Subjects with diabetes were excluded from the normal hepatic function group. Exclusion criteria specific to subjects with hepatic impairment included acute exacerbation of hepatic insufficiency, clinical signs of acute hepatitis, biliary obstruction and/or other causes of hepatic impairment not related to parenchymal disorders and/or diseases, history or presence of severe hepatic encephalopathy (≥grade 3), advanced ascites, ascites that require emptying and albumin supplementation, esophageal variceal bleeding within 3 months of Visit 2/Day 1, and a prothrombin prolongation time >18 s.
2.2 Study Design and Pharmacokinetic Sampling
This was a single-center (University Hospital Bratislava, Dérer’s Hospital, Bratislava, Slovakia), single-dose, open-label, parallel-group trial (ClinicalTrials.gov identifier: NCT00976326). The sample size of six subjects in each of the four groups (total 24 subjects) was selected to meet the primary objective of this trial and is in accordance with current guidelines from the US FDA and the European Medicines Agency [21, 22]. The sample size was calculated under the assumption of equal sample size in each group to correspond to a power of 80 % in a test for monotonous trend in the primary endpoint—log[area under the 120-h serum insulin degludec concentration–time curve (AUC120 h)]—in patients with decreasing hepatic function, with the significance level set to 5 %. The study consisted of three visits: a screening visit (Visit 1), followed 2–21 days later by a single dosing visit (Visit 2). A follow-up visit (Visit 3) was conducted 7–21 days after the dosing visit. The study was approved by the Slovak health authority, the State Institute for the Control of Drugs, Bratislava, and by an independent ethics committee (Ethics Committee, University Hospital Bratislava, Dérer’s Hospital, Bratislava) and performed in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines (July 2002, CPMP/ICH/135/95), as defined by the International Conference on Harmonisation, in force at the time of study initiation.
Subjects were admitted to the clinic the day before dosing (approximately 2000 hours) and remained in-house for the first 48 h after insulin degludec injection, or longer if deemed necessary by the investigator. At the dosing visit each subject received a single 0.4 U/kg subcutaneous dose of insulin degludec administered into a lifted skinfold on the anterior surface of the thigh. Administration occurred between 0800 and 1000 hours in the morning.
Pharmacokinetic blood samples were collected pre-dose (−15 min and at 0 h), at 1- or 2-h intervals until 24 h post-dose, and then at 30, 36, and 48 h. Blood glucose samples were taken pre-dose, every 30 min for the first 7 h post-dose, then hourly until 24 h post-dose and then at 30, 36, and 48 h, after which subjects could leave the clinic. More frequent blood glucose sampling (every 5–15 min) was undertaken when a subject had low blood glucose (<4 mmol/L). In case of signs of hypoglycemia, oral carbohydrate and/or intravenous glucose were given. Subjects returned to the clinic for additional blood sample collections for pharmacokinetic and blood glucose analysis at 72, 96, and 120 h. A baseline urine sample was collected pre-dose, and fractionated urine collection was performed to determine insulin degludec concentration/excretion at predefined intervals post-dose at Visit 2 (0–8 h, 8–16 h, and 16–24 h). The total duration of the study ranged from 10 to 43 days for an individual subject.
2.3 Bioanalytical Assay
Serum and urine concentrations of insulin degludec were measured using a validated, sandwich enzyme-linked immunosorbent assay (ELISA), specific for insulin degludec with a lower limit of quantification of 20 pM for both serum and urine. For the assay, the capture antibody was a mouse monoclonal antibody specific for human insulin (HUI 001) and the detection antibody was a biotin-labeled monoclonal mouse antibody (NN454-1 F31) [9].
2.4 Data and Statistical Analyses
The primary aim of this study was to investigate the pharmacokinetic exposure of insulin degludec after a single dose in subjects with mild, moderate, or severe hepatic impairment and in subjects with normal hepatic function. Secondary objectives were to characterize further the pharmacokinetic and safety profiles of insulin degludec. Steady-state pharmacokinetic profiles were also simulated for these subject populations.
All pharmacokinetic analyses were based on the full analysis set, and safety analyses were based on the safety analysis set. Both analysis sets included all subjects exposed to insulin degludec during the course of the trial.
The primary endpoint was AUC120 h, and secondary endpoints included maximum insulin degludec concentration (C max) and apparent insulin degludec clearance (CL/F); all endpoints were derived from the individual serum insulin degludec concentration–time curves after a single dose. AUC120 h was derived using the linear trapezoidal technique based on observed values and actual measurement times between 0 and 120 h, with missing values interpolated. CL/F was calculated as dose/area under the serum concentration-time curve from time zero to infinity (AUC∞).
To assess the effect of the degree of hepatic impairment on insulin degludec pharmacokinetic parameters, the endpoints were log-transformed and analyzed using an analysis of variance (ANOVA) model with hepatic function group, sex, and age at baseline as fixed effects. Estimates were then transformed to the normal scale. Estimated mean ratios among the three impaired hepatic function groups compared with the normal hepatic function group were calculated with two-sided 90 % confidence intervals (CIs). A test for monotonous trend in AUC120 h as a function of severity of impaired hepatic function was also conducted in the same ANOVA model described above. In order to investigate the effect of serum albumin concentrations on total exposure of insulin degludec, serum albumin was included as an extra explanatory variable in the same ANOVA model.
To simulate the mean steady-state pharmacokinetic profile of insulin degludec from this single-dose study, a population pharmacokinetic model was used. The model consisted of an absorption part with a depot compartment, a delay compartment, an absorption-rate parameter, and a delay-rate parameter; and a disposition part with one compartment, a clearance parameter and a volume of distribution parameter. The parameters of the model were estimated in a population pharmacokinetic setting using a non-linear mixed–effects approach, which allowed individual sets of the four parameters for each of the subjects included in the trial to be obtained. The values of the absorption rate parameter were subsequently calibrated based on information from the comprehensive clinical pharmacology program with insulin degludec. The same calibration factor was applied for all subjects. Using the individual parameters, a simulation of once-daily multiple dosing was carried out to obtain a mean steady-state profile. More specifically, multiple once-daily dosing for 6 days at a dose level of 0.4 U/kg was simulated by extrapolating the profile for each of the subjects, and subsequently calculating the mean of the profiles on Day 6.
Safety and tolerability of insulin degludec were assessed through adverse events, physical examination, vital signs, electrocardiogram, hypoglycemic events, and clinical laboratory tests (biochemistry, hematology, and urinalysis). Adverse events were classified as mild, moderate, or severe, and judged as having a probable, possible, or unlikely relationship to the trial product by the investigator. Hypoglycemia episodes were defined as ‘confirmed’ if the plasma glucose concentration was <3.1 mmol/L (56 mg/dL), irrespective of symptoms, or if classified as ‘severe’ (requiring assistance) by American Diabetes Association (ADA) guidelines [23]. Hypoglycemic events were classified as ‘treatment emergent’ if the onset of the episode was on or after the time of trial product administration and no later than 7 days after trial product administration.
3 Results
3.1 Subjects
A total of 34 subjects were screened, and 24 subjects (six in each group) were allocated to one of the four hepatic function groups depending on the extent of hepatic impairment. All 24 subjects were allocated to both safety and efficacy populations.
Baseline demographic characteristics were similar among subjects across hepatic function groups (Table 1). Each hepatic function group comprised two males and four females. All subjects were white and were aged between 23 and 62 years. In the severe hepatic impairment group, three subjects (50 %) had type 2 diabetes (mean duration 2.6 years) and were taking oral antidiabetic drug treatment only; no subjects in the other three hepatic function groups had diabetes.
Subjects in the normal hepatic function group had no abnormal physical examination findings. Subjects with hepatic impairment had abnormal findings, mostly related to hepatic disease (such as hepatomegaly at screening visit). None of these abnormalities were considered to conflict with the purposes of this study.
3.2 Pharmacokinetics
No difference was observed in pharmacokinetic parameters (AUC120 h, C max, and CL/F) for subjects with impaired versus normal hepatic function after a single dose of insulin degludec (Table 2). Moreover, a test of monotonous trend between grade of hepatic impairment and AUC120 h was not significant (p = 0.63). Geometric mean [coefficient of variation (CV) in percentage] AUC120 h, C max, and CL/F values for each group are reported in Table 3. Including serum albumin as an extra explanatory variable in the analysis showed that AUC120 h was not significantly affected by differences in serum albumin concentration (p = 0.19).
3.3 Simulated Mean Steady-State Insulin Degludec Concentrations
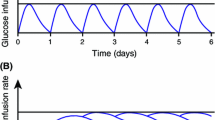
Mean steady-state insulin degludec pharmacokinetic profiles for once-daily 0.4 U/kg subcutaneous administration were simulated and showed an even distribution of exposure across a 24-h dosing interval regardless of hepatic function status (Fig. 1).

Simulated mean insulin degludec concentrations at steady state in subjects with normal hepatic function and in subjects with hepatic impairment (once-daily insulin degludec 0.4 U/kg). IDeg insulin degludec
3.4 Safety
No adverse events (other than hypoglycemia) or medical events of special interest were reported during the course of the study. A total of 12 treatment-emergent hypoglycemic episodes were reported in eight subjects (ten episodes in seven subjects without diabetes and two episodes in one subject with type 2 diabetes); none of these episodes were categorized as severe. No injection-site reactions were reported among the study cohort and no clinically significant changes in laboratory parameters, vital signs, physical examination, or electrocardiogram were observed from initial screening to follow-up.
4 Discussion
This study demonstrates that the ultra-long pharmacokinetic properties of insulin degludec were preserved in subjects with mild, moderate, or severe hepatic impairment. Notably, total exposure (AUC120 h), C max, and CL/F of insulin degludec were comparable for subjects with normal and varying degrees of impaired hepatic function. Furthermore, total exposure of insulin degludec was not significantly affected by the degree of hepatic impairment. This was evidenced by the results of the trend analysis of AUC120 h with decreasing hepatic function, as well as the comparisons between subjects with varying degrees of hepatic function and subjects with normal hepatic function. Moreover, hepatic impairment had no statistically significant effect on C max and CL/F of insulin degludec. Insulin degludec was well-tolerated in subjects with normal or impaired hepatic function; no unexpected safety issues were identified in any subject.
As mentioned previously, limited information is available concerning the effect of hepatic impairment on insulin analogs. For insulin detemir, no clinically relevant difference in pharmacokinetics for subjects with hepatic impairment has been reported [16]. Studies conducted on shorter-acting insulin analogs have indicated that hepatic impairment may not adversely affect their pharmacokinetic properties [17, 18]. However, this limited evidence should be considered in the broader context. Hepatic fat content has been reported as a potential cause of heterogeneity in insulin requirements among patients with type 2 diabetes, through effects on the sensitivity of endogenous glucose production to insulin [19]. Furthermore, the rate at which endogenous insulin is degraded in the liver is reduced in patients with liver cirrhosis [25]. Therefore, while the current study indicates the pharmacokinetics of insulin degludec are not affected by hepatic impairment, the relationship between hepatic disease and insulin requirements remains complex. As a result, treatment with insulin degludec (and indeed any insulin) should be based on individual requirements.
In accordance with regulatory standards [21, 22], the present study included both subjects with and without diabetes. This was not expected to affect the study results as the pharmacokinetics of insulin degludec are similar between healthy subjects and subjects with type 2 diabetes. Due to the inclusion of non-diabetic subjects, it was not possible to conduct a multiple-dose study using a clinically relevant dose level, as this would have imposed an unacceptable risk of hypoglycemia on these non-diabetic subjects. Therefore, to address the steady-state pharmacokinetics of insulin degludec in subjects with hepatic impairment, a population pharmacokinetic model was used to simulate the pharmacokinetic profiles of insulin degludec to steady state based on data from the current trial. The simulations showed that the flat, stable, and ultra-long pharmacokinetic properties shown in other studies [6, 9] are preserved among groups regardless of hepatic function, and with comparable exposure to insulin degludec between subjects with hepatic impairment and subjects with normal hepatic function. In addition to these findings, a study examining the effects of varying degrees of impaired renal function on the pharmacokinetics of insulin degludec found that pharmacokinetic properties were also preserved in subjects with renal impairment [24].
5 Conclusion
In conclusion, the results reported herein support that the ultra-long pharmacokinetic profile of insulin degludec is preserved in patients with hepatic impairment, and that hepatic impairment had no clinically relevant effect on the exposure of insulin degludec.
References
Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2012;5:1577–96.
Peyrot M, Rubin RR, Lauritzen T, et al. Resistance to insulin therapy among patients and providers: results of the cross-national Diabetes Attitudes, Wishes, and Needs (DAWN) study. Diabetes Care. 2005;28:2673–9.
Peyrot M, Barnett AH, Meneghini LF, Schumm-Draeger PM. Factors associated with injection omission/non-adherence in the Global Attitudes of Patients and Physicians in Insulin Therapy study. Diabetes Obes Metab. 2012. doi:10.1111/j.1463-1326.2012.01636.x.
Donnelly LA, Morris AD, Evans JM. Adherence to insulin and its association with glycaemic control in patients with type 2 diabetes. QJM. 2007;100:345–50.
Molyneaux LM, Constantino MI, McGill M, et al. Better glycaemic control and risk reduction of diabetic complications in type 2 diabetes: comparison with the DCCT. Diabetes Res Clin Pract. 1998;42:77–83.
Kurtzhals P, Heise T, Strauss HM, et al. Multi-hexamer formation is the underlying basis for the ultra-long glucose-lowering effect of insulin degludec. Diabetologia. 2011;54:S426.
Jonassen I, Havelund S, Hoeg-Jensen T, et al. Design of the novel protraction mechanism of insulin degludec, an ultra-long-acting basal insulin. Pharm Res. 2012;9:2104–14.
Heise T, Hermanski L, Nosek L, et al. Insulin degludec: four times lower pharmacodynamic variability than insulin glargine under steady-state conditions in type 1 diabetes. Diabetes Obes Metab. 2012;14:859–64.
Heise T, Nosek L, Bøttcher SG, et al. Ultra-long-acting insulin degludec has a flat and stable glucose-lowering effect in type 2 diabetes. Diabetes Obes Metab. 2012;14:944–50.
Meneghini LF, Atkin SL, Gough SCL, et al. The efficacy and safety of insulin degludec given in variable once-daily dosing intervals compared with insulin glargine and insulin degludec dosed at the same time daily: a 26-week, randomized, open-label, parallel group, treat-to-target trial in individuals with type 2 diabetes. Diabetes Care. 2013;36:858–64.
Ratner RE, Gough SC, Mathieu C, et al. Hypoglycaemia risk with insulin degludec compared with insulin glargine in type 2 and type 1 diabetes: a pre-planned meta-analysis of phase 3 trials. Diabetes Obes Metab. 2013;15:175–84.
Morello CM. Pharmacokinetics and pharmacodynamics of insulin analogs in special populations with type 2 diabetes mellitus. Int J Gen Med. 2011;4:827–35.
Quintana JO, García-Compean D, González JA, et al. The impact of diabetes mellitus in mortality of patients with compensated liver cirrhosis-a prospective study. Ann Hepatol. 2011;10:56–62.
Tolman KG, Fonseca V, Dalpiaz A, Tan MH. Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care. 2007;30:734–43.
European Medicines Agency. Sanofi. Lantus® EPAR. October 2012. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000284/WC500036082.pdf. Accessed 28 Jun 2013.
European Medicines Agency. Novo Nordisk A/S. Levemir® EPAR. October 2012. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000528/WC500036662.pdf. Accessed 28 Jun 2013.
Holmes G, Galitz L, Hu P, Lyness W. Pharmacokinetics of insulin aspart in obesity, renal impairment, or hepatic impairment. Br J Clin Pharmacol. 2005;60:469–76.
European Medicines Agency. Eli Lilly and Company. Humalog® EPAR. October 2012. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000088/WC500050332.pdf. Accessed 28 Jun 2013.
Ryysy L, Häkkinen AM, Goto T, et al. Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes. 2000;49:749–58.
Pugh RNH, Murray-Lyon IM, Dawson JL, et al. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–9.
U.S. Department of Health and Human Services & Food and Drug Administration. Guidance for Industry. Pharmacokinetics in patients with impaired hepatic function: study design, data analysis and impact on dosing and labeling. 2003. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072123.pdf. Accessed 26 Jun 2013.
European Medicines Agency. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with impaired hepatic function. CPMP/EWP/2339/02. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003122.pdf. Accessed Jan 2012.
American Diabetes Association Workgroup on Hypoglycemia. Defining and reporting hypoglycemia in diabetes. Diabetes Care. 2005;28:1245–9.
Kiss I, Arold G, Roepstorff C, et al. Insulin degludec: Pharmacokinetics in patients with renal impairment. Clin Pharmacokinet. 2013. doi:10.1007/s40262-013-0113-2.
Narita R, Abe S, Kihara Y, et al. Insulin resistance and insulin secretion in chronic hepatitis C virus infection. J Hepatol. 2004;41:132–8.
Acknowledgments
The involvement of the University Hospital Bratislava, Dérer’s Hospital, Bratislava, Slovakia, is gratefully acknowledged. Medical writing support was provided by ApotheCom ScopeMedical Ltd, funded by Novo Nordisk A/S. This study was funded by Novo Nordisk A/S. Carsten Roepstorff, Malene Højbjerre, Søren Klim, and Hanne Haahr are employees of Novo Nordisk A/S. Viera Kupčová and Gerhard Arold have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
Trial registration: ClinicalTrials.gov identifier: NCT00976326.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Kupčová, V., Arold, G., Roepstorff, C. et al. Insulin Degludec: Pharmacokinetic Properties in Subjects with Hepatic Impairment. Clin Drug Investig 34, 127–133 (2014). https://doi.org/10.1007/s40261-013-0154-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-013-0154-1