
Abstract
Background
MB02 (bevacizumab biosimilar) showed similar structural, functional, and pharmacokinetic properties to reference bevacizumab (Avastin®; EU-bevacizumab).
Objectives
To confirm clinical similarity between MB02 and EU-bevacizumab, a comparability study was undertaken in the first-line treatment of stage IIIB/IV non-squamous non-small cell lung cancer (NSCLC).
Patients and Methods
This multinational, double-blind, randomized, phase III study (STELLA) compared MB02 or EU-bevacizumab (15 mg/kg) administered with chemotherapy (paclitaxel 200 mg/m2 and carboplatin AUC6) on Day 1 of every 3-week cycle for 6 cycles (Week 18), followed by MB02/EU-bevacizumab in blinded monotherapy until disease progression, unacceptable toxicity, death, withdrawal of consent or end of study (Week 52). The primary efficacy endpoint was objective response rate (ORR) evaluated by an independent radiological review committee (IRC) at Week 18 (intent-to-treat population). Secondary endpoints included progression-free survival (PFS), overall survival (OS), safety and immunogenicity.
Results
A total of 627 subjects were randomized 1:1 to MB02 (n = 315) or EU-bevacizumab (n = 312). ORR, assessed by the IRC at Week 18, was comparable in MB02 (40.3%) and EU-bevacizumab (44.6%) groups. ORR risk ratio of 0.910 (90% CI 0.780 to 1.060; 95% CI 0.758 to 1.092) and ORR risk difference of −4.02 (90% CI −10.51 to 2.47; 95% CI −11.76 to 3.71) were within the similarity predefined margins. There were no significant differences between MB02 and EU-bevacizumab groups in median PFS (36.0 vs 37.3 weeks, respectively; HR 1.187; 95% CI 0.98 to 1.44) and median OS (not achieved; HR 1.108; 95% CI: 0.83 to 1.49) at the end of study. The safety profile of MB02 and EU-bevacizumab regarding nature, frequency and severity of the adverse events (AE) was comparable. The most frequent grade ≥3 investigational-product-related AEs were hypertension and anemia, with a difference between treatment groups of <5%. Anti-drug antibodies (ADA) and neutralizing ADA (NAb) incidence were similar in both treatment groups.
Conclusion
MB02 demonstrated similar efficacy to EU-bevacizumab, in combination with carboplatin and paclitaxel, in subjects with advanced non-squamous NSCLC, with comparable safety and immunogenicity profiles.
Clinical trial registration
EudraCT No. 2017-001769-26; ClinicalTrials.gov: NCT03296163.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
MB02, a bevacizumab biosimilar, has demonstrated analytical similarity to reference bevacizumab on a comprehensive chemistry, manufacturing, and control (CMC) and bioanalytical similarity program. PK similarity has been further confirmed in three bioequivalence studies comparing the pharmacokinetic profiles of MB02 and reference bevacizumab following the administration of a single dose (3 mg/kg IV) in more than 276 healthy male subjects. |
The STELLA clinical equivalence study compared both drugs in the first-line treatment of advanced non-squamous non-small cell lung cancer (NSCLC) patients as the last step in biosimilarity assessment. Results from this study provide reassurance that clinical activity, and hence efficacy, clinical safety and immunogenicity of MB02 and reference bevacizumab are comparable. |
The results contribute to the totality of evidence demonstrating similarity of MB02 bevacizumab candidate with the marketed reference product. The incorporation of MB02 into the therapeutic armamentarium of bevacizumab biosimilar drugs would increase the options for cancer patients, whether alone, in combination with standard chemotherapy or with novel immunotherapy. |
1 Introduction
Non-small cell lung cancer (NSCLC), the most common type of lung cancer, is among the leading causes of death worldwide and contributes significantly to growing health care costs [1, 2]. Avastin®, the reference bevacizumab, has been approved for use in many cancer indications and settings, including first-line treatment of advanced NSCLC in combination with other chemotherapeutic agents [3, 4]. Several international guidelines recommend the use of bevacizumab in association with chemotherapy in first-line and maintenance settings in advanced NSCLC [5, 6]. In addition, recent evidence points to novel combinations of bevacizumab with new molecular therapies or immuno-oncology drugs, as well as for maintenance beyond disease progression [7, 9].
Bevacizumab is a recombinant humanized monoclonal antibody that specifically binds to human vascular endothelial growth factor (VEGF), preventing its interaction with VEGF receptors (VEGFRs) on the surface of endothelial cells, and limiting angiogenesis as a result. Through this mechanism, bevacizumab can potentially reduce tumor size (by promoting regression of existing tumor vasculature) and inhibit tumor growth (by inhibiting the formation of new tumor blood vessels) [10].
MB02 was developed by mAbxience Research SL as a biosimilar to the reference bevacizumab following the recommendations of the existing international guidelines [11,12,13,14]. A biosimilar is a medicine similar to another biological medicine (the reference product) already marketed, in terms of its physical, chemical and biological properties. Its approval follows the same strict standards of quality, safety and efficacy that apply to any other biological medicine [15]. The comparability exercise at the quality and functional level forms the basis of the biosimilarity demonstration and, in this sense, MB02 has demonstrated similarity to reference bevacizumab in a comprehensive program of drug chemistry, manufacturing and controls (CMC), and analytical similarity. A full comparison of the in vitro pharmacodynamic properties of MB02 versus the reference product was conducted as part of the comparability exercise. This exercise demonstrated comparable binding affinities to all VEGF isoforms, similar neutralization potencies and similar mode of action [16]. This most important foundation of biosimilarity had been further confirmed by another highly sensitive model, the investigation of clinical equivalence in pharmacokinetics (PK). PK similarity between MB02 and bevacizumab has been demonstrated in three bioequivalence studies comparing the PK profiles of MB02 with reference bevacizumab (US- or EU-approved) following the administration of a single dose (3 mg/kg IV) in 276 healthy male subjects (ClinicalTrials.gov identifiers: NCT04238663; NCT03293654 and NCT04238650).
To contribute to the totality of evidence and support the similarity of MB02 already demonstrated in preclinical and PK clinical development, a properly conducted confirmatory clinical trial demonstrating comparable efficacy was performed as the last step of the similarity exercise according to guidelines issued by the international authorities worldwide [11,12,13,14,15].
This clinical study was performed in first-line treatment in subjects with advanced (stage IIIB/IV) non-squamous NSCLC with the aim of showing comparable efficacy, safety and immunogenicity of the proposed biosimilar MB02 to EU-approved reference bevacizumab (EU-bevacizumab), when used in combination with chemotherapy (carboplatin and paclitaxel).
2 Material and Methods
2.1 Study Design and Participants
This multinational, double-blind, randomized, parallel group, phase III clinical comparability study (STELLA) was conducted in 93 centers in the following 16 countries: Brazil, Bulgaria, Chile, Georgia, Greece, Hungary, India, Lebanon, Malaysia, Mexico, Philippines, Russia, Serbia, Thailand, Turkey, and Ukraine. This study is registered with EudraCT (No. 2017-001769-26) and ClinicalTrials.gov (NCT03296163).
Eligible subjects included adult (≥18 years) patients with newly diagnosed or recurrent stage IIIB/IV non-squamous NSCLC not amenable to curative intent surgery with at least one unidimensional measurable lesion as per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) [17]; with an Eastern Cooperative Oncology Group (ECOG) status of 0 or 1. Subjects must have not received any previous systemic chemotherapy, immunotherapy, targeted or anti-VEGF therapy, or biological therapy for advanced disease; previous adjuvant treatment <6 months or previous radiation ≤4 weeks before randomization and have adequate hepatic, renal, hematological function and coagulation parameters. Key exclusion criteria included known malignant central nervous system disease except for treated brain metastases; small cell carcinoma or squamous cell carcinoma of the lung; known tumors that harbor activating epidermal growth factor receptor mutations and anaplastic lymphoma receptor tyrosine kinase translocations; or previous malignancy within 3 years of randomization.
2.2 Randomization and Masking
Subjects were randomly assigned (1:1 ratio) by Interactive Web Response System (IWRS) to receive MB02 or EU-bevacizumab according to a pre-specified blocked randomization scheme. The IWRS was managed by an unblinded third-party provider, and the randomization schedule was provided by a separate unblinded statistical team. Subjects, investigators, all other study staff, laboratories, and the rest of the Sponsor study team were blinded to subject treatment assignment until the database was locked at the end of the study. Randomization was stratified by sex (male/female), smoking status (smoker/non-smoker), disease diagnosis (newly diagnosed/recurrent disease) and disease stage (Stage IIIB/Stage IV). For stratification purposes, former smokers were classified as smokers if they had stopped <5 years prior to the study entry and as non-smokers if they had not smoked for the last 5 years or more, or had stopped prior to study entry and could not provide information on when they stopped smoking.
2.3 Procedures
MB02 or EU-bevacizumab (15 mg/kg) were administered as an IV infusion in combination with chemotherapy (paclitaxel 200 mg/m2 and carboplatin AUC6) on Day 1 of every 3-week cycle for six cycles (Week 18) unless there was evidence of disease progression or unacceptable toxicity to study treatment. The first MB02 or EU-bevacizumab treatment was administered as a 90-minute infusion. If the study drug was well tolerated, the next infusion was given over a 60-minute period. Thereafter, the drug was given as a 30-minute IV infusion.
After six cycles (i.e., at the start of Cycle 7), subjects received monotherapy treatment with the investigational product (IP; MB02 or EU-bevacizumab) under blinded conditions every 3 weeks until evidence of disease progression, unacceptable toxicity, death, withdrawal of consent or end of study (Week 52). Until the Week 18 assessment, reduction in MB02/EU-bevacizumab dose was not permitted, but was allowed after Week 18, if clinically necessary, to a dose level of 7.5 mg/kg. Dose reductions were allowed for paclitaxel/carboplatin according to the indications in the corresponding effective product information.
Anti-tumor activity was assessed using computed tomography (CT) and/or magnetic resonance imaging (MRI). RECIST version 1.1 response criteria was used to define all responses as follows: complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD). Tumor assessments were performed at intervals of 6 weeks, from Cycle 1, Day 1 until the end of Cycle 6 and, thereafter, at intervals of 9 weeks until evidence of PD and/or the start of new antitumor treatment, death, or end of study (Week 52). The tumor response for the primary efficacy analysis was based on the data from independent central review. An independent central review committee (IRC) provided an independent assessment of radiographic response and progression for each subject enrolled in the study up to Week 18. Target, non-target and new lesion assessments were performed by the IRC, as described in the protocol, according to a pre-defined protocol (IRC Charter) and RECIST version 1.1. All baseline and subsequent on-study images required by the protocol were collected by the sites and submitted to the IRC as soon as possible after the scans were performed. Radiological assessments were followed by the treating investigator to assess disease status and survival outcomes.
For safety assessments, ECOG performance status was established, and physical examinations, clinical laboratory tests, 12-lead electrocardiogram and left ventricular ejection fraction were performed at screening visit and at designated timepoints throughout the study. Treatment-emergent adverse events (TEAEs) were recorded during the study and up to 30 days after the end of study treatment. Beyond this date, only TEAEs and serious TEAEs considered related to study drugs were collected/reported. TEAEs were coded according to the Medical Dictionary for Regulatory Activities (MedDRA; version 20.1) and graded on the basis of the US National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE, version 4.03).
The immunogenicity of MB02 and EU-bevacizumab were determined by detections of anti-MB02 and anti-bevacizumab antibodies (anti-drug antibodies; ADA) in serum. For immunogenicity assessment, blood samples were collected at specified study cycles through Week 52. ADA incidence, titers and its neutralizing activity were assessed using a validated semi-quantitative immunoassay. The data were generated using Meso Scale Discovery (MSD; electrochemiluminescence [ECL]) platform. The immune response was evaluated by a three-tiered approach which comprised an immunogenicity assay for the screening, confirmation and titration. All samples were subjected to an initial screening assay (Tier 1), and those falling above a specific pre-determined screening cut-point were tested in the confirmation assay (Tier 2). Samples that confirmed positive in the confirmatory assay were deemed positive and further analyzed in the titer tier (Tier 3), and for the presence of neutralizing antibodies. A validated qualitative ligand binding assay was used to detect neutralizing anti-MB02/reference bevacizumab antibodies (neutralizing antibodies; NAb) in human serum using streptavidin magnetic beads and read on the MSD ECL platform. The signal produced was inversely proportional to the concentration of neutralizing anti-MB02/anti-bevacizumab antibodies present.
Subjects were followed up for survival until death or end of study. After study completion, all subjects were offered the opportunity to continue receiving biosimilar MB02 monotherapy until disease progression, unacceptable toxicity, initiation of any new treatment or death.
2.4 Outcomes
The primary efficacy endpoint was the objective response rate (ORR), defined as the rate of either CR or PR according to RECIST v1.1 at Week 18 as assessed by an IRC. Best objective response rate (BORR) was also assessed by the IRC, considering the best overall response (BOR) of either CR or PR achieved at any post-baseline time point up to, and including, Week 18. Any subjects who discontinue study treatment before Week 18 were classed as non-responders in the final analysis of the primary efficacy endpoint.
Secondary efficacy endpoints included progression-free survival (PFS), defined as the time from randomization to subsequent progression per RECIST version 1.1 or death (whichever occurred first), and overall survival (OS), defined as the time from randomization to subsequent death, as assessed by the treating investigator up to the end of study. Duration of overall response (OR) (time from the first documentation of OR to the first documentation of PD or death), and time to OR (from randomization until the first documentation of OR) were secondary exploratory efficacy endpoints. Safety and immunogenicity assessments were also assessed as secondary endpoints.
2.5 Statistical Analysis
To ascertain clinical equivalence of ORR between MB02 and EU-bevacizumab, the risk ratio (RR) and risk difference (RD) of ORR, with their corresponding 90% and 95% confidence intervals (CIs), were compared in the intention-to-treat (ITT) population (all randomized patients). Equivalence was demonstrated if the two-sided 90% (US Food and Drug Administration [FDA]) and 95% (Japan’s Pharmaceuticals and Medical Devices Agency [PMDA]) CIs of the ORR RR were entirely contained within a predefined equivalence margin of 0.73–1.36, or if the two-sided 95% CI (European Medicines Agency [EMA] and PMDA) of ORR RD was entirely contained within a predefined equivalence margin of −12% to 12%. Non-responder imputation (i.e., subjects not completing Week 18 treatment) was primarily employed to address missing data at Week 18.
Based on a fixed effects meta-analysis of five historical reference studies selected for their applicability to this study, a sample size of 300 randomized subjects per group (600 total) was chosen to provide adequate power for the proposed analyses.
The ORR estimate was stratified using the Cochran–Mantel–Haenszel estimate of the RR and RD, and the corresponding two-sided 90% and 95% CIs based on the Mantel–Haenszel method (RR) or Wald asymptotic method (RD) were presented. BOR and BORR per IRC review were also analyzed, utilizing the same procedures as described above. Additional sensitivity analyses were conducted on ORR and BORR, implementing a multiple imputation (MI) process for imputation of missing data, analyzed using the same Cochran–Mantel–Haenszel procedure as above, with results for each imputation combined using Rubin’s rule, applied using SAS Proc Mianalyze [18].
Kaplan-Meier estimates of OS and PFS were presented stratified by treatment group and were compared using the log-rank test. For PFS, clinical progression (i.e., treatment discontinuation due to progression of disease) was also included as an event. Hazard ratio was estimated using the Cox proportional hazards model, including treatment group, with sex, smoking status, disease diagnosis and disease stage as covariates. Analyses of duration of OR and time to OR followed the same statistical approaches as for PFS and OS.
Safety and immunogenicity analyses were assessed in the safety population, consisting of all randomized subjects who received at least one dose of MB02 or EU-bevacizumab. Incidence of TEAEs was summarized using descriptive statistics and frequencies were compared between treatments using inferential statistical analyses. Safety data was reviewed on an ongoing basis during the study by an independent data monitoring committee (IDMC). The frequency of treatment-induced ADA (TI-ADA) and treatment-induced NAb (TI-NAb) (defined as an ADA or NAb that developed in a subject who had tested negative at baseline), to MB02 or EU-bevacizumab, was summarized for each treatment group and compared between them. Efficacy and safety profiles were also described for TI-ADA and TI-NAb positive subjects.
3 Results
3.1 Subjects and Exposure
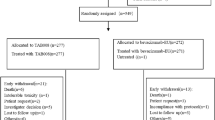
Between January 23, 2018 and March 5, 2019, 804 subjects were screened and 627 were randomized in a 1:1 ratio (ITT population; MB02: 315 subjects; EU-bevacizumab: 312 subjects). Of those randomized, 621 subjects were treated with investigational product (IP). Six of the randomized subjects (four in MB02; two in EU-bevacizumab) did not receive treatment (Fig. 1).

Subject disposition
Subject demographics and baseline disease characteristics were well balanced between the treatment groups (Table 1). Overall, the median subject age was 61 years, 383 (61.1%) subjects were males, and the majority were designated as White or Caucasian (469 [74.8%]) or Asian (125 [19.9%]). Most subjects had newly diagnosed NSCLC (576 [91.9%]) compared with those with recurrent disease (51 [8.1%]). As prior interventions, most patients had surgery including biopsies (440; 70.2%), 56 (8.9%) subjects had prior radiotherapy and 18 (2.9%) subjects had prior chemotherapy mainly in the adjuvant setting. A total of 318 (50.7%) subjects were randomized as non-smokers and 309 (49.3%) subjects were randomized as smokers as defined in the study protocol.
At study completion (February 27, 2020), a median of 9.0 cycles (range 1–18) per subject were administered to subjects in the MB02 group compared with 10.0 cycles (range 1–18) administered to those in the EU-bevacizumab group. Carboplatin and paclitaxel exposure were comparable between treatment groups, with subjects receiving a median of 6.0 cycles (range 1–6) in each treatment group.
3.2 Efficacy
On July 3, 2019, all randomized subjects had completed the combination treatment (IP plus chemotherapy) up to Week 18 or had discontinued early from the study. At Week 18, ORR, as assessed by the IRC (ITT population), was comparable for subjects receiving MB02 (ORR 40.3%; 95% CI 34.9 to 46.0) or EU-bevacizumab (ORR 44.6%; 95% CI 39.0 to 50.3) in combination with carboplatin and paclitaxel. For the primary efficacy endpoint of ORR, clinical efficacy equivalence of MB02 and EU-bevacizumab was demonstrated in the ITT population as per the margins established per protocol. An RR of 0.910 (90% CI 0.780 to 1.060; 95% CI 0.758 to 1.092) was entirely contained within the boundaries of the predefined similarity margin (0.73–1.36). For RD, the point estimate for the difference between treatments of −4.02 (90% CI −10.51 to 2.47; 95% CI −11.76 to 3.71) was also entirely contained within the predefined similarity margin of ±12% (Table 2). The primary endpoint was based on non-responder imputation of missing data at Week 18; sensitivity analyses based on an MI approach reflected the primary analysis and supported the observed outcomes of the primary analysis.
Results from secondary endpoints and sensitivity analyses reflected those of the primary analysis. Similar efficacy between MB02 and EU-bevacizumab was supported by the ad-hoc analyses of BORR at Week 18 based on IRC assessments in the ITT population (RR 0.926; 90% CI 0.818 to 1.049; RD −4.04%; 95% CI −11.86 to 3.78) (Table 2).
No significant difference was observed in subjects treated with MB02 or EU-bevacizumab in median PFS (36.00 weeks [95% CI 33.00 to 36.43] vs 37.29 weeks [95% CI 36.14 to 45.14], respectively; HR 1.187; 95% CI 0.98 to 1.44); median OS (not achieved in both treatment arms) (HR 1.108; 95% CI 0.83 to 1.49) at the end of study; median duration of OR (30.29 vs 37.14 weeks, respectively [HR 1.195; 95% CI 0.92 to 1.56]); and median time to OR (12.43 vs 12.29 weeks, respectively; HR 0.949; 95% CI 0.768 to 1.172) (Fig. 2a–2d).


Kaplan–Meier plots of (a) progression-free survival, (b) overall survival, (c) duration of response and (d) time to overall response in the intention-to-treat population
3.3 Safety and Immunogenicity
In the safety population (621 subjects), a similar incidence of TEAEs were reported during the study in the MB02 and EU-bevacizumab groups (288 subjects [92.6%] vs 288 subjects [92.9%]; p = 0.89), and most of them were classified as NCI-CTCAE severity Grade 1 or 2 events (Table 3). Between treatment groups at each severity level, the distribution of events was similar, with a risk difference in subjects reporting TEAEs of <5%. Incidence of Grade 3 or 4 TEAEs reported in the MB02 and EU-bevacizumab groups were comparable, observing a difference that was not statistically significant (p = 0.65), with anemia (26 subjects [8.4%] vs 21 subjects [6.8%]) and neutropenia (16 subjects [5.1%] vs 21 subjects [6.8%]) as the most frequently reported events.
Most TEAEs were considered by the investigator as related to any study treatment (bevacizumab or chemotherapy) and were reported in a similar number of subjects with MB02 (264 [84.9%]) and with EU-bevacizumab (270 [87.1%]), observing a risk difference of <5% between treatment groups. Overall, 189 (30.4%) subjects had Grade 3 or 4 study drug-related TEAEs, with a similar distribution observed in each treatment group (p = 0.56). No differences were noted for IP-related TEAEs (p = 0.97).
When assessed by study period (up to Week 18 in combination with chemotherapy and, after Week 18 and through Week 52 in monotherapy), the incidence of subjects reporting events was also comparable between treatment groups (Table 3). The most common (in ≥5% subjects) TEAEs reported in the MB02 and EU-bevacizumab groups during combination therapy were those commonly seen with chemotherapy (carboplatin and paclitaxel), including alopecia (155 [49.8%] and 163 [52.6%] subjects, respectively), anemia (91 [29.3%] and 77 [24.8%] subjects) and nausea (41 [13.2%] subjects in each group) (Table 4). During monotherapy, anemia (24 [7.7%] and 41 [13.2%] subjects, respectively) and thrombocytopenia (19 [6.1%] and 15 [4.8%] subjects) were the most common TEAEs. During this period, IP-related TEAEs reported were those commonly reported with the use of bevacizumab: proteinuria (9 [2.9%] and 15 [4.8%] subjects, respectively) and hypertension (7 [2.3%] and 11 [3.5%] subjects).
Other events commonly described with bevacizumab (TEAEs of special interest), such as gastrointestinal perforations and fistulae, hypertension, thromboembolism, proteinuria or hemorrhage were also reported in similar rates in both treatment groups. Treatment-related TEAEs of special interest occurring in >1% of subjects in MB02 and EU-bevacizumab groups included hypertension (19 [6.1%] and 21 [6.8%], respectively) and pulmonary embolism (5 [1.6%] vs 4 [1.3%]); leukopenia (4 [1.3%] vs 2 [0.6%]), epistaxis (13 [4.2%] vs 9 [2.9%]) and hemoptysis (8 [2.6%] vs 4 [1.3%]) that were more common in the MB02-treated subjects; and neutropenia (5 [1.6%] vs 10 [3.2%]) and proteinuria (12 [3.9%] vs 18 [5.8%]) that were more common in EU-bevacizumab-treated subjects.
Serious TEAEs were reported in similar frequency with no statistically significant difference between the treatment groups (p = 0.69) (Table 4). Serious TEAEs were considered related to MB02 in 21 subjects (6.8%) and to EU-bevacizumab in 18 subjects (5.8%). The most common grade 3 or 4 IP-related serious TEAE in the MB02 group was pulmonary embolism, and in the EU-bevacizumab group the most common were pulmonary embolism, fatigue and pneumonia. No clear treatment-group-related trends were observed for IP-related serious TEAEs.
A total of 47 subjects died as a result of a TEAE (MB02: 23 subjects [7.4%]; EU-bevacizumab: 24 subjects [7.7%]; p = 0.87). Death was considered IP-related in nine subjects (MB02: six subjects [1.9%]; EU-bevacizumab: three subjects [1.0%]). Fatal TEAEs considered related to IP included pulmonary hemorrhage (two subjects, one in each treatment group); nephrotic syndrome, hemoptysis, gastric ulcer, acute myocardial infarction and cardio-respiratory arrest (one subject each) in the MB02 group; and acute kidney injury and pulmonary embolism (one subject each) in EU-bevacizumab group.
No clinically meaningful differences were noted in the rates, severity, and type of TEAEs between the treatment groups and the safety profile of MB02 was consistent with that described for the reference product, bevacizumab.
After 52 weeks of study, most subjects tested negative for ADAs at all time points. TI-ADAs were observed in 53 subjects (17%) in the MB02 group and 50 subjects (16.1%) in the EU-bevacizumab group. TI-NAb responses were also similar between both groups, occurring in 10 subjects (3.2%) in the MB02 group and 13 subjects (4.2%) in the EU-bevacizumab group in the ITT population. Most of these positive TI-ADA results were transient, occurring most often at a single time point and with no neutralizing capacity. The occurrence of antibodies against MB02 or EU-bevacizumab did not appear to impact the efficacy results or correlate with any safety concern.
4 Discussion
In this confirmatory phase III clinical study, the efficacy, safety, and immunogenicity of MB02 and EU-bevacizumab, both in combination with chemotherapy, were compared in patients with advanced non-squamous NSCLC. The primary objective of efficacy, RD of the ORRs (MB02 minus EU-bevacizumab) and RR of the ORRs (\(\text{MB02} / \text{EU-bevacizumab}\)) as assessed by an IRC at Week 18 in the ITT population, met the criteria applied for similarity as discussed with the main international regulatory authorities, demonstrating equivalence of clinical efficacy between MB02 and EU-bevacizumab.
The percentage of subjects achieving ORR for MB02 in the ITT population (40.3%) was completely in line with the historical response rates reported for the reference bevacizumab in the same population of patients (32.2%, 53.9%) [19,20,21,22,23,24,25,26]. Moreover, the sensitivity analysis using imputation of missing data (MI approach) provided additional support for similar efficacy between MB02 and EU-bevacizumab under the ITT population, and supports the robustness of the primary analysis.
Secondary efficacy endpoints (PFS, OS, duration of OR and time to OR) and ad-hoc endpoints (BORR) were also comparable between treatment groups and were consistent with the observed results of the primary endpoint. In particular, BORR was assessed to confirm primary endpoint ORR results. BORR reduces potentially confounding factors of diverse cycles and delayed administration due to toxicity, and is commonly used in an oncological clinical setting. When comparing the analysis based on BORR up to Week 18 to that of primary ORR analysis, there was almost no difference, which is considered reassuring.
The safety profile of MB02 was comparable to that of EU-bevacizumab and results were those expected for the reference product in an equivalent study population and with the same concomitant therapies. Overall, the number, type, and severity of TEAEs, including those of special interest (such as gastrointestinal perforations, hypertension, thromboembolism or hemorrhage) were consistent with the safety profile reported for NSCLC patients in the reference bevacizumab product information [3, 4]. No new safety signals or observable trends were identified in either treatment group in the study. No impact on the safety in general, and no immune-related safety risks in particular, appear to be correlated with treatment-related antibodies. Similarly, from the analyses performed, the effect of treatment-related antibodies does not appear to account for any differences in efficacy between the products.
Similarity of MB02 to EU-bevacizumab was demonstrated in the relevant characteristics assessed by and founded on a comprehensive CMC and bioanalytical similarity program, and was further confirmed by the investigation of clinical equivalence in PK. The next step in the program of biosimilar clinical development was to confirm comparable clinical performance of MB02 and the reference bevacizumab, rather than demonstrate patient benefit per se, which has already been demonstrated for the reference bevacizumab in numerous clinical trials and published studies [8]. Due to the absence of pharmacodynamic markers for bevacizumab that can be related to patient outcome, a comparative study designed to demonstrate similar clinical efficacy between MB02 and EU-bevacizumab was required to confirm efficacy. The choice of non-squamous NSCLC patients as the study population was made in accordance with the relevant regulatory guidelines and endorsed by the main international regulatory competent authorities, as a sensitive model with known effect sizes to test for potential differences in efficacy between MB02 and EU-bevacizumab [11,12,13,14,15]. Likewise, the primary efficacy endpoint, ORR at study Week 18, was considered the most sensitive endpoint for the detection of differences in clinical efficacy between MB02 and EU-bevacizumab, as it primarily measures activity and, unlike other endpoints such as PFS and OS, is not likely to be influenced as much by factors not attributable to product differences such as underlying tumor burden, performance status, previous treatments and underlying clinical conditions. In the current study, the primary analysis in the ITT population met the predefined criteria for demonstrating equivalence, and results from sensitivity analyses support similarity of MB02 to EU-bevacizumab with respect to the primary efficacy endpoint ORR, with comparable safety and immunogenicity profiles.
A possible limitation for the study was that the study protocol definition used for smoking-status classification differed from the new standard definition currently in use in NSCLC clinical study protocols. The current definition regards smokers as subjects who had smoked >100 cigarettes in a lifetime and non-smokers as subjects who had never smoked or had smoked <100 cigarettes in a lifetime [27]. In consequence, the proportion of subjects included in smoker/non-smoker categories according to the study protocol definition used in the study protocol (smokers: 309 [49.3%]; non-smokers: 318 [50.7%]) is slightly different to that reported in recent NSCLC publications [28]. After the study completion, the proportion of smokers/non-smokers was re-assessed using the current new standard definition (smokers: 390 [62.2%]/non-smokers: 237 [37.8%]), observing that the percentage of smokers in the study is in line with recent publications. Efficacy analysis conducted for the primary endpoint with inclusion of this definition for smoker status was similarly in line with the reported results of the study. Thus, the smoking status definition as defined in the protocol was considered valid, especially considering that subjects were randomized under this stratification factor as per study protocol.
Fifteen years after its first approval for clinical use, bevacizumab still remains a useful tool as part of the standard of care in the treatment of advanced cancers, observing consistent efficacy across indications and a well-established clinical efficacy and safety profile. Moreover, recent studies indicate some immunomodulatory properties of bevacizumab, and treatment outcomes seem to be further improved when bevacizumab is used in combination with novel targeted cancer therapies [8]. In particular, the results of combining bevacizumab with the immune checkpoint inhibitor atezolizumab have shown benefit in NSCLC (a reduction in the risk of progression by 38% [HR 0.62; p < 0.001]) and in hepatocellular carcinoma and had led to the approval of this combination in both cancer indications [7, 9]. This combination has also shown to be effective in advanced renal carcinoma [29]. Bevacizumab biosimilars are more affordable therapeutic options that can override the cost barrier of reference bevacizumab for many patients who might otherwise benefit from its use, and the incorporation of the proposed biosimilar, MB02, to the therapeutic armamentarium increases the available options for these patients.
Based on the extensive analytical data package and the PK studies performed for MB02, no differences have been identified that give rise to uncertainty over the expected clinical performance. Together with a comprehensive set of comparative safety and immunogenicity data, the STELLA phase III clinical comparability study provides reassurance that clinical activity (and hence efficacy), and clinical safety of MB02 and EU-bevacizumab are comparable, contributing to the totality of evidence approach required by regulatory agencies for biosimilar product development, further demonstrating the biosimilarity of MB02 to its reference product bevacizumab.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021. https://doi.org/10.3322/caac.21660.
Desai A, Gyawali B. Financial toxicity of cancer treatment: moving the discussion from acknowledgement of the problem to identifying solutions. E Clin Med. 2020;20:100269.
EMA. Avastin (bevacizumab). Summary of product characteristics 2021. https://www.ema.europa.eu/en/documents/product-information/avastin-epar-product-information_en.pdf. Accessed 24 Feb 2021.
FDA. Avastin® (bevacizumab) prescribing information. Genentech, Inc, 2021. https://www.gene.com/download/pdf/avastin_prescribing.pdf. Accessed 24 Feb 2021.
National Comprehensive Cancer Network. Clinical practice guidelines in oncology. Non-small cell lung cancer. Version 8.2020. https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Accessed 18 Sep 2020.
Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):iv192–237.
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894–905.
Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, et al. Bevacizumab (Avastin®) in cancer treatment: a review of 15 years of clinical experience and future outlook. Cancer Treat Rev. 2020;86:102017.
Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–301.
Tobelem G. VEGF: a key therapeutic target for the treatment of cancer-insights into its role and pharmacological inhibition. Target Oncol. 2007;2(3):153–64.
EMA. Guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues. European Medicines Agency (EMA/CHMP/BMWP/403543/2010), 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf. Accessed 24 Feb 2021.
EMA. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. European Medicines Agency (EMEA/CHMP/BMWP/42832/2005 Rev1); 2014. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf. Accessed 24 Feb 2021.
FDA. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Guidance for Industry, 2015. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291128.pdf. Accessed 11 Jun 2018.
WHO. Guidelines on evaluation of monoclonal antibodies as similar biotherapeutic products (SBPs). Sixty seventh report. WHO Expert Committee on Biological Standardization WHO Technical Report series, 2017. http://apps.who.int/iris/bitstream/10665/255657/1/9789241210133-eng.pdf. Accessed 24 Feb 2021.
EMA. Biosimilars in the EU. Information guide for healthcare professionals. Prepared jointly by the European Medicines Agency and the European Commission, 2017. http://www.ema.europa.eu/docs/en_GB/document_library/Leaflet/2017/05/WC500226648.pdf. Accessed 24 Feb 2021.
Sacristán D, Beydon ME, Ruppen I, Almeida AM, Miriyala SK. Analytical Similarity Assessment of Bevacizumab Biosimilar MB02 Using Multiple State-of-the-art Assays., DIA Europe, 2021.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1·1). Eur J Cancer. 2009;45(2):228–47.
Rubin DB. Multiple imputation for nonresponse in surveys. New York: Wiley; 1987.
Barlesi F, Scherpereel A, Rittmeyer A, Pazzola A, Ferrer Tur N, Kim JH, et al. Randomized phase III trial of maintenance bevacizumab with or without pemetrexed after first-line induction with bevacizumab, cisplatin, and pemetrexed in advanced nonsquamous non-small-cell lung cancer: AVAPERL (MO22089). J Clin Oncol. 2013;31(24):3004–11.
Johnson DH, Fehrenbacher L, Novotny WF, Herbst RS, Nemunaitis JJ, Jablons DM, et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2004;22(11):2184–91.
Niho S, Kunitoh H, Nokihara H, Horai T, Ichinose Y, Hida T, et al. Randomized phase II study of first-line carboplatin and paclitaxel with or without bevacizumab in Japanese patients with advanced non-squamous non-small-cell lung cancer. Lung Cancer. 2012;76(3):362–7.
Patel JD, Socinski MA, Garon EB, Reynolds CH, Spigel DR, Olsen MR, et al. PointBreak: a randomized phase III study of pemetrexed plus carboplatin and bevacizumab followed by maintenance pemetrexed and bevacizumab versus paclitaxel plus carboplatin and bevacizumab followed by maintenance bevacizumab in patients with stage IIIB or IV nonsquamous non-small-cell lung cancer. J Clin Oncol. 2013;31(34):4349–57.
Reck M, von Pawel J, Zatloukal P, Ramlau R, Gorbounova V, Hirsh V, et al. Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non-small-cell lung cancer: AVAil. J Clin Oncol. 2009;27(8):1227–34.
Reck M, von Pawel J, Zatloukal P, Ramlau R, Gorbounova V, Hirsh V, et al. Overall survival with cisplatin-gemcitabine and bevacizumab or placebo as first-line therapy for nonsquamous non-small-cell lung cancer: results from a randomised phase III trial (AVAiL). Ann Oncol. 2010;21(9):1804–9.
Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–50.
Zhou C, Wu YL, Chen G, Liu X, Zhu Y, Lu S, et al. BEYOND: a randomized, double-blind, placebo controlled, multicenter, phase III study of first-line carboplatin/paclitaxel plus bevacizumab or placebo in Chinese patients with advanced or recurrent nonsquamous nonsmall-cell lung cancer. J Clin Oncol. 2015;33(19):2197–204.
Centers for Disease control and Prevention. https://www.cdc.gov/nchs/nhis/tobacco/tobacco_glossary.htm. Accessed 28 Jan 2021.
Lee SJ, Lee J, Park YS, Lee CH, Lee SM, Yim JJ, et al. Impact of smoking on mortality of patients with non-small cell lung cancer. Thorac Cancer. 2014;5(1):43–9.
Rini BI, Powles T, Atkins MB, Escudier B, McDermott DF, Suarez C, et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet. 2019;393:2404–15.
Acknowledgements
We thank all patients and investigators who participated in the STELLA study. We would like to thank Nick Wells (Senior Principal Biostatistician at Syneos Health) for the statistical review of the manuscript, and the members of the IDMC (Dr Pere Gascon, Hospital Clinic, Barcelona, Spain; Dr Enriqueta Felip, Hospital P. Vall d´Hebron, Barcelona, Spain; Dr Federico Cappuzzo, Ospedale S. Maria delle Croci, Ravenna, Italy and Eckhard Pecher, MSc, Cytel France SARL, Paris, France).
STELLA Investigators: Luiz Henrique de Lima Araujo, Instituto Nacional de Cancer Jose de Alencar Gomes da Silva/Coordenacao de Pesquisa Clinica, Centro Rio de Janeiro, Brazil. Carla Maria de Oliveira Ferreira, Fundacao Faculdade Regional de Medicina de Sao Jose do Rio Preto Sao Jose do Rio Preto, Brazil. Helio Pinczowski, Instituto de Ensino e Pesquisa Sao Lucas – IEP Sao Paulo, Brazil. Maria Marcela Fernandes Monteiro, Instituto do Cancer do Ceara, Fortaleza, Brazil. Assen Dudov, Acibadem City Clinic UMHAT EOOD, Sofia, Bulgaria. Janeta Syrova, Specialized Hospital for Active Treatment of Oncology Diseases Sofia, Bulgaria. Francisco Javier Orlandi Jorquera, Orlandi Oncología, Santiago, Region Metropolitana, Chile. Carlos Eduardo Gallardo Arenada, Fundacion Arturo Lopez Perez (FALP) Providencia Santiago, Region Metropolitana, Chile. Christian Lorenzo Caglevic Medina, Fundacion Arturo Lopez Perez (FALP) Providencia Santiago, Region Metropolitana, Chile. Davit Giorgadze, LTD Multiprofile Clinic Consilium Medulla Tbilisi, Georgia. Nino Mchedlidze, LTD Aversi Clinic Tbilisi, Georgia. Vladimer Kuchava, LTD Institute of Clinical Oncology Tbilisi, Georgia. Tamar Melkadze, Ltd Academician Fridon Todua Medical Center- Ltd Research Institute of Clinical Medicine Tbilisi, Georgia. Amiran Matitashvili, LTD Cancer Research Centre 4, Tbilisi, Georgia. Nana Chikhladze, LEPL First University Clinic of Tbilisi State Medical University Tbilisi, Georgia. Epaminodas Samantas, General Oncology Hospital of Kifissia “Oi Agii Anargiri” Athens, Greece. Theodoros Kontakiotis, G. Papanikolaou General Hospital of Thessaloniki Thessaloniki, Greece. Beatrix Balint, Csongrad Megyei Mellkasi Betegsegek Szakkorhaza Deszk, Hungary. Balazs Medgyasszay, Veszprem Megyei Tudogyogyintezet Farkasgyepu, Hungary. Eszter Csanky, Borsod-Abauj-Zemplen Megyei Kozponti Korhaz es Egyetemi Oktatokorhaz Semmelweis Tagkorhaz, Tudogyogyaszati osztaly Miskolc, Hungary. Shailesh Arjun Bondarde, Shatabdi Hospital, Suyojit City Centre Maharashtra, India. Lovenish Goyal, Aadhar Health Institute Hisar, Haryana, India. Ajay Sharma, Action Cancer Hospital New Delhi, Delhi, India. Baijumon Balan, PVS Hospital (P) Ltd. Kerala, India. Prabrajya Narayan Mohapatra, Apollo Gleneagles Hospitals Kolkata, West Bengal, India. Kaushalkumar Babubhai Patel, Nirmal Hospital Pvt Ltd Surat, Gujarat, India. Sachin Sharadchandra Hingmire, Lata Mangeshkar Medical Foundation’s, Deenanath Mangeshkar Hospital & Research Centre, Pune, Maharashtra, India. Mithun Satish Shah, Zydus Hospitals & Healthcare Research Pvt. Ltd. Ahmedabad, Gujarat, India. Kartikeya Jain, Shree Himalaya Cancer Hospital and Research Institute, Vadodara, Gujarat, India. Ashish Agrawal, Kiran Hospital Surat, Gujarat, India. Prashant Kumbhaj, G.P. Shekhawati Hospital & Research Centre Jaipur, Rajasthan, India. Asis Mukhopadhyay, Netaji Subhash Chandra Bose Cancer Hospital, Kolkata, West Bengal, India. Paul Khoueiry , Centre Hospitalier Universitaire Notre Dame de Secours. Jbeil, Lebanon. Suhana Yusak, National Cancer Institute No. 4, Jalan P7, Presint 7 Putrajaya, Wilayah Persekutuan, Malaysia. Yong Kek Pang, University Malaya Medical Centre Lembah Pantai, Kuala Lumpur, Malaysia. Voon Pei Jye, Hospital Umum Sarawak Jalan Hospital, Kuching, Sarawak, Malaysia. Prathepamalar Yehgambaram, Hospital Kuala Lumpur Jalan Pahang, Kuala Lumpur, Malaysia. Fuad bin Ismail, Department of Radiotherapy & Oncology, Universiti Kebangsaan Malaysia Medical Centre, Jalan Yaacob Latif, Bandar Tun Razak Cheras, Kuala Lumpur, Malaysia. Gokula Kumar Appalanaido, Institut Perubatan & Pergigian Termaju Universiti Sains Malaysia, Bertam Kepala Batas, Pulau Pinang, Malaysia. Feliciano Barron Barron, Instituto Nacional de Cancerologia Tlalpan, Ciudad de Mexico, Mexico. Ma. Noemi Uy, Cebu Doctors’ University Hospital Cebu, Philippines. Felycette Gay Martinez- Lapus, Davao Doctors Hospital, Department of Internal Medicine Davao, Davao del Sur, Philippines. Maria Belen Tamayo, Makati Medical Center, Makati, Metro Manilla, Philippines. Jennifer Sandoval-Tan, Philippine General Hospital Manilla, Philippines. Jamela Anne Osorio Sanchez, Perpetual Succour Hospital, 12Th Floor The Research Instiute Lahug Cebu, Philippines. Christina G. Galvez, St. Luke’s Medical Center – Global City Taguig, Metro Manilla, Philippines. Josephine Contreras-Tolentino, The Medical City, Room 306, Medical Arts Tower Building Pasig City, Metro Manilla, Philippines. Evgeny Ledin, PhD, LLC University Headache Clinic Moscow, Russia. Daniil Stroyakovskiy, PhD, State Budgetary Healthcare Institution of Moscow Moscow City Oncological Hospital #62 of Moscow Healthcare Department, Moscow Region, Russia. Igor Kudryavtsev, State Budgetary Healthcare Institution of the Kaluzhsky Region Kaluzhsky Region Oncology Dispensary, Kaluga, Russia. Vladimir Vladimirov, SBHI of the Stavropol Region Pyatigorsk Interdistrict Oncology Dispensary, Pyatigorsk, Stavropol Region, Russia. Evgeniy Gotovkin, Regional Budgetary Healthcare Institution Ivanovo Regional Oncology Dispensary Ivanovo, Russia. Marina Shomova, State Budgetary Institution Ryazan Region Regional Clinical Dispensary, Ryazan, Russia. Guzel Mukhametshina, SAHI Republican Clinical Oncology Dispensary of the Ministry of Health of the Republic of Tatarstan, Kazan, Russia. Igor Lifirenko, PhD, Regional Budgetary Healthcare Institution Kursk Regional Clinical Oncology Dispensary of Healthcare Committee of Kursk Region Ryshkovsky rural council Kursk region, Russia. Nina Karaseva, St. Petersburg State Budgetary Healthcare Institution City Clinical Oncology Dispensary, Saint-Petersburg, Russia. Marina Nechaeva, SBHI Arkhangelsk Region, Arkhangelsk Clinical Oncological Dispensary Arkhangelsk, Arkhangelsk Region, Russia. Anna Tarasova, State Budgetary Healthcare Institution, Samara Regional Clinical Oncology Dispensary 50 Samara, Russia. Alexander Luft, State Budgetary Healthcare Institution Leningrad Regional Clinical Hospital,St. Petersburg, Vyborgskij Region, Russia. Lyudmila Kuzina, OGBUZ Belgorod Oncology Dispensary Belgorod, Russia. Marina Petrovic, Clinical Center Kragujevac Kragujevac, Serbia. Milan Rancic, Clinical Center Nis, Clinic for Pulmonary Diseases Nis, Serbia. Borjan Zaric, Institute for Pulmonary Diseases of Vojvodina Sremska Kamenica, Serbia. Manoch Buranachokphaisan, Buddhachinaraj Phitsanulok Hospital Muang, Phitsanulok, Thailand. Sarayut Lucien Geater, Songklanagarind Hospital, Faculty of Medicine, Prince of Songkla University Hat Yai, Songkla, Thailand. Ekkapong Tharavichitkul, Chiangrai Prachanukroh Hospital Chiangrai, Muang, Chiangrai, Thailand. Mahmut Gumus, Istanbul Medeniyet University Goztepe Training and Research Hospital, Department of Medical Oncology Istanbul, Kadikoy, Turkey. Igor Bondarenko, M I Dnipropetrovsk City Multi-field Clinical Hospital #4 of Dnipropetrovsk Regional Council, Chemotherapy Department Dnipro, Ukraine. Yaroslav Shparyk, Lviv State Oncological Regional Treatment and Diagnostic Center, Department of Chemotherapy, Lviv, Ukraine. Serhii Shevnia, Vinnytsia Regional Clinical Oncology Dispensary, Chemotherapy Department, Vinnytsia, Ukraine. Iryna Lytvyn, Municipal Institution Clinical Oncological Dispensary of Dnipropetrovsk Regional Council, Department of Chemotherapy Dnipro, Ukraine. Oleksii Kolesnik, Communal Institution Zaporizhzhya Regional Clinical Oncological Dispensary of Zaporizhzhya Regional Council Thoracic Department Zaporizhzhya, Ukraine. Yevhen Hotko, Central Municipal Clinical Hospital, City Oncology Centre Uzhhorod, Ukraine. Ivan Sinielnikov, Healthcare Facility Volyn Regional Oncological Dispensary, Department of Chemotherapy, Lutsk, Ukraine. Hryhoriy Adamchuk, Municipal Institution Kryviy Rih Oncology Dispensary of Dnipropetrovsk Regional Council, Chemotherapy Department Kriviy Rih, Ukraine. Grygorii Ursol, Private Enterprise Private Manufacturing Company Acinus, Medical and Diagnostic Centre, Kropyvnytskyi, Ukraine. Oleksandr Ivashchuk, Municipal Insitution Chernivtsy Regional Clinical Oncological Dispensary, Daily Hospital Department, Chernivtsi, Ukraine. Yuriy Ostapenko, National Cancer Institute, Department of Minimally Invasive and Endoscopic Surgery, Interventional Radiology, Kyiv, Ukraine. Tetiana Popovska, State Institution Grigoriev Institute for Medical Radiology National Academy of Medical Science of Ukraine, Kharkiv, Ukraine.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Funding
The study was supported by mAbxience Research SL. Employees of the sponsor had a role in study design, data analysis and manuscript preparation. Employees of the funder had no role in data collection.
Competing interests
SM, AP, AF and ADCG are employees at mAbxience Research SL.
All the other authors declared that they have no conflicts of interest.
Availability of data
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
Ethics approval
The study was conducted in accordance with the Declaration of Helsinki, the International Council for Harmonization Good Clinical Practice (ICH-GCP) guidelines and the appropriate regulatory requirements in the countries where the study was conducted. Ethical approval was sought and granted for each center. The protocol, its amendments and informed consent documentation were reviewed and approved by the institutional review board(s) or independent ethics committee(s) at each center.
Consent to participate
All patients signed an Institutional Review Board/Independent Ethics Committee-approved informed consent form before any study-specific procedures were performed.
Consent for publication
Not applicable.
Authors contributions
SM and AP were involved in the conception and design of the study. DT, EP, ZA, TM, FJOJ, BRS, CC, AF, HAIA, KS, NK, IR, FB, JI, PK and RDCC were involved in the provision of study material and patients and acquisition of data. SM, AP, AF and ADCG did the analysis and/or interpretation of data. All authors wrote the report and/or revised it critically for important intellectual content. All authors approved the final version.
Additional information
The members of the STELLA study investigators are listed in Online Resource 1 and in the Acknowledgements section.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Trukhin, D., Poddubskaya, E., Andric, Z. et al. Efficacy, Safety and Immunogenicity of MB02 (Bevacizumab Biosimilar) versus Reference Bevacizumab in Advanced Non-Small Cell Lung Cancer: A Randomized, Double-Blind, Phase III Study (STELLA). BioDrugs 35, 429–444 (2021). https://doi.org/10.1007/s40259-021-00483-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-021-00483-w