
Abstract
Background
There is an increasing body of evidence supporting a more flexible approach in clinical data requirements for the approval of more complex biosimilar substances such as monoclonal antibodies (mAbs).
Objective
The aim of this paper is to further analyse the role of quality/chemistry, manufacturing and controls (CMC) and clinical data for the conclusion on biosimilarity and the decision on marketing authorisation (MA).
Methods
In the present study, we analysed the MA applications (MAAs) of all 33 mAbs and three fusion proteins evaluated by the European Medicines Agency (EMA) between July 2012 and November 2022 with special emphasis on all submitted rituximab (four products) and trastuzumab (seven products) biosimilar candidates, including withdrawn applications. For the two withdrawn applications, the comparative efficacy trials suggested biosimilarity, but the quality/CMC package was not accepted by EMA. We therefore investigated whether a negative MAA outcome could have been predicted based on the evidence generated in the quality/CMC packages, regardless of clinical trial data. For this purpose, we reviewed the respective European Public Assessment Reports (EPARs) or withdrawal assessment reports, and the first regulatory assessments for all these 36 MAAs (i.e. day 120 of the centralized procedure), which are not publicly available. During EMA review, where significant issues are identified which would preclude a marketing authorisation, these issues are raised as questions to the applicant and are classified as major objections (MO).
Results
In 67% of cases, the outcome of the quality and clinical assessment was the same, i.e. both the quality and clinical assessments either supported approval or did not support approval. In 11% of cases, MO were identified in the quality part of the submission but not in the clinical data. In 22% of cases, MO were raised on the clinical data package but not on the quality data. However, we found no instance where seemingly negative clinical data, including failed efficacy trials, led to a negative overall decision. In each instance, the failure to confirm similar clinical performance in all investigated aspects was eventually viewed as not being related to the biosimilar per se but as being due to imbalances in the trial arms, immaturity of secondary endpoint results, change in the reference product, or even chance findings. Furthermore, when performing an in-depth analysis of the quality and clinical packages of trastuzumab and rituximab biosimilars, we found that in no case were clinical trial data necessary to resolve residual uncertainties regarding the quality part.
Conclusion
The results further support the argument that sufficient evidence for biosimilarity can be obtained from a combination of analytical and functional testing and pharmacokinetic studies which may also generate immunogenicity data. This calls into question the usefulness of comparative efficacy studies for the purposes of regulatory decision-making when approving biosimilar mAbs and fusion proteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In the regulatory assessment of 33 mAbs and three fusion proteins evaluated by EMA, we found no instance where seemingly negative clinical data, including failed efficacy trials, led to a negative overall decision. |
In the analysis of quality and clinical packages of trastuzumab and rituximab biosimilar candidates, in no case were clinical trial data necessary to resolve residual uncertainties regarding the quality part. |
Our analyses suggest that the quality/CMC part of the dossier appears to be predictive for the marketing authorisation of a biosimilar mAb or fusion protein candidate, irrespective of the outcome of the clinical trial. |
In the authors’ opinion, the findings of this study may allow a reduction of the clinical development program for regulatory review before marketing authorisation. |
1 Introduction
Biologic medicines are effective treatment options for complex conditions such as cancer and an increasingly important component of health care solutions.
However, patient access to highly effective biological medicines is still unequally distributed across countries, often due to the high cost of these medicines.
Biologics represent 35% of medicine spending in Europe at list prices and have been growing at an 11.3% compound annual growth rate over the past 5 years [1]. The expenditure on cancer medicines is growing at rates higher than the growth rates of the patient population and overall health expenditure [2].
Biosimilar competition, i.e. having multiple suppliers of the same active substance, is necessary to curtail overall healthcare costs and to avoid supply shortages. Although the savings from current biosimilar competition in the European Union (EU) market and patient access are improving, a growing disparity is occurring across countries [1].
The past 5 years have shown a maturation of the biosimilars market. However, it is estimated that in the period 2023–2027, 55% of biologics with loss of market exclusivity will be without competitors [1]. Products with low sales value are unattractive to biosimilar manufacturers due to clinical development costs, largely driven by large and lengthy clinical trials and procurement costs of reference product (RP) comparator batches. Cost estimates for developing a biosimilar range from 100 to 300 million US dollars [3], compared with 1–5 million US dollars for a small molecule generic, largely due to clinical development costs [4,5,6]. This cannot be in the interest of stakeholders, including regulators, and contradicts the strategic priorities of the European Medicines Agency (EMA) [7], which has set out to allow for more rational use of clinical resources.
Recent efforts have resulted in alternatives to costly comparative clinical studies for certain biosimilars, where the extent of clinical data required can vary depending on the complexity and characterisation of the molecule [8,9,10]. Also, several initiatives have been launched that could impact regulatory decision making and lead to revised guidelines such as omitting or reducing the size of studies involving human subjects for a larger set of biosimilar products [11, 12].
In a recent publication [13], the degree of analytical similarity and the role of clinical data were analysed for authorised adalimumab and bevacizumab biosimilars. It was argued that clinical trial requirements for monoclonal antibody (mAb)-biosimilars can be further reduced, or such trials even omitted, where a robust and convincing analytical biosimilarity package is available in conjunction with an appropriately powered pharmacokinetic (PK) study that also provides safety and immunogenicity data.
The remaining question is whether product candidates with good or promising quality data could nevertheless translate into poor (i.e. those with high uncertainty) or failed clinical trials, which may prevent approval of the product where otherwise it would have been approved.
The aim of this paper is to further analyse the role of quality/chemistry, manufacturing and controls (CMC) and clinical data for the conclusion on biosimilarity in a broader setting and in more depth, including all classes of approved biosimilar mAbs and refused or withdrawn biosimilar candidates, which likely would have failed marketing authorisation (MA).
We therefore analysed the final outcome of the submitted marketing authorisation applications (MAAs) of all 36 mAbs and fusion protein biosimilar candidates evaluated by EMA up to November 2022, including those for which a withdrawal/refusal assessment report (AR) is available on the EMA website, i.e. one trastuzumab and one rituximab biosimilar candidate, and contextualised these findings by analyses of all approved rituximab and trastuzumab biosimilar products.
We also reviewed the regulatory assessment during the first phase of these MAAs [i.e. the day 120 assessment reports (D120 AR) of the centralised procedure], which are not publicly available, to analyse whether quality data are predictive for clinical outcome and how clinical assessment impacts the decision on MA.
2 Methods
2.1 Analysis of MAA Outcome
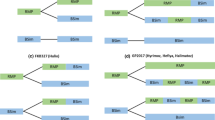
We analysed the outcome of the MAAs, as well as the available regulatory ARs of all biosimilar mAbs and fusion proteins, i.e. adalimumab, bevacizumab, etanercept, infliximab, ranibizumab, rituximab, and trastuzumab, which included 33 mAbs and three fusion proteins evaluated by the EMA between July 2012 and November 2022. This included biosimilar candidates, which received a MA or a negative opinion by the Committee for Medicinal Products for Human Use (CHMP) or which were withdrawn by applicants prior to a CHMP opinion; a European Public Assessment Report (EPAR) or a withdrawal AR for these candidates is available on the EMA website [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49]. Concerns regarding quality/CMC (biosimilarity, general quality) or clinical aspects [PK/pharmacodynamic (PD), efficacy (E)/safety (S)/immunogenicity (I)], as indicated in the EPAR or withdrawal AR, were analysed. We categorised the 36 mAbs and fusion protein biosimilar candidates according to five possible scenarios (Fig. 1) indicating whether the quality/CMC, PK and clinical aspects of the biosimilar MAA dossier were acceptable to the EMA (shown in green/no pattern) or not acceptable (shown as horizontal red stripes). Vertical green stripes indicate some remaining uncertainties that were discussed within the EMA scientific committees and working parties, but not severe enough to prevent MA. Furthermore, the outcome of the MAA is indicated as well as the active substance and the IgG class.

Analysis of MAA outcome. Fulfilment of EMA requirements and outcome of marketing authorisation applications (MAAs) for monoclonal antibody and fusion protein biosimilar candidates based on information provided in the European Public Assessment Reports or Withdrawal Assessment Reports. Green (no pattern) indicates fulfilment of EMA requirements. Vertical green stripes indicate some remaining uncertainties not precluding marketing authorisation (MA). Horizontal red stripes indicate failure to meet EMA requirements. EMA European Medicines Agency, E/S/I efficacy, safety and immunogenicity, IgG Immunoglobulin G, MA marketing authorisation, PD pharmacodynamics, PK pharmacokinetics, Q quality
2.2 Analysis of First Regulatory Assessment Reports
We analysed the list of questions (LoQ) raised by the CHMP in the D120 ARs of all above-mentioned 36 mAbs and fusion protein biosimilar candidates. D120 ARs are discussed and agreed by the CHMP during the first phase of the centralised MA procedure. They are shared with the applicant but are usually not publicly available [50].
They include formal regulatory aspects (Table 1) and the scientific evaluation of all quality/CMC, non-clinical and clinical data and the risk management plan that led to a LoQ regarding concerns and uncertainties that must be addressed by applicants before MA [51]. These questions are classified as major objections (MO) and other concerns (OC). MO are defined as critical issues that will preclude authorisation if not resolved [52], while OC should be resolved but are not severe enough to preclude authorisation per se.
We counted the number of MO and OC and classified those as either related to quality or clinical aspects, in the same manner as for the MAA outcome analyses (Fig. 2). Formal regulatory aspects were not included in the analysis because these are not of direct scientific concern in the context of this study.

Analysis of questions raised in the first assessment report of the MAA procedure. MO are classified in same manner as for the MAA outcome analyses. In case of MO within multidisciplinary aspects, the specific itemised questions were analysed and distributed according to their content to the quality/CMC or clinical (PK/PD, E/S/I) categories. MO regarding formal aspects were not included in the analysis because these are not of direct scientific concern. a For the comparison of the percentage of MO raised with regard to quality/CMC or clinical aspects of the MAAs, the sum of MO (quality/CMC, clinical PK/PD and clinical E/S/I) was calculated and normalised to the number of all MO. b MO related to the quality/CMC of the biosimilar candidate were analysed in more detail. Here the number of MO with regard to general quality/CMC or biosimilarity aspects was divided by the sum of all quality/CMC MO. c Based on the D120 AR, biosimilar candidates were categorised to four different cases with no MO (green/no pattern) or at least one MO (horizontal red stripes) in the respective area. The percentage of candidates represented by the different cases is indicated. CMC chemistry manufacturing and control, D120 AR day 120 assessment report, E/S/I efficacy, safety, immunogenicity, MAA marketing authorisation application, MO major objections, PD pharmacodynamics, PK pharmacokinetics
2.3 Evaluation of Analytical and Clinical Biosimilarity for Rituximab and Trastuzumab Biosimilars
We analysed the quality/CMC and clinical data packages for all submitted rituximab and trastuzumab biosimilars to contextualise the results observed for the two withdrawn products [43, 44]. The analysis included four rituximab biosimilars [26,27,28, 43, 53, 54] and seven trastuzumab biosimilars [36,37,38,39, 44, 48, 49]. The data lock point for the analysis was February 2023.
Comparison of analytical biosimilarity [quality attributes (QA)] for approved biosimilars was performed using the methodology of our previous paper. Briefly, we extracted raw data from the biosimilar product dossiers, anonymised and colour-categorised them depending on the percentage of analysed biosimilar batches with values within the similarity range of the RP (Online Resource 1). For cases where less than 100% of batches were within the reference range it was analysed how the resulting uncertainty was resolved (Table 2).
For full details, see Guillen et al. [13].
For withdrawn applications, we also looked at quality issues of the biosimilar itself affecting performance and consistency of the manufacturing process, which must be ensured in line with current guidance [55]. Therefore, we constructed a figure (Fig. 3) that covers all these aspects, following the key quality/CMC requirements from Bielsky et al. as a reference [56]. Analytical data were extracted from the withdrawal ARs, which contain public information that can be found on the EMA website and therefore anonymisation is not necessary [57].

Analysis of quality requirements for biosimilars withdrawn by the applicant during the review process. For the withdrawn biosimilar applications key relevant quality requirements (and if they were met) were analysed (applications withdrawn for commercial reasons not included). BS biosimilar candidate, CQA critical quality attributes, MoA mechanism of action, QTPP quality target product profile, RP reference product
Comparison of clinical biosimilarity is presented in Online Resources 2–5 and employs the same methodology as Guillen et al. [13]. For cases where discrepancies were observed in clinical attributes, it was analysed how the resulting uncertainty was resolved (Table 3).
3 Results
3.1 MAA Evidence and Results
For 36 mAbs and fusion protein biosimilar candidates (mostly IgG1), the quality/CMC (i.e. general quality aspects and analytical comparability exercise), clinical PK/PD and clinical efficacy, safety and immunogenicity (E/S/I) aspects were analysed based on the information provided in the EPAR. Results are shown in Fig. 1 according to five possible scenarios.
For more than 80% of the biosimilar candidates analysed (29/36), the quality/CMC part of the dossier, the clinical PK/PD as well as the E/S/I results all unambiguously supported biosimilarity (Fig. 1, Scenario 1). For two biosimilar candidates, differences in some QAs and functional assays were observed [14, 15], but these differences were not seen in PK/PD and clinical E/S/I studies. One candidate had higher immunogenicity [15], later deemed irrelevant (see Discussion). All these biosimilar candidates listed for Scenario 1 obtained a MA.
Scenario 2 applies to two cases with an unsatisfactory quality/CMC package but with overall acceptable clinical trial results (Fig. 1, Scenario 2). In these two cases [43, 44], major concerns were raised regarding the biosimilarity exercise as well as regarding the comparability of the clinical batches and the commercial batches of the biosimilar. The clinical PK and efficacy trials formally met their primary endpoints. However, uncertainties remained for the clinical efficacy trial regarding secondary and subgroup analyses for the rituximab biosimilar candidate [43]. Both applications were withdrawn by the companies owing to major remaining uncertainties expressed in unresolved quality MO.
Scenario 3 was defined as those product candidates having an acceptable quality/CMC package but indicating differences in the clinical PK/PD profile or remaining questions regarding representativeness of test material used in the PK study, while all other clinical data demonstrated comparability (Fig. 1, Scenario 3). Two of the biosimilar candidates analysed [45, 46] had an initially failed PK study. In both instances, it was argued that the observed differences in glycan structures known to affect PK (high mannose content) were too small to explain the initially observed PK differences [58]. The conduct of a second PK trial with improved design features was requested and led to successful demonstration of similar PK profiles [59]. For a third biosimilar candidate, PK results were not accepted because the test product was not deemed representative of the commercial product [47].
Scenario 4 lists those cases with an acceptable quality/CMC package and successful PK trial but with issues regarding the clinical E/S/I package (Fig. 1, Scenario 4). For both affected trastuzumabs [48, 49] the primary efficacy endpoint was formally not met as the upper limit of the confidence interval (CI) was not contained within the pre-defined equivalence margin. For both trastuzumabs, a MA was granted based on the convincing quality/CMC, PK, safety and immunogenicity data packages, despite a failed primary endpoint.
The last hypothetical scenario would be unconvincing quality/CMC data and failed clinical trials (PK and efficacy trial), which was not observed in any of the 36 cases (Fig. 1, Scenario 5).
3.2 Analysis of First Regulatory Assessment Reports
For the majority of biosimilar candidates analysed (34/36), the LoQ raised by the CHMP in the D120 AR was adequately addressed by the applicants and thus led to the final approval.
Analysing the number of MO for the 36 biosimilar candidates concerning scientific issues indicates that 56% of MO were related to quality/CMC, 19% to clinical PK/PD and 25% to clinical E/S/I issues, respectively (Fig. 2a). Within the quality/CMC part, the majority of MO dealt with general pharmaceutical issues rather than biosimilarity aspects (Fig. 2b).
Analysis of OC revealed a similar distribution with 64% of OC pertaining to the quality of the biosimilar candidates, 12% to PK/PD and 24% to E/S/I (data not shown).
When categorising the 36 biosimilar candidates based on where MO were raised, i.e. quality versus PK/PD versus E/S/I, we differentiated four cases, depending on whether MO were identified and knowing that any unresolved MO would prevent approval. We differentiated case 1 when assessment of quality and clinical parts of the dossier led to no MO (positive alignment) in 42% (15/36) of the MAAs analysed, thus supporting biosimilarity. Case 2 was when the quality assessment led to MO that, if not resolved, would lead to rejection of the filing. This applies to 11% (4/36) of MAAs analysed. For case 3, when quality assessment supported biosimilarity but clinical queries challenged the validity of the package, 22% (8/36) of cases were identified (8% of cases with MO regarding PK/PD, 11% with MO regarding E/S/I and 3% regarding both PK/PD and E/S/I). And finally, case 4, when both quality and clinical packages raised concerns (negative alignment), with 25% (9/36) of MAAs analysed (Fig. 2c).
The main reasons for MO are summarised in Table 1.
3.3 Evaluation of Analytical Biosimilarity and Clinical Comparability for Rituximab and Trastuzumab Biosimilars
Rituximab and trastuzumab biosimilar products were selected for further in-depth analysis of quality/CMC (Online Resource 1, Table 2; Fig. 3) and clinical data (Online Resource 2–5, Table 3) as these included withdrawn applications.
3.3.1 Comparison of Analytical Biosimilarity Across Products
The number of biosimilar batches analysed per product varied between 3 and 40, for most QAs. The analytical comparability packages of the rituximab and trastuzumab biosimilars comprised between 35 and 85 individual assays per product. For most of the QAs, orthogonal analytical methods were used.
Rituximab is an IgG1 kappa type mAb directed against CD20 expressed on the surface of pre-B and mature B lymphocytes, but not on hematopoietic stem cells and terminally differentiated antibody-producing plasma cells or other tissues. Upon binding to CD20, rituximab mediates B cell lysis (leading to B cell depletion) by three distinct mechanisms of action (MoAs): complement dependent cytotoxicity (CDC), antibody dependent cellular cytotoxicity (ADCC) and apoptosis [60]. Therefore, the biological activity of rituximab is determined by a combination of CD20 binding assay and an apoptosis induction assay, together with fragment crystallisable (Fc) functionality. Besides activating the pathways of CDC and ADCC, binding of rituximab to its target (CD20 expressed on B cells) also triggers apoptosis via the caspase signalling pathway [61]. Antibody-dependent cellular phagocytosis (ADCP) has been further implicated as plausible MoA of rituximab in its killing of chronic lymphocytic leukaemia cells [60, 62].
Trastuzumab is an IgG1 mAb which binds to human epidermal growth factor receptor 2 (HER2), a transmembrane oncoprotein overexpressed in approximately 20–25% of invasive breast cancers [63]. Binding of trastuzumab to HER2 inhibits ligand-independent HER2 signalling and prevents the proteolytic cleavage of its extracellular domain, an activation mechanism of HER2. As a result, trastuzumab inhibits the proliferation of human tumour cells that overexpress HER2. Therefore, the biological activity of trastuzumab is determined by the combination of HER2 binding assay and an inhibition of cellular proliferation assay, together with Fc functionality. However, in contrast to rituximab, CDC activation is not thought of as a MoA of trastuzumab [64].
For other fragment antigen binding (Fab) mediated assays, glycan and purity profile and charge variants we followed a similar categorisation as in our previous paper [13]. Additional assays include, for example, ADCP for both rituximab and trastuzumab, and inhibition of vascular endothelial growth factor (VEGF) secretion for trastuzumab.
Online Resource 1 provides a summary of the analytical biosimilarity results for approved rituximab (products A–C) and trastuzumab (D–I) biosimilars, and Table 2 provides a summary of the instances where less than 100% of batches were within the reference range and how the resulting uncertainty was resolved.
High similarity [≥ 90% of batches within range (solid dark and light-green horizontal stripes)] was found for protein content, biological activity (CD20 binding and apoptosis induction for rituximab, and HER2 binding and inhibition of cellular proliferation assay for trastuzumab), FcγRIIIa binding, neonatal Fc Receptor (FcRn) and C1q binding, ADCC and CDC for almost all rituximab and trastuzumab biosimilars. Exceptions included inhibition of cellular proliferation for one trastuzumab (product I), FcRn for one rituximab (product C) and one trastuzumab (product I) biosimilar and the high affinity FcγRIIIa v/v genotype for one rituximab (product B). However, as seen in Table 2, in most cases these differences were considered within the method variability or viewed as sufficiently justified based on high similarity found in other critical QAs (CQA) (i.e., ADCC for FcγRIIIa v/v), the results from PK comparability studies and regulatory experience. None of the authorised trastuzumab biosimilars displayed CDC activity (represented as dark-green vertical stripes in Online Resource 1), which is expected.
More variability was found for binding to other Fcγ receptors, purity and glycosylation profile, charged variants and additional assays (Online Resource 1). Again, as seen in Table 2 the observed differences in Fc binding assays and the glycan profiles were accepted because similarity was confirmed in biological assays. Moreover, afucosylation was 100% within range for all except one trastuzumab biosimilar (product G). Differences in purity and charge variants were seen as negligible based on regulatory experience and product understanding, and differences in additional assays were accepted based on the totality of the evidence presented for similarity.
3.3.2 Comparison of Analytical Biosimilarity for Withdrawn Products
Figure 3 represents the key quality/CMC requirements and whether these were met for the two withdrawn biosimilar applications [43, 44]. These key requirements were categorised following the classification from Bielsky et al. [56].
Of the quality/CMC requirements included, less than half were met for either of the products. Regarding the RP characterisation, both applicants failed to demonstrate two out of four of the prerequisites, demonstrating in both cases an in-depth knowledge of MoA and CQA of the RP but failing to analyse enough representative RP batches or to adequately establish the quality target product profile (QTTP). Regarding the biosimilar candidate attributes, out of the three prerequisites, only one was met for each product. The quality/CMC package included suitable and qualified analytical methods for the withdrawn rituximab and an adequate manufacturing process for the trastuzumab. However, none of the other requirements were met, including the representativeness of clinical and commercial batches or the use of additional orthogonal assays. Finally, only the withdrawn rituximab included an adequate overall approach for demonstrating biosimilarity.
3.4 Results of Clinical Comparability Studies
Clinical data are presented as raw data in Online Resource 2–5 (the product rows are not in the same order as Online Resource 1 to maintain anonymity). Table 3 provides a summary on all the uncertainties in clinical data, and how these were resolved.
3.4.1 PK Studies
3.4.1.1 Rituximab
For all rituximab biosimilars, PK studies were performed in patients with rheumatoid arthritis (RA) with supportive PK data from oncology patients as part of the efficacy studies. With regard to the withdrawn rituximab application, a comparative efficacy study (in RA) that included PK similarity as a secondary objective was conducted prior to a dedicated comparative PK study [in non-Hodgkin’s lymphoma (NHL)] [43]. Length of follow-up ranged from 24 to 25 weeks (26 weeks in case of the withdrawn application). Primary endpoints [area under the curve to infinity (AUCinf), maximum concentration (Cmax) and AUC from time of administration up to the time of the last quantifiable concentration (AUClast) were contained within the pre-specified acceptance range for all approved biosimilars and secondary endpoints supported biosimilarity. Although for the withdrawn rituximab the pre-defined equivalence margin was 70-143% in the PK comparability study, the 90% CIs of the primary endpoints also met the standard equivalence margin of 0.8–1.25. As seen in Table 3, in two cases [26, 27, 53, 54, 65], results of a secondary endpoint were found outside of the standard acceptance limits, but deviations were seen as minor and not clinically relevant. Detailed information on the PK studies is available in Online Resource 2.
3.4.1.2 Trastuzumab
For all trastuzumab biosimilar candidates, PK studies were performed in healthy subjects with supportive PK data obtained in clinical trials in oncology patients. For the withdrawn trastuzumab application, an additional PK similarity study in healthy subjects was submitted. Length of follow-up of the PK studies ranged from 56 to 99 days (53 days in case of the withdrawn application). In all cases, the primary endpoints (AUCinf, Cmax and AUClast) were contained within the pre-specified acceptance range and secondary endpoints supported biosimilarity. Detailed information on the PK studies is available in Online Resource 4.
3.4.1.3 Population PK
Population PK (PopPK) was performed for some products, using different approaches [28, 37,38,39, 49]. Absence of PopPK analysis was accepted where PK similarity had been demonstrated in the dedicated PK study and PopPK was seen as supportive in the other cases.
3.4.2 Clinical Efficacy Studies
3.4.2.1 Rituximab
Rituximab is currently approved in seven indications, both autoimmune and oncological [66]. Two applicants, including the one for the withdrawn rituximab MAA [27, 43, 53, 65], chose to compare efficacy in RA subjects as a model indication and the remaining two [26, 28, 54] chose follicular lymphoma (FL). Length of follow-up was up to 3 years. Overall response rate (ORR) was chosen as primary endpoint in FL and disease activity score using 28 joint counts (DAS28) or American College of Rheumatology Response (ACR 20) for RA. Detailed information on the efficacy studies is available in Online Resource 3.
3.4.2.2 Trastuzumab
Trastuzumab is currently approved in three indications [67]. For three biosimilars [37,38,39] metastatic breast cancer (MBC) was chosen as model indication in the pivotal clinical trial and for the remaining four, including the withdrawn MAA [36, 44, 48, 49], early breast cancer (EBC) was used. Length of follow-up was up to 3 years.
Three applicants chose ORR and the remaining four pathologic complete response (pCR) as the primary endpoint. Pre-specified equivalence margins for risk difference (RD) varied even though patient populations were the same as different reference studies were used for clinical and statistical justifications [37, 39, 48, 49]. Detailed information on the efficacy studies is available in Online Resource 5.
Table 3 shows those instances where some differences were found and how the remaining uncertainties were resolved. For two products [48, 49], the 95% CI of the difference in the pCR rates between treatments was not fully contained within the pre-defined equivalence margin, thus superiority of the biosimilar cannot be excluded.
3.4.2.3 Safety and Immunogenicity
The overall safety and immunogenicity profiles were compared descriptively and appeared similar between the biosimilars and the RP, as reviewed in detail by Kurki et al. [68].
With regard to the withdrawn rituximab biosimilar candidate application [43], the overall safety profile appeared to be similar in patients with RA but imbalances in adverse events (AEs), serious adverse events (SAEs), severity and deaths were observed in the comparative PK study in patients with NHL. Eight patients died in the product arm versus none in the reference arm; investigators assessed the causal relationship as not (6/8) or unlikely (2/8) related to study drug for all fatal SAEs.
4 Discussion
When considering any change in the current requirements for comparative efficacy studies for biosimilar mAb and fusion protein developments, a fundamental concern of stakeholders including patients, physicians and regulators is that biosimilar product candidates with demonstrated analytical/functional comparability could nevertheless translate into failed clinical comparability. The concern is that in the absence of such clinical trial data, a biosimilar might be inappropriately approved based on quality data only.
Our study shows that this concern is not supported by data and that regulatory decision-making follows a totality-of-the-evidence approach with the main focus on the pharmaceutical quality/CMC (biosimilarity, general quality) and PK similarity aspects. In our opinion, a clinical efficacy study may not need to be routinely requested. In the following parts, we discuss evidence obtained from different analyses performed and its implications.
4.1 Discussion of MAA Evidence and Results
We analysed the MAA reviews of biosimilar mAbs and fusion proteins performed by the EMA CHMP and found that in most cases (29/36 cases) good quality/CMC packages were matched with successful clinical trials leading to MA. Interestingly, good quality/CMC packages could also be paired with formally failed efficacy studies, which were evaluated to be due to reasons not related to the biosimilar candidate, thus permitting MA (see discussion of analysis of clinical comparability for rituximab and trastuzumab biosimilars including withdrawn biosimilar candidates below).
On the contrary, unconvincing quality/CMC data paired with successful clinical trials precluded MA, primarily due to the lack of demonstration of sufficient pharmaceutical quality and/or analytical/functional similarity with the RP [43, 44].
In our analysis, there was only one case where clinical data analysis led to a MO with a divergent position published by the CHMP [15] (Annex) which nevertheless received MA. The issue was later resolved by more mature follow-up data, which did not confirm the objections regarding potentially increased immunogenicity of the biosimilar [69].
For three of the biosimilar candidates analysed [45,46,47] PK studies were deemed insufficient, which led to the conduct of a new, acceptable PK trial. The reasons for repeating the PK trials were due to methodological issues and differences in the formulation buffer [59] or because of representativeness of the test product used [47]. For two biosimilar candidates the PK data submitted in the initial MAA raised major questions regarding biosimilarity. However, re-analysis of the data, which was already pre-specified in the statistical analysis plan, led to the conclusion that PK similarity was shown [18, 21]. Taking this into account, our analysis indicates that, where biosimilarity was shown at the quality/CMC level, this always translated into PK similarity of the biosimilar candidates with the RP.
4.2 Discussion of Analysis of First Regulatory Assessment Reports
We investigated whether the MAA outcome could have been predicted based on the evidence generated solely in the quality dossier. We found that the quality and clinical assessments aligned in 67% of cases, i.e. both quality and clinical data packages, were considered sufficient to support a MA, or both the quality and clinical data packages were not accepted. In 11% of cases, MO were identified in quality parts of the submission, whereas the clinical data supported biosimilarity.
Of particular interest are those 22% of cases (11% E/S/I only) where no MO were observed in the quality part, but MO were raised on the clinical data. Without further regulatory deliberation and additional justifications from the applicants, these cases could have resulted in false negative conclusions, i.e. a true biosimilar being rejected due to issues with clinical studies. However, even in those cases where the efficacy trials formally failed, biosimilarity was ultimately accepted by EMA based on the demonstration of analytical/functional comparability and comparable PK profiles. In each instance, identified issues in the clinical package were eventually accepted as the result of unanticipated problems such as imbalances in trial arms, immaturity of secondary endpoint data at the time of MAA submission, changes in the QA of the RP or even chance findings. In some cases, a further in-depth sensitivity analysis improved the understanding of the clinical data and facilitated a positive conclusion. These cases highlight that for biosimilar mAbs and fusion proteins, the analytical and functional characterisation data are the most critical for decision making and regulatory approval.
In summary, our analyses of MAAs and first regulatory assessment reports show that the quality/CMC part of the dossier is predictive for the MA of a biosimilar candidate.
4.3 Discussion of Analysis of Analytical Biosimilarity for Rituximab and Trastuzumab Biosimilars
Since our analyses included the two withdrawn MAs, we reviewed to what degree thorough analysis of the quality/CMC package could have been predictive for the clinical outcome. These two substance classes are particularly interesting, as the two originator mAbs are used in oncology (rituximab, trastuzumab) and/or in autoimmune indications (rituximab). Based on information provided in the MAA, and following the scientific evaluation carried out by EMA CHMP, for approved rituximab and trastuzumab biosimilars, we found that over 90% (and in most cases 100%) of the biosimilar batches met the EU reference product similarity range for CQAs such as protein content, biological activity (CD20 binding and apoptosis induction for rituximab and HER2-binding and inhibition of cellular proliferation assay for trastuzumab), FcγRIIIa binding, FcRn and C1q binding, ADCC and CDC.
A lower percentage of biosimilar batches were within the similarity range for QAs which are considered less critical (glycosylation profile, charge variants or additional assays). Furthermore, as seen in Table 2, in all instances where uncertainties were raised, these were resolved considering the close similarity demonstrated in CQAs, the results of PK studies and overall, the totality of the evidence.
In no case were clinical trial data necessary to resolve residual uncertainties regarding the quality part. As seen in Fig. 1 (Scenario 4), for the withdrawn MAs, clinical data was also not sufficient to justify the differences in the quality/CMC package. This is in line with previous findings [13, 56] and further demonstrates that the array of orthogonal methods that are submitted in the quality/CMC package are robust and predictive of clinical outcome.
4.4 Discussion of Analysis of Clinical Comparability for Rituximab and Trastuzumab Biosimilars Including Withdrawn Biosimilar Candidates
For all rituximab and trastuzumab biosimilar candidates studied, a comprehensive clinical program was submitted according to the relevant EMA guidelines, i.e. consisting of at least one PK study and a clinical efficacy study, which confirmed biosimilarity in all but two instances [36,37,38,39, 44, 48, 49].
For all rituximab biosimilars included in the analysis, PK results were obtained in one therapeutic area and confirmed in a subset of patients in the second therapeutic indication either by supportive PK analysis [26, 27, 43, 53, 54, 65] or by PopPK analyses [28]. In no instance were deviating results observed. One applicant [26] removed PopPK in a protocol amendment which was acceptable to the regulators. In our opinion, this redundancy of PK evaluations should no longer be necessary, as besides CD20 no other antigen or target is involved in rituximab’s binding and MoA. This has been demonstrated by the tissue specificity in several human tissue cross reactivity studies [66, 70].
In our study, in all cases except for the withdrawn applications, remaining uncertainties regarding clinical data were resolved based on a strong quality/CMC package, together with demonstrated PK similarity and considering that the studies were not powered to demonstrate similarity with regard to secondary endpoints. Moreover, regarding safety, it should be noted that clinical trials are not powered for safety endpoints, since this is considered unnecessary according to EMA guidelines and would usually require several thousand study participants.
Therefore, the value of extensive analyses of secondary endpoints in efficacy trials remains questionable, as they were either viewed as inconclusive [27, 53, 65], or immature [26, 54].
For both trastuzumab cases [48, 49], EMA concluded that it was likely that the apparent difference was caused by a noted downward “shift” in ADCC activity in some of the RP batches and did not preclude approval. These findings have been discussed in the literature [56, 71, 72] and also demonstrate that physicochemical methods and functional assays are able to detect differences in functional attributes with predictive character. With regards to the withdrawn trastuzumab biosimilar [44], a conclusion on biosimilarity from a safety point of view was precluded as the clinical batches were not considered representative for the commercial product.
Several authors [56, 59, 73,74,75] pointed out limitations of indiscriminative clinical efficacy studies in light of technical advances in analytical methods, which provide more discriminative research tools sparing patients from entering unnecessary and redundant clinical trials.
It is known from the literature that the recommended doses of many mAbs are in the flat part of the dose-response curve, e.g. half of the administered dose of rituximab would result in the same clinical outcome, suggesting the overall equivalence of 2 × 500 mg with the licensed dose of 2 × 1000 mg for clinical efficacy outcomes [76], thus rendering clinical trials insensitive tools to assess biosimilarity. Some authors argue that treatment response to rituximab in RA is only determined by the level of B cell depletion, regardless of how it is achieved [76,77,78,79], yet it is important to measure binding and all functional activity. In our analysis we found that rituximab binding to the CD20 receptor plus all four important MoAs of rituximab [60] were adequately measured by appropriate analytical testing and that for the successful quality/CMC dossiers > 90% of batches met the required similarity ranges.
4.5 General Discussion on Current Flexibility in Clinical Trial Requirements
Our analysis revealed that there is regulatory flexibility in the acceptability of clinical data packages. This is, for example, evidenced by the acceptance of different primary endpoints such as DAS28 versus ACR20 in RA trials, different methods of analyses such as RD versus risk ratio (RR), or different equivalence margins for the same study population depending on the reference study chosen, provided appropriate scientific justification is given [29, 37, 38, 48].
Currently, most mAb developers are planning to evaluate S/I over one year, as advised by guidelines. However, the primary efficacy analysis is usually performed at a much earlier timepoint [26,27,28, 37,38,39]. Based on our analyses of the submitted clinical trial data, as well as on the raw data analysis of Kurki et al. [68], it is clear that most dossiers are submitted with preliminary 4–6 months S/I data, depending on the timepoint of primary efficacy analysis, while the full one-year dataset is submitted later during the evaluation period. In no instance have the conclusions reached after the initial data submission changed after completion of the study after 12 months, suggesting that such long trials are unnecessary. Other authors have concluded that pertinent comparative safety and immunogenicity data will be obtained from the PK trials, which in all instances were shown to support results of the E/S study [68].
We conclude that a sufficiently robust analytical/functional similarity package, together with a PK trial capturing data on safety and immunogenicity would be sufficient for the purpose of regulatory decision making for biosimilar mAbs and fusion proteins. In some case, if it were deemed necessary (e.g. when the MoA of the biologic is poorly understood), a shorter efficacy trial ending at or near the timepoint of primary efficacy analysis could provide additional safety and immunogenicity information.
4.6 Limitation of the Study
Since our study was limited to mAb and fusion protein biosimilars of IgG class, our conclusions may not be applicable to more complex biologics. Furthermore, our analysis is restricted to products that have undergone regulatory assessment and appraisal. Whether there have been biosimilars that failed quality/CMC and/or clinical development and were therefore never submitted for regulatory evaluation or publication has not been scrutinised in our study. Also, overall, the sample size of 36 was limited by the available submissions to EMA in the previous 10 years. However, to our knowledge this is the largest set of biosimilar MAs analysed to date.
5 Conclusions of Our Study
The results of our analysis show that a comprehensive and convincing quality/CMC package demonstrating high analytical/functional similarity of the biosimilar with the RP is essential for MA. Since the first approval of less complex biosimilars, the analytical techniques have advanced markedly resulting in very sensitive assays for the structural and functional characterisation of even complex mAb molecules.
The concern, that in the absence of comparative efficacy and safety results, a biosimilar candidate might be inappropriately approved based on quality data only, is not supported by our findings. The analytical and biological results can be considered predictive for the clinical performance of the biosimilar candidates.
Based on the combination of modern analytics, control and pharmacovigilance systems in place, as well as requirements on comparability assessment in case of manufacturing changes, clinical performance of IgG biopharmaceuticals is ensured throughout the lifecycle of the product. As shown in our analysis, the CQA that are known to impact clinical efficacy and safety, including immunogenicity, must be closely monitored.
In the authors’ opinion, these findings allow a reduction of the clinical development program for regulatory review before MA. This conclusion is further supported by the positive experience in the market gained for biosimilar mAbs approved in the last ten years. Consequently, a revision of the respective regulatory biosimilars guidelines in Europe should be considered, to allow a more rational use of clinical resources and improve the access to innovative and affordable medicines for patients.
References
IQVIA. White paper: the impact of biosimilar competition in Europe 2022. Dec 2022. https://www.iqvia.com/library/white-papers/the-impact-of-biosimilar-competition-in-europe-2022. Accessed 30 Apr 2023.
Prasad V. Do cancer drugs improve survival or quality of life? BMJ. 2017;359: j4528. https://doi.org/10.1136/bmj.j4528.
The Center for Biosimilars. Scott Gottlieb, MD: phase 3 trials and biosimilars [Interview Transcript]. AJMC. 2019.
Blackstone EA, Joseph PF. The economics of biosimilars. Am Health Drug Benefits. 2013;6:469–78.
Federal Trade Commission. Emerging health care issues: Follow-on biologic drug competition. 2009. https://www.ftc.gov/sites/default/files/documents/reports/emerging-health-care-issues-follow-biologic-drug-competition-federal-trade-commission-report/p083901biologicsreport.pdf. Accessed 30 Apr 2023.
IQVIA. The prospects for biosimilars of orphan drugs in Europe. 2020. https://www.iqvia.com/insights/the-iqvia-institute/reports/the-prospects-for-biosimilars-of-orphan-drugs-in-europe. Accessed 30 Apr 2023.
EMA. EMA regulatory science to 2025 EMA/110706/2020. 2020. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/ema-regulatory-science-2025-strategic-reflection_en.pdf. Accessed 30 Apr 2023.
EMA. Guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant human insulin and insulin analogues EMEA/CHMP/BMWP/32775/2005_Rev.1. 2015. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-non-clinical-clinical-development-similar-biological-medicinal-products-containing_en-0.pdf. Accessed 30 Apr 2023.
EMA. Guideline on non-clinical and clinical development of similar biological medicinal products containing low-molecular-weight-heparins EMEA/CHMP/BMWP/118264/2007 Rev. 1. 2016. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-non-clinical-clinical-development-similar-biological-medicinal-products-containing-low_en.pdf. Accessed 30 Apr 2023.
EMA. Guideline on similar biological medicinal products containing recombinant granulocyte-colony stimulating factor (rG-CSF) EMEA/CHMP/BMWP/31329/2005 Rev 1. 2018. https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-similar-biological-medicinal-products-containing-recombinant-granulocyte-colony_en.pdf. Accessed 30 Apr 2023.
U.S. Food and Drug Administration. BsUFA III regulatory research pilot program: research roadmap. https://www.fda.gov/media/164751/download. Accessed 30 Apr 2023.
Vandekerckhove K, Seidl A, Gutka H, Kumar M, Gratzl G, Keire D, et al. Rational selection, criticality assessment, and tiering of quality attributes and test methods for analytical similarity evaluation of biosimilars. AAPS J. 2018;20:68. https://doi.org/10.1208/s12248-018-0230-9.
Guillen E, Ekman N, Barry S, Weise M, Wolff-Holz E. A data driven approach to support tailored clinical programs for biosimilar monoclonal antibodies. Clin Pharmacol Ther. 2023;113:108–23. https://doi.org/10.1002/cpt.2785.
EMA. EPAR Remsima EMA/CHMP/589317/2013. 2013. https://www.ema.europa.eu/en/medicines/human/EPAR/remsima. Accessed 30 Apr 2023.
EMA. EPAR Flixabi EMA/CHMP/272283/2016. 2016. https://www.ema.europa.eu/en/medicines/human/EPAR/flixabi. Accessed 30 Apr 2023.
EMA. EPAR Zessly EMA/223369/2018. 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/zessly. Accessed 30 Apr 2023.
EMA. EPAR Benepali EMA/CHMP/819219/2015. 2015. https://www.ema.europa.eu/en/medicines/human/EPAR/benepali. Accessed 30 Apr 2023.
EMA. EPAR Erelzi EMA/CHMP/302222/2017. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/erelzi. Accessed 30 Apr 2023.
EMA. EPAR Amgevita EMA/106922/2017. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/amgevita. Accessed 30 Apr 2023.
EMA. EPAR Imraldi EMA/CHMP/559383/2017. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/imraldi. Accessed 30 Apr 2023.
EMA. EPAR Hulio EMA/541826/2018. 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/hulio. Accessed 30 Apr 2023.
EMA. EPAR Idacio EMA/124342/2019. 2019. https://www.ema.europa.eu/en/medicines/human/EPAR/idacio. Accessed 30 Apr 2023.
EMA. EPAR Amsparity EMA/2756/2020. 2019. https://www.ema.europa.eu/en/medicines/human/EPAR/amsparity. Accessed 30 Apr 2023.
EMA. EPAR Yuflyma EMA/47907/2021. 2020. https://www.ema.europa.eu/en/medicines/human/EPAR/yuflyma. Accessed 30 Apr 2023.
EMA. EPAR Hukyndra EMA/555939/2021. 2021. https://www.ema.europa.eu/en/medicines/human/EPAR/hukyndra. Accessed 30 Apr 2023.
EMA. EPAR Rixathon EMA/303207/2017. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/rixathon. Accessed 30 Apr 2023.
EMA. EPAR Blitzima EMA/CHMP/421793/2017. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/blitzima. Accessed 30 Apr 2023.
EMA. EPAR Ruxience EMA/CHMP/155201/2020. 2020. https://www.ema.europa.eu/en/medicines/human/EPAR/ruxience. Accessed 30 Apr 2023.
EMA. EPAR Mvasi EMA/798844/2017. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/mvasi. Accessed 30 Apr 2023.
EMA. EPAR Zirabev EMA/97237/2019. 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/zirabev. Accessed 30 Apr 2023.
EMA. EPAR Aybintio EMA/380645/2020. 2020. https://www.ema.europa.eu/en/medicines/human/EPAR/aybintio. Accessed 30 Apr 2023.
EMA. EPAR Equidacent EMA/427966/2020. 2020. https://www.ema.europa.eu/en/medicines/human/EPAR/equidacent. Accessed 30 Apr 2023.
EMA. EPAR Alymsys EMA/112177/2021. 2021. https://www.ema.europa.eu/en/medicines/human/EPAR/alymsys. Accessed 30 Apr 2023.
EMA. EPAR Abevmy EMA/176217/2021. 2021. https://www.ema.europa.eu/en/medicines/human/EPAR/abevmy. Accessed 30 Apr 2023.
EMA. EPAR Vegzelma EMA/CHMP/636045/2022. 2022. https://www.ema.europa.eu/en/medicines/human/EPAR/vegzelma. Accessed 30 Apr 2023.
EMA. EPAR Herzuma EMA/44005/2018. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/herzuma. Accessed 30 Apr 2023.
EMA. EPAR Trazimera EMA/414095/2018. 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/trazimera. Accessed 30 Apr 2023.
EMA. EPAR Ogivri EMA/810499/2018. 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/ogivri. Accessed 30 Apr 2023.
EMA. EPAR Zercepac EMA/327248/2020. 2020. https://www.ema.europa.eu/en/medicines/human/EPAR/zercepac. Accessed 30 Apr 2023.
EMA. EPAR Byooviz EMA/446448/2021. 2021. https://www.ema.europa.eu/en/medicines/human/EPAR/byooviz. Accessed 30 Apr 2023.
EMA. EPAR Ranivisio EMA/640346/2022. 2022. https://www.ema.europa.eu/en/medicines/human/EPAR/ranivisio. Accessed 30 Apr 2023.
EMA. EPAR Ximluci EMA/CHMP/795998/2022. 2022. https://www.ema.europa.eu/en/medicines/human/EPAR/ximluci. Accessed 30 Apr 2023.
EMA. Withdrawal assessment report Rituximab Mabion EMA/CHMP/141953/2020. 2019. https://www.ema.europa.eu/en/medicines/human/withdrawn-applications/rituximab-mabion. Accessed 30 Apr 2023.
EMA. Withdrawal assessment report Tuznue EMA/CHMP/902445/2022. 2022. https://www.ema.europa.eu/en/medicines/human/withdrawn-applications/tuznue. Accessed 30 Apr 2023.
EMA. EPAR Cyltezo EMA/CHMP/750187/2017. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/cyltezo. Accessed 30 Apr 2023.
EMA. EPAR Hyrimoz EMA/CHMP/404076/2018. 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/hyrimoz. Accessed 30 Apr 2023.
EMA. EPAR Nepexto EMA/212524/2020. 2020. https://www.ema.europa.eu/en/medicines/human/EPAR/nepexto. Accessed 30 Apr 2023.
EMA. EPAR Ontruzant EMA/CHMP/9855/2018. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/ontruzant. Accessed 30 Apr 2023.
EMA. EPAR Kanjinti EMA/CHMP/261937/2018. 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/kanjinti. Accessed 30 Apr 2023.
EMA. Guide to information on human medicines evaluated by EMA. https://www.ema.europa.eu/en/documents/other/guide-information-human-medicines-evaluated-european-medicines-agency-what-agency-publishes-when_en.pdf. Accessed 30 Apr 2023.
EMA. The evaluation of medicines, step-by-step. 2020. https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/evaluation-medicines-step-step. Accessed 30 Apr 2023.
EMA. How EMA evaluates medicines for human use. https://www.ema.europa.eu/en/about-us/what-we-do/authorisation-medicines/how-ema-evaluates-medicines#:~:text=If%20unresolved%2C%20major%20objections%20preclude,seriousness%20of%20the%20side%20effects. Accessed 30 Apr 2023.
EMA. EPAR Truxima EMA/CHMP/75695/2017. 2016. https://www.ema.europa.eu/en/medicines/human/EPAR/truxima. Accessed 30 Apr 2023.
EMA. EPAR Riximyo EMA/440905/2017. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/riximyo. Accessed 30 Apr 2023.
EMA. Guideline on similar biological medicinal products containing biotechnology-derived protiens as active substance: quality issues EMA/CHMP/BWP/247713/2012. 2014. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-0.pdf. Accessed 30 Apr 2023.
Bielsky MC, Cook A, Wallington A, Exley A, Kauser S, Hay JL, et al. Streamlined approval of biosimilars: moving on from the confirmatory efficacy trial. Drug Discov Today. 2020. https://doi.org/10.1016/j.drudis.2020.09.006.
EMA. Find medicines. https://www.ema.europa.eu/en/medicines. Accessed 30 Apr 2023.
Kim J, Luo H, White W, Rees W, Venkat R, Albarghouthi M. Impact of Fc N-linked glycans on in vivo clearance of an immunoglobulin G1 antibody produced by NS0 cell line. MAbs. 2020;12:1844928. https://doi.org/10.1080/19420862.2020.1844928.
Wolff-Holz E, Tiitso K, Vleminckx C, Weise M. Evolution of the EU biosimilar framework: past and future. BioDrugs. 2019;33:621–34. https://doi.org/10.1007/s40259-019-00377-y.
Jaglowski SM, Alinari L, Lapalombella R, Muthusamy N, Byrd JC. The clinical application of monoclonal antibodies in chronic lymphocytic leukemia. Blood. 2010;116:3705–14. https://doi.org/10.1182/blood-2010-04-001230.
Byrd JC, Kitada S, Flinn IW, Aron JL, Pearson M, Lucas D, Reed JC. The mechanism of tumor cell clearance by rituximab in vivo in patients with B-cell chronic lymphocytic leukemia: evidence of caspase activation and apoptosis induction. Blood. 2002;99:1038–43. https://doi.org/10.1182/blood.v99.3.1038.
Kuhns S, Shu J, Xiang C, de Guzman R, Zhang Q, Bretzlaff W, et al. Differential influence on antibody dependent cellular phagocytosis by different glycoforms on therapeutic monoclonal antibodies. J Biotechnol. 2020;317:5–15. https://doi.org/10.1016/j.jbiotec.2020.04.017.
Pohlmann PR, Mayer IA, Mernaugh R. Resistance to trastuzumab in breast cancer. Clin Cancer Res. 2009;15:7479–91. https://doi.org/10.1158/1078-0432.CCR-09-0636.
FDA. FDA briefing document: Oncologic drugs advisory committee meeting. BLA 761074. MYL-1401O, a proposed biosimilar to Herceptin (trastuzumab) Applicant: Mylan Pharmaceuticals. 2017. https://www.fda.gov/media/106566/download. Accessed 30 Apr 2023.
EMA. EPAR Ritemvia EMA/CHMP/421799/2017. 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/ritemvia. Accessed 30 Apr 2023.
EMA. EPAR MabThera. https://www.ema.europa.eu/en/medicines/human/EPAR/mabthera. Accessed 30 Apr 2023.
EMA. Herceptin | European Medicines Agency. https://www.ema.europa.eu/en/medicines/human/EPAR/herceptin. Accessed 30 Apr 2023.
Kurki P, Barry S, Bourges I, Tsantili P, Wolff-Holz E. Safety, immunogenicity and interchangeability of biosimilar monoclonal antibodies and fusion proteins: a regulatory perspective. Drugs. 2021;81:1881–96. https://doi.org/10.1007/s40265-021-01601-2.
EMA. Flixabi: Procedural steps taken and scientific informaiton after the authorisation. https://www.ema.europa.eu/en/medicines/human/EPAR/flixabi. Accessed 30 Apr 2023.
Tedder TF, Engel P. CD20: a regulator of cell-cycle progression of B lymphocytes. Immunol Today. 1994;15:450–4. https://doi.org/10.1016/0167-5699(94)90276-3.
Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29:310–2. https://doi.org/10.1038/nbt.1839.
Kim S, Song J, Park S, Ham S, Paek K, Kang M, et al. Drifts in ADCC-related quality attributes of Herceptin®: impact on development of a trastuzumab biosimilar. MAbs. 2017;9:704–14. https://doi.org/10.1080/19420862.2017.1305530.
McCamish M, Woollett G. The continuum of comparability extends to biosimilarity: how much is enough and what clinical data are necessary? Clin Pharmacol Ther. 2013;93:315–7. https://doi.org/10.1038/clpt.2013.17.
Schiestl M, Ranganna G, Watson K, Jung B, Roth K, Capsius B, et al. The path towards a tailored clinical biosimilar development. BioDrugs. 2020;34:297–306. https://doi.org/10.1007/s40259-020-00422-1.
Webster CJ, Wong AC, Woollett GR. An efficient development paradigm for biosimilars. BioDrugs. 2019;33:603–11. https://doi.org/10.1007/s40259-019-00371-4.
Vital EM, Rawstron AC, Dass S, Henshaw K, Madden J, Emery P, McGonagle D. Reduced-dose rituximab in rheumatoid arthritis: efficacy depends on degree of B cell depletion. Arthritis Rheum. 2011;63:603–8. https://doi.org/10.1002/art.30152.
Emery P, Fleischmann R, Filipowicz-Sosnowska A, Schechtman J, Szczepanski L, Kavanaugh A, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54:1390–400. https://doi.org/10.1002/art.21778.
Rubbert-Roth A, Tak PP, Zerbini C, Tremblay J-L, Carreño L, Armstrong G, et al. Efficacy and safety of various repeat treatment dosing regimens of rituximab in patients with active rheumatoid arthritis: results of a Phase III randomized study (MIRROR). Rheumatology (Oxford). 2010;49:1683–93. https://doi.org/10.1093/rheumatology/keq116.
Tak PP, Rigby WF, Rubbert-Roth A, Peterfy CG, van Vollenhoven RF, Stohl W, et al. Inhibition of joint damage and improved clinical outcomes with rituximab plus methotrexate in early active rheumatoid arthritis: the IMAGE trial. Ann Rheum Dis. 2011;70:39–46. https://doi.org/10.1136/ard.2010.137703.
Acknowledgements
The authors thank Iordanis Gravanis, Steffen Thirstrup and Malgorzata Zienowicz (EMA) and Jan Müller-Berghaus (PEI and Member of CHMP and CAT, EMA) for their valuable suggestions and comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was funded by the German Federal Ministry of Health (Grant no. 2522FSB450).
Conflict of interest
This study was funded by the German Federal Ministry of Health. The authors report no potential conflicts of interest or financial disclosures that are pertinent to this article.
Disclaimer
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the regulatory agencies with which the authors are affiliated.
Ethics approval
Ethics approval is not applicable to this article as no human or animal subjects were analysed.
Consent to participate
Consent to participate is not applicable to this article as no human or animal subjects were analysed.
Consent for publication
Consent for publication is not applicable to this article as no human or animal subjects were analysed.
Availability of data and material
The datasets analysed for clinical comparability during the current study are available in the EMA repository, which are publicly available at: https://www.ema.europa.eu/en/medicines/field_ema_web_categories%253Aname_field/Human. The datasets generated and/or analysed during the current study for analytical biosimilarity, marketing authorization applications and regulatory assessment reports are not publicly available due to confidentiality issues.
Code availability
Not applicable.
Author contributions
NK-S, EG, NE, SB, VK, SK, MW, and EW-H wrote the manuscript. NK-S, EG, NE, and EW-H designed the research. NK-S, EG, NE, VK, and EW-H performed the research and analysed the data. NK-S and EG contributed equally to the manuscript. All authors read and approved the final manuscript.
Additional information
The work contributed by EW-H has been carried out while she was an employee of PEI, member of the Scientific Advice Working Party and chair of the Biosimilar Medicinal Products Working Party.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kirsch-Stefan, N., Guillen, E., Ekman, N. et al. Do the Outcomes of Clinical Efficacy Trials Matter in Regulatory Decision-Making for Biosimilars?. BioDrugs 37, 855–871 (2023). https://doi.org/10.1007/s40259-023-00631-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-023-00631-4