
Abstract
Objective
The aim of this study was to investigate long-term clinical outcomes of extended treatment with CT-P10, a rituximab biosimilar, compared with rituximab reference products sourced from the USA and the EU (US-RTX and EU-RTX) in rheumatoid arthritis (RA) for up to 48 weeks.
Methods
In this multinational, randomized, double-blind trial, adults with active RA received up to two courses of CT-P10, US-RTX, or EU-RTX alongside methotrexate. Efficacy endpoints included Disease Activity Score 28-joint count (DAS28) and American College of Rheumatology (ACR) response rates. Pharmacokinetics, pharmacodynamics, immunogenicity, and safety were also assessed.
Results
Of 372 patients randomized to the study drug, 330 (88.7%) completed the second treatment course. Mean change from baseline to week 48 in DAS28-C-reactive protein was comparable in the CT-P10 and combined rituximab (US-RTX and EU-RTX) groups (− 2.7 and − 2.6, respectively). ACR20, ACR50, and ACR70 response rates at week 48 indicated no differences between groups (80.6%, 55.4%, and 31.7% vs. 79.8%, 53.9%, and 33.7% in the CT-P10 and combined rituximab groups, respectively). Similar improvements in the Health Assessment Questionnaire Disability Index and all medical outcomes in the Short Form 36-Item Health Survey, including physical and mental health, were seen in all groups. At week 48, antidrug antibodies were detected in 4.9%, 9.4%, and 8.6% of patients in the CT-P10, US-RTX, and EU-RTX groups, respectively. CT-P10 and rituximab displayed similar pharmacokinetic, pharmacodynamic, and safety profiles.
Conclusion
CT-P10 was similar to EU-RTX and US-RTX in terms of efficacy, pharmacokinetics, pharmacodynamics, immunogenicity, and safety up to week 48.
ClinicalTrials.gov identifier
NCT02149121.
Similar content being viewed by others


This randomized phase III study demonstrated that the long-term efficacy of CT-P10 after two courses was comparable to that of reference rituximab from the USA and the EU in patients with rheumatoid arthritis. |
Pharmacokinetic, pharmacodynamic, immunogenicity, and safety findings were also comparable between groups up to week 48. |
These data provide evidence for the long-term use of CT-P10 in terms of its efficacy and safety in clinical practice, supporting the recent approval of CT-P10 for the treatment of patients with rheumatoid arthritis. |
1 Introduction
B-cells play a major part in the pathogenesis of rheumatoid arthritis (RA) via antibody-dependent and -independent mechanisms [1]. Rituximab is an anti-cluster of differentiation (CD)-20 monoclonal antibody that exerts therapeutic effects in RA through the depletion of B-cells that express the CD20 surface antigen [2]. Randomized controlled trials (RCTs) of rituximab in patients with active RA have demonstrated that the biological drug reduces the progression of joint damage and improves physical function [3, 4]. Based on such evidence, rituximab was approved for use with methotrexate in patients with RA who have had an inadequate response or shown intolerance to anti-tumor necrosis factor (TNF) agents [5]. Rituximab is currently available as MabThera® (Roche, Welwyn Garden City, UK) in Europe (EU-RTX) and as Rituxan® (Genentech, Inc. South San Francisco, CA, USA) in the USA (US-RTX) and is also approved for use in antineutrophil cytoplasmic antibody vasculitis and in certain B-cell-related hematological cancers.
A biosimilar is a highly similar version of an approved innovator biologic, or reference product (RP). Given the variability that is inherent in manufacturing biologics such as monoclonal antibodies, a biosimilar will never be entirely identical to its RP. Therefore, for a biosimilar to gain regulatory approval, comparative nonclinical and clinical data demonstrating biosimilarity are required. A stepwise approach is typically taken that starts with structural/functional analysis, followed by pharmacokinetic/pharmacodynamic studies and then clinical assessment, with each step informing the type and extent of data required at the next step. The totality of evidence is considered in the approval decision; however, generally, the biosimilar must show statistical equivalence to the RP in terms of pharmacokinetics and efficacy, as well as comparable safety [6, 7].
CT-P10 (CELLTRION, Inc., Incheon, Republic of Korea) is a biosimilar of rituximab that shares an identical primary structure with its RP and highly similar higher order structures and biological activities [8]. CT-P10 is currently approved in Europe, Australia, and South Korea for the same indications as rituximab (RA, granulomatosis with polyangiitis and microscopic polyangiitis, non-Hodgkin’s lymphoma, and chronic lymphocytic leukemia).
In a phase I study, CT-P10 showed pharmacokinetics that were equivalent to, and efficacy, pharmacodynamics, immunogenicity, and safety that were comparable to, EU-RTX in patients with RA [8]. Furthermore, a randomized, double-blind, active-controlled, three-arm phase III study compared CT-P10 with both EU-RTX and US-RTX in patients with RA. The first phase of this study (up to week 24) was divided into two parts, both of which achieved their primary endpoints [9]. The first part of the study demonstrated equivalent pharmacokinetics of CT-P10, US-RTX, and EU-RTX over 24 weeks; the second part demonstrated comparable efficacy of CT-P10 versus a combined rituximab group (i.e., pooled data from patients who received US-RTX or EU-RTX) at week 24. In addition, similar pharmacodynamics, immunogenicity, and safety profiles were demonstrated between CT-P10 and the combined rituximab group up to week 24 [9]. Additionally, pharmacokinetic equivalence up to week 12, and non-inferior efficacy of CT-P10 and US-RTX up to week 24 (and comparable B-cell kinetics, immunogenicity, and safety) has been shown in patients with advanced follicular lymphoma [10].
To investigate the long-term efficacy, pharmacodynamics, immunogenicity, and safety of extended treatment with CT-P10, the phase III study in RA was continued for a further 24 weeks [9]; the results up to week 48 are reported here.
2 Methods
2.1 Study Design
This was a randomized, double-blind, active-controlled, parallel-group phase III study conducted in 76 centers in Europe, Asia Pacific, and Latin America. The study was performed in accordance with the Declaration of Helsinki [11] and Good Clinical Practice guidelines [12]. The study protocol was reviewed and approved by the relevant independent ethics committee at each site, and all patients provided written informed consent. The study was registered with ClinicalTrials.gov (NCT02149121). Full methodological details of this study, including the primary pharmacokinetics and efficacy analysis, have been described previously [9].
2.2 Patients
Patients were aged 18–75 years with active RA diagnosed per the revised 1987 American College of Rheumatology (ACR) classification criteria [13] ≥ 6 months before randomization. Active disease was defined by the presence of six or more swollen joints and six or more tender joints, and serum C-reactive protein (CRP) ≥ 1.5 mg/dL or an erythrocyte sedimentation rate (ESR) ≥ 28 mm/h. Patients had experienced an inadequate response or were intolerant to anti-TNF agents and had received oral or parenteral methotrexate 7.5–25 mg/week for at least the past 12 weeks, with a stable dose over the 4 weeks immediately before screening. Patients with a history of any inflammatory or rheumatic disease other than RA were excluded from the study. Full inclusion and exclusion criteria have been reported previously [9].
2.3 Procedures
Patients received a first course of study drug administered as two intravenous infusions of CT-P10, US-RTX, or EU-RTX 1000 mg given 2 weeks apart (at weeks 0 and 2). Patients could receive a second course irrespective of clinical response (with infusions given at weeks 24 and 26) provided they met predefined safety criteria for each infusion (absolute neutrophil count ≥ 1.5 × 109 cells/L, platelet count ≥ 75 × 109 cells/L, aspartate aminotransferase or alanine aminotransferase ≤ 2.5 times the upper limit of normal, and levels of immunoglobulin G ≥ 500 mg/dL at the last blood sample analysis; and the patient had not developed any condition which, in the investigator’s opinion, precluded the patient receiving further courses of treatment). Oral or parenteral methotrexate 7.5–25 mg/week and folic acid ≥ 5 mg/week were coadministered with the study drug. Patients also received methylprednisolone, an antipyretic, and an antihistamine 30–60 minutes before each infusion.
2.4 Study Endpoints and Assessments
Disease Activity Score 28-joint count (DAS28) and ACR response criteria were assessed at baseline, before the first drug infusion, and at 4-weekly intervals up to week 24, and then at 8-weekly intervals up to week 48. Efficacy parameters reported are mean change from baseline in DAS28-ESR and DAS28-CRP up to week 48, and response rates defined by ACR 20% improvement criteria (ACR20), ACR 50% improvement criteria (ACR50), and ACR 70% improvement criteria (ACR70) criteria, as well as ACR hybrid score up to week 48. Additional efficacy measures included the change from baseline to week 48 in joint damage score using the van der Heijde modification of the Sharp scoring system (range 0–448) and the Short Form 36-Item Health Survey (SF-36) score, and the proportion of patients with a good/moderate European League Against Rheumatism (EULAR) response at weeks 24 and 48. The Health Assessment Questionnaire Disability Index (HAQ-DI), Clinical Disease Activity Index (CDAI) and Simplified Disease Activity Index (SDAI) were also assessed until week 48.
Pharmacokinetic parameters, including maximum serum concentration after the first (Cmax, 1) and second (Cmax, 2) infusions of each treatment course, time to maximum concentration in the first (Tmax 1st course) and second (Tmax 2nd course) course, predose concentrations at weeks 24 (Cmin W24) and 48 (Cmin W48), and concentration before the second infusion of the first (Ctrough 1st course) and second (Ctrough 2nd course) treatment courses, were assessed up to week 48.
Pharmacodynamics were assessed via the monitoring of B-cell kinetics over time, including depletion and recovery at week 48, using absolute numbers of CD19+ and CD20+ cells with a FACScalibur flow cytometer (Becton Dickinson and Company; San Jose, CA, USA). Blood samples for measurement of CRP, ESR, rheumatoid factor, and anticyclic citrullinated peptide were collected at baseline, week 24, and week 48.
Immunogenicity (i.e., presence or absence of antidrug antibodies [ADAs] and neutralizing antibodies [NAbs]) was measured at baseline, week 24, and week 48. ADAs were detected in diluted serum using an electrochemiluminescence bridging assay using biotinylated and sulfo-tagged CT-P10 as the capture and detection antibody, respectively. To detect the presence of NAbs against CT-P10 or rituximab, diluted serum samples were tested for inhibition of CT-P10-induced complement-dependent cytotoxicity in WIL2-S cells, as described previously [8, 9].
Safety was monitored throughout the study, including adverse events (AEs), AEs of special interest (infections, infusion-related reactions [IRRs], malignancy and progressive multifocal leukoencephalopathy), and clinical laboratory assays. Reported AEs were treatment-emergent AEs that were not present before exposure to study drug or that worsened in intensity or frequency after exposure to study drug.
Biomarker was assessed via collecting blood samples for evaluation of Fc gamma receptor (Fcγ) genotype between randomization and the first study drug infusion only in patients who signed a separate informed consent form.
2.5 Statistical Analysis
The target sample size of the study (n = 361 for the efficacy population) was determined to allow for a drop-out rate of 10% and provide 82% power for demonstration of similarity in the primary endpoint, change in DAS28 score from baseline at week 24 between CT-P10 and rituximab groups. Full details of the sample size calculations have been reported previously [9].
Continuous data—including mean change from baseline and actual values for disease activity measured by DAS28-ESR and DAS28-CRP, SF-36 (mental and physical component score), joint damage progression, and B-cell counts—were summarized using descriptive statistics. Categorical data—including the proportion of patients achieving ACR20, ACR50, ACR70, and EULAR responses—were summarized using counts and percentages. Exact binomial analyses were performed to assess any treatment differences in ACR response rates. Efficacy, pharmacokinetic, and pharmacodynamic analysis populations included all patients who received at least one full dose of study drug and provided at least one post-treatment efficacy, pharmacokinetics, or pharmacodynamics result, respectively. The “2nd treatment course subset” of each of these populations consisted of all patients who received at least one full dose of study drug and provided at least one post-treatment result (efficacy, pharmacokinetics, or pharmacodynamics) during the second treatment course. Patient data from weeks 0 to 24 are presented as the efficacy/pharmacokinetic/pharmacodynamic populations and after week 24 as the efficacy/pharmacokinetic/pharmacodynamic—2nd treatment course subsets. All patients who received at least one dose of study drug were included in the safety population.
All analyses were performed using SAS software version 9.1.3 or higher.
3 Results
3.1 Patient Recruitment and Baseline Characteristics
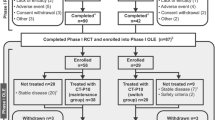
A total of 372 patients were randomly assigned to study drug (CT-P10, n = 161; US-RTX, n = 151; EU-RTX, n = 60; [combined rituximab, n = 211]). The first patient was recruited on 6 August 2014, and the last week 48 visit took place on 5 July 2016. Patient disposition is summarized in Fig. 1. Of the 372 patients administered study drug at week 0, 330 (88.7%) completed the second treatment course (CT-P10: 140/161 [87.0%]; US-RTX: 134/151 [88.7%]; EU-RTX 56/60 [93.3%]).

Patient disposition. * indicates two patients in the CT-P10 group were discontinued as both had not previously received tumor necrosis factor-antagonist treatment (major protocol deviation). ** indicates three patients discontinued after week 24 (one from the CT-P10 group due to withdrawal of patient consent, and two from the US-RTX group—one due to lack of efficacy and one due to signs of disease progression). *** indicates four patients (two each from the CT-P10 and US-RTX groups) did not satisfy the retreatment criteria after the first treatment course and were monitored up to week 48. RTX rituximab
Overall, demographics and baseline disease characteristics were similar across groups (Table 1) [9]. The median age of patients was 53.0 years for CT-P10 and US-RTX and 51.5 years for EU-RTX, and most patients were female (83.3–86.1%). Baseline DAS28-ESR and DAS28-CRP scores were similar between the three groups, as were the duration of disease activity, and prior anti-TNF exposure.
3.2 Efficacy
3.2.1 Disease Activity Score 28-Joint Count: Erythrocyte Sedimentation Rate (DAS28-ESR)
Mean decreases from baseline in DAS28-ESR were similar across groups up to week 48 (Fig. 2a). At week 24, the mean ± standard deviation (SD) decrease from baseline in DAS28-ESR was − 2.6 ± 1.1 for the CT-P10 group and − 2.5 ± 1.2 for the combined rituximab group. At week 48, mean decreases in DAS28-ESR from baseline were − 2.9 ± 1.3 and − 2.8 ± 1.4 for the CT-P10 and combined rituximab groups, respectively. Similar decreases were observed for both the US-RTX and the EU-RTX groups throughout the study (week 24: − 2.5 ± 1.1 and − 2.3 ± 1.3, respectively; week 48: − 2.8 ± 1.4 and − 2.9 ± 1.3, respectively; Fig. S1a in the electronic supplementary material [ESM]).

Efficacy of CT-P10 and combined rituximab over 48 weeks (efficacy population)a: a mean change from baseline in DAS28-ESR; b DAS28-CRP; c proportion of patients achieving ACR20, ACR50, and ACR70 criteria; and d mean hybrid ACR score. aData after week 24 are from the efficacy population–2nd treatment course subset. ACR American College of Rheumatology, CRP C-reactive protein, DAS28 Disease Activity Score 28-Joint Count, ESR erythrocyte sedimentation rate, RTX rituximab, SD standard deviation, SE standard error
3.2.2 DAS28-C-Reactive Protein (CRP)
Improvements in disease activity as revealed by mean decreases from baseline in DAS28-CRP were similar in the CT-P10 and combined rituximab groups up to week 48 (Fig. 2b). The primary efficacy endpoint was the mean ± SD decrease from baseline in DAS28-CRP at week 24; this was similar in the CT-P10 and combined rituximab groups (CT-P10: − 2.3 ± 1.1; combined rituximab: − 2.3 ± 1.2). This similarity was maintained throughout the study, such that, at week 48, the mean ± SD decrease in DAS28-CRP from baseline was − 2.7 ± 1.2 for CT-P10 and − 2.6 ± 1.3 for the combined rituximab groups. Similar decreases were observed with US-RTX and EU-RTX throughout the study (week 24: − 2.3 ± 1.1 and − 2.3 ± 1.3, respectively; week 48: − 2.6 ± 1.3 and − 2.7 ± 1.3, respectively; Fig. S1b in the ESM).
3.2.3 American College of Rheumatology (ACR) Responses
A similar proportion of patients achieved a clinical response according to ACR criteria in the CT-P10 and combined rituximab groups over the 48 weeks (Fig. 2c). ACR20, ACR50, and ACR70 response rates at week 48 indicated no differences between groups; 80.6%, 55.4%, and 31.7% of patients in the CT-P10 group achieved ACR20, ACR50, and ACR70 versus 79.8%, 53.9%, and 33.7% in the combined rituximab group, respectively. Estimated treatment differences in ACR20, ACR50, and ACR70 response rates at week 48 between the two groups were as follows: ACR20: 0.01 (95% confidence interval [CI] − 0.07 to 0.09); ACR50: 0.02 (95% CI − 0.09 to 0.13); ACR70: − 0.02 (95% CI − 0.13 to 0.09). Similar outcomes were observed in the US-RTX and EU-RTX groups (Fig. S2 in the ESM). Comparable mean ACR hybrid scores (Fig. 2d) and individual ACR components (Table S1 in the ESM) were also demonstrated between groups.
3.2.4 European League Against Rheumatism (EULAR) Responses
The proportion of patients with a good or moderate EULAR response was similar between the CT-P10 and combined rituximab groups at week 24 (82.6 and 83.8%) and week 48 (89.9 and 87.0%), respectively (Fig. S3 in the ESM).
3.2.5 Low Disease Activity (LDA) and Remission Rates by DAS28
At week 24, similar proportions of patients in the CT-P10 and combined rituximab groups achieved low disease activity (LDA) (12.3 and 11.3%) or remission (10.3 and 13.3%) using the DAS28-ESR. At week 48, a total of 17.3 and 11.4% achieved LDA and 17.3 and 22.8% achieved remission in the CT-P10 and combined rituximab groups, respectively. Similar results were obtained using DAS28-CRP in the US-RTX and EU-RTX groups (Table 2). The proportion of patients achieving sustained LDA (including remission) for at least 6 months was also similar between groups (Table 2).
3.2.6 Remission Rates by ACR-EULAR Boolean Criteria
Remission rates were also assessed according to ACR-EULAR Boolean criteria (tender joint count, swollen joint count, CRP, and patient global assessment; all at levels ≤ 1) [14]. The proportions of patients achieving remission at week 24 were 7.7 and 10.8% in the CT-P10 and combined rituximab groups, respectively. At week 48, the proportions were 11.5 and 15.5%, respectively (Table 2).
3.2.7 Disease Activity
Mean CDAI and SDAI scores were reduced from baseline to week 48 (Fig. S4 in the ESM). The reductions observed were of similar magnitude across the CT-P10 and rituximab groups.
3.2.8 Patient-Reported Outcomes
The mean HAQ-DI score decreased from baseline to week 48 in all groups (Fig. 3a). Similarly, the mean SF-36 score increased from baseline to week 24 in all groups and then remained stable to week 48. Scores were comparable between groups for physical, mental, and other components of the SF-36 (Fig. 3b).

Effects of CT-P10 and combined rituximab on patient-reported outcomes (efficacy population)a: a HAQ-DI over 48 weeks and b SF-36 at week 48. aData after week 24 are from the efficacy population–2nd treatment course subset. HAQ-DI Health Assessment Questionnaire Disability Index, RTX rituximab, SD standard deviation, SF-36 Short Form 36-Item Health Survey
3.2.9 Radiologic Progression
At baseline, the mean Van der Heijde modified total sharp score was 78.9 ± 59.1 in the CT-P10 group compared with 85.8 ± 70.3 in the combined rituximab group. At week 48, the mean increase from baseline in each group was 1.8 ± 8.9 and 1.2 ± 3.0, respectively. Analyses of the cumulative probability distributions of total joint space narrowing and total erosion scores also revealed similar disease progression between the two groups (Fig. S5 in the ESM).
3.2.10 Additional Efficacy Analysis According to Genotype
Polymorphisms in FcγRIIa and FcγRIIIa genes did not affect the response to CT-P10 or rituximab, with similar changes from baseline to weeks 24 and 48 in DAS28-ESR and DAS28-CRP evident in all subtypes across groups (Table S2 in the ESM).
3.3 Pharmacokinetics
No notable differences were demonstrated between groups in any of the secondary pharmacokinetic parameters, with comparable outcomes observed after the first and second treatment courses. Maximum concentration after the second infusion of the first treatment course (Cmax 1st course) was 438.0, 432.6, and 474.2 μg/mL in the CT-P10, US-RTX, and EU-RTX groups, respectively, and 418.9, 420.3, and 464.5 μg/mL, respectively, after the second infusion of the second treatment course (Cmax 2nd course; Table S3 in the ESM).
3.4 Pharmacodynamics
Before the first treatment course, the median B-cell count at baseline in the pharmacodynamic population was 169.5, 178.0, and 124.0 cells/μL in the CT-P10, US-RTX, and EU-RTX groups, respectively. Median B-cell counts decreased to below the lower limit of quantification (LLoQ; 20 cells/μL) immediately after the first infusion and remained below this level up to week 48. B-cell counts were similar between all three groups at every visit up to week 48. Of the patients with detectable B-cell levels at baseline that showed depletion after treatment, the proportions of patients with B-cell counts above the LLoQ was 13/118 (11.0%), 14/110 (12.7%), and 4/45 (8.9%) at predose of week 48 in the CT-P10, US-RTX, and EU-RTX groups, respectively.
3.5 Immunogenicity
The majority of patients in all three groups tested negative for ADAs throughout the study. The number of patients with detectable ADAs at baseline was 19 (11.8%), 13 (8.6%) and 7 (11.7%) in the CT-P10, US-RTX, and EU-RTX groups, respectively. Of these patients, ten (52.5%), four (30.8%), and three (42.9%) were ADA positive at one or more post-baseline visit up to week 48, respectively, and three (15.8%), two (15.4%), and zero (0.0%) were positive at week 48, respectively. The number of patients who tested positive for ADAs was 24 (14.9%), 33 (21.9%), and 16 (26.7%) at week 24 in the CT-P10, US-RTX, and EU-RTX groups, respectively, and 7 (4.9%), 13 (9.4%), and 5 (8.6%), respectively, at week 48. NAbs were detected in one patient (US-RTX group) at week 24 and in two patients (one each in the CT-P10 and US-RTX groups) at week 48.
3.6 Safety
All patients who received at least one full or partial dose of study drug were included in the safety analysis. A total of 740 AEs were reported in 261 (70.2%) patients by week 48, and AEs occurred in a broadly similar proportion of patients in each group (Table 3). Most AEs were grade 1 or grade 2. The most common AE was upper respiratory tract infection followed by IRRs (Table S4 in the ESM). A total of 34 serious AEs were reported in 31 (8.3%) patients (Table S5 in the ESM), with those in six patients (five in the US-RTX group and one in the EU-RTX group) considered to be related to the study drug. A similar proportion of patients in each group experienced AEs leading to study drug discontinuation (Table 3), with the most common reason for discontinuation being IRR (two [1.2%], three [2.0%], and one [1.7%] of all patients in the CT-P10, US-RTX, and EU-RTX groups, respectively). Overall, 62 patients (38.5%) in the CT-P10 group developed infections compared with 71 patients (33.6%) in the combined rituximab group (54 [35.8%] with US-RTX and 17 [28.3] with EU-RTX; Table 3). Up to week 48, there were no cases of progressive multifocal leukoencephalopathy and no hepatitis B reactivation. Four malignancies were reported in four patients: two (1.3%) from the US-RTX group and two (3.3%) from the EU-RTX group. For US-RTX, one patient had bladder cancer and one had breast cancer. For EU-RTX, one patient had adenocarcinoma of the colon and one had breast cancer. One patient treated with CT-P10 died because of cellulitis, although the event was not considered related to the study drug.
4 Discussion
This phase III study demonstrated that CT-P10 and two rituximab RPs displayed comparable efficacy and impact on quality of life, as well as comparable B-cell kinetics, immunogenicity, and safety for up to 48 weeks. These results support those from week 24 [9], which revealed pharmacokinetic and therapeutic equivalence of CT-P10 versus both US-RTX and EU-RTX, as well as similar pharmacodynamics and safety between CT-P10 and the combined rituximab group.
This is the first full report on the long-term efficacy and safety of the rituximab biosimilar CT-P10. A key strength of the study is that it allowed the three-way comparison of CT-P10 and the two licensed rituximab RPs in the 48-week analyses of pharmacokinetics, efficacy, pharmacodynamics, and safety. Debate continues as to whether EU-RTX and US-RTX can be regarded as the same drug given the slight differences in their manufacturing processes [15]. However, part 1 of this study demonstrated the equivalence of EU-RTX and US-RTX (and CT-P10) [9]. Data for CT-P10 were equivalent to those obtained in the US-RTX and EU-RTX groups individually as well as in the combined rituximab group. Additional strengths included the randomized design of the study, the use of well-established outcome measures, and the high levels of patient retention at 48 weeks.
Key historical RCTs of rituximab such as the REFLEX (Randomized Evaluation oF Long-term Efficacy of Rituximab in RA) and DANCER (Dose-ranging Assessment iNternational Clinical Evaluation of rituximab in RA) studies demonstrated the efficacy of this treatment up to week 24 in patients with active RA [3, 4]. The REFLEX trial was extended over a 5-year period and data collected after multiple courses of treatment. Efficacy outcomes at 24 weeks after the second course of rituximab revealed ACR20, ACR50, and ACR70 response rates of 72.8%, 41.2%, and 19.4%, respectively, and a mean change in DAS28-ESR from baseline of − 2.7 [16]. Furthermore, in the SERENE (Study Evaluating rituximab’s Efficacy in methotrexate iNadequate rEsponders) trial, efficacy measures were maintained or improved throughout the 48-week study period, during which most (~ 90%) patients with RA received two courses of rituximab 1000 mg (ACR20: 50.6% at week 24, 57.6% at week 48; mean change in DAS28-ESR from baseline: − 1.7 at week 24, − 2.0 at week 48) [17]. However, direct comparison with historical data should be performed with caution because of differences in study protocol and baseline patient characteristics. Results from the current study demonstrate that responses to CT-P10 at week 48 following a second course of treatment were similar to those with rituximab and to data from previous studies of rituximab in combination with methotrexate.
The incidence of IRRs and infections in historical clinical trials of rituximab has ranged from 22 to 71% for IRRs, and from 30 to 55% for infections [3,4,5, 17]. The proportion of patients reporting IRRs or infections in the present study up to week 48 were within the ranges previously described. None of these cases were considered serious, all resolved without issue, and most occurred after the first infusion [9], in accordance with previous reports [5, 17]. No unexpected safety concerns were raised in the current study, and safety profiles remained comparable for CT-P10 and rituximab after retreatment, consistent with the 24-week data [9] and with previous rituximab trials [17].
One patient who showed clinical improvement following treatment with CT-P10 displayed incomplete depletion of B-cells at week 48. This is likely to be due to a combination of immunogenicity (the patient was positive for ADAs and NAbs up to week 48), an FF FcγRIIIa genotype, and underlying Sjögren’s syndrome in this patient. Similar cases have been reported in patients who do not experience B-cell depletion following rituximab treatment but still show a clinical response [18].
Approximately 10% of patients in this study had detectable ADAs at baseline; these data can be considered as “not product related”. Since false positives were found to occur as a result of interference from circulating CD20-expressing B-cell membrane fragments (CMFs) [19], ofatumumab was used in this study to improve the confirmatory and titration methods to effectively inhibit the signals produced by CMFs [19]. However, the possibility remains of further non-specific binding by other unknown factors. It should be noted that there was no noticeable trend in pharmacokinetic/pharmacodynamic profiles in patients who were ADA positive at baseline in this study compared with those who were ADA negative.
Among 73 patients who were ADA positive at week 24, a total of 50 (68.5%) were negative at week 48. Those 50 patients can be considered as transient ADA positive. No negative effect was found in terms of efficacy and pharmacokinetics in transient ADA positive patients, and a similar finding has been presented in other studies of biologic agents in autoimmune diseases [20, 21].
A number of other rituximab biosimilar candidates are currently in development, but all are at a less advanced stage of testing than CT-P10. Therefore, this is the first published study to demonstrate long-term equivalence of a rituximab biosimilar to its RP.
The results of a 24-week extension study will provide further data relating to the longer-term use of CT-P10, during which patients who had received CT-P10 initially remained on the biosimilar. Those who received EU-RTX for the first course were switched to CT-P10, and those who received US-RTX were randomized 1:1 to either remain on US-RTX or switch to CT-P10. This extension will provide valuable information regarding the efficacy and safety of switching from RP to CT-P10. Biosimilars are associated with lower developmental costs, and, as such, may be available at a reduced price. Furthermore, the availability of biosimilars can also instigate price reductions through competition between pharmaceutical companies [22]. It is expected that switching patients from RP to a biosimilar, and the introduction of biosimilars into clinical practice in general, will likely result in financial savings for healthcare systems. This, in turn, may increase availability and patient access to these clinically effective drugs.
5 Conclusion
This phase III RCT demonstrated the long-term similarity of CT-P10 and two approved rituximab RPs in terms of efficacy, pharmacokinetics, pharmacodynamics, immunogenicity, and safety for up to 48 weeks. These results provide further support for the recent approval of CT-P10 for the treatment of RA and other CD20+ B-cell-related diseases.
References
Marston B, Palanichamy A, Anolik JH. B cells in the pathogenesis and treatment of rheumatoid arthritis (review). Curr Opin Rheumatol. 2010;22:307–15.
Bugatti S, Vitolo B, Caporali R, Montecucco C, Manzo A. B cells in rheumatoid arthritis: from pathogenic players to disease biomarkers (review). Biomed Res Int. 2014;2014:681678.
Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793–806.
Emery P, Fleischmann R, Filipowicz-Sosnowska A, Schechtman J, Szczepanski L, Kavanaugh A, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54:1390–400.
European Medicines Agency (EMA). MabThera (rituximab) [summary of product characteristics]. London, UK: EMA; 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000165/WC500025821.pdf. Accessed 30 Nov 2016.
European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf. Accessed 18 Dec 2016.
U.S. Food and Drugs Administration. Scientific considerations in demonstrating biosimilarity to a reference product: Guidance for industry. 2015. https://www.fda.gov/downloads/drugs/guidances/ucm291128.pdf. Accessed 31 May 2018.
Yoo DH, Suh CH, Shim SC, Jeka S, Cons-Molina FF, Hrycaj P, et al. A multicentre randomised controlled trial to compare the pharmacokinetics, efficacy and safety of CT-P10 and innovator rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76:566–70.
Park W, Božić-Majstorović L, Milakovic D, Berrocal Kasay A, Chalouhi El-Khouri E, Irazoque-Palazuelos F, et al. Comparison of biosimilar CT-P10 and innovator rituximab in patients with rheumatoid arthritis: a randomised controlled phase III trial. MAbs. 2018;10:934–43.
Kim WS, Buske C, Ogura M, Jurczak W, Sancho JM, Zhavrid E, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4:e362–73.
World Medical Association (WMA). WMA Declaration of Helsinki: Ethical principles for medical research involving human subjects. Adopted by the 18th WMA General Assembly, Helsinki, Finland, June 1964 and amended (latest) by the 64th WMA General Assembly, Fortaleza, Brazil; 2013 October. https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/. Accessed 18 January 2017.
International Conference on Harmonisation (ICH). Tripartite guideline. Guidance for good clinical practice E6 (R1). ICH; 1996. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. Accessed 18 Jan 2017.
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24.
Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LH, Funovits J, et al. American College of Rheumatology/European League against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Ann Rheum Dis. 2011;70:404–13.
International Conference on Harmonisation (ICH). Comparability of biotechnological/biological products subject to changes in their manufacturing process. ICH; 2014. http://www.ich.org/products/guidelines/quality/quality-single/article/comparability-of-biotechnologicalbiological-products-subject-to-changes-in-their-manufacturing-proc.html. Accessed 18 Jan 2017.
Keystone EC, Cohen SB, Emery P, Kremer JM, Dougados M, Loveless JE, et al. Multiple courses of rituximab produce sustained clinical and radiographic efficacy and safety in patients with rheumatoid arthritis and an inadequate response to 1 or more tumor necrosis factor inhibitors: 5-year data from the REFLEX study. J Rheumatol. 2012;39:2238–46.
Emery P, Deodhar A, Rigby WF, Isaacs JD, Combe B, Racewicz AJ, et al. Efficacy and safety of different doses and retreatment of rituximab: a randomised, placebo-controlled trial in patients who are biological naive with active rheumatoid arthritis and an inadequate response to methotrexate (Study Evaluating Rituximab’s Efficacy in MTX iNadequate rEsponders (SERENE)). Ann Rheum Dis. 2010;69:1629–35.
Vital EM, Rawstron AC, Dass S, Henshaw K, Madden J, Emery P, et al. Reduced-dose rituximab in rheumatoid arthritis: Efficacy depends on degree of B cell depletion. Arthritis Rheum. 2011;63:603–8.
Chen K, Page JG, Schwartz AM, Lee TN, DeWall SL, Sikkema DJ, et al. False-positive immunogenicity responses are caused by CD20+ B cell membrane fragments in an anti-ofatumumab antibody bridging assay. J Immunol Methods. 2013;394:22–31.
Sandborn WJ, Wolf DC, Kosutic G, Parker G, Schreiber S, Lee SD, et al. Effects of transient and persistent anti-drug antibodies to certolizumab pegol: longitudinal data from a 7-year study in Crohn’s disease. Inflamm Bowel Dis. 2017;23:1047–56.
Siljehult F, Ärlestig L, Eriksson C, Rantapää-Dahlqvist S. Concentrations of infliximab and anti-drug antibodies in relation to clinical response in patients with rheumatoid arthritis. Scand J Rheumatol. 2018;47:345–50.
Erwin A. The economics of biosimilars. Am Health Drug Benefits. 2013;6:468–78.
Acknowledgements
Medical writing support (including development of a draft outline and subsequent drafts in consultation with the authors, assembling tables and figures, collating author comments, copyediting, fact checking, and referencing) was provided by Louisa Lyon, PhD, at Aspire Scientific (Bollington, UK), and funded by CELLTRION, Inc. (Incheon, Republic of Korea).
Author information
Authors and Affiliations
Contributions
Contribution to study conception and design: CHS, DHY, WP, SCS, SJL, SYL. Contribution to acquisition of data: CHS, DHY, ABK, ECEK, FFCM, PS, PM, FGMR, PW, SJ, JCC, TL, PH, MAB, IH, JJ, MP, MB, MK, SS, AC, DA, WP, SCS. Contribution to analysis and interpretation of data: CHS, DHY, WP, SCS, SJL, SYL. Drafting the article or revising it critically for important intellectual content: CHS, DHY, ABK, ECEK, FFCM, PS, PM, FGMR, PW, SJ, JCC, TL, PH, MAB, IH, JJ, MP, MB, MK, SS, AC, DA, WP, SCS, SJL, SYL. Approval of the version of the article to be published: CHS, DHY, ABK, ECEK, FFCM, PS, PM, FGMR, PW, SJ, JCC, TL, PH, MAB, IH, JJ, MP, MB, MK, SS, AC, DA, WP, SCS, SJL, SYL. Guarantor for the overall content: CHS.
Corresponding author
Ethics declarations
Funding
This work was supported by CELLTRION, Inc. (Incheon, Republic of Korea). Employees of the funder had a role in the study design, data analysis, data interpretation, and writing of the report and had access to the raw data. Employees of the funder had no role in data collection. The authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Conflicts of interest
Chang-Hee Suh received consulting fees (< US$10,000) during the conduct of the study and grants outside the submitted work from CELLTRION, Inc. Dae Hyun Yoo is a scientific consultant (no fee), is on the speaker’s bureau of CELLTRION, Inc., and has received research grants not related to this clinical study. Pedro Miranda received fees from CELLTRION for conducting the trial and has performed trials for CELLTRION outside the submitted work. Won Park and Seung Cheol Shim have received consulting fees (< US$10,000) from CELLTRION, Inc. Sang Joon Lee and Sung Young Lee are employees of CELLTRION, Inc. Alfredo Berrocal Kasay, Elias Chalouhi El-Khouri, Francisco Fidencio Cons Molina, Pavel Shesternya, Francisco G. Medina-Rodriguez, Piotr Wiland, Slawomir Jeka, Jose Chavez-Corrales, Thomas Linde, Pawel Hrycaj, Mauricio Abello-Banfi, Ihor Hospodarskyy, Janusz Jaworski, Mariusz Piotrowski, Marek Brzosko, Marek Krogulec, Sergii Shevchuk, Armando Calvo, and Daina Andersone have no conflicts of interest that are directly relevant to the content of this article.
Ethics approval and consent to participate
The study was performed in accordance with the Declaration of Helsinki [11] and Good Clinical Practice guidelines [12]. The study protocol was reviewed and approved by the relevant independent ethics committee at each site, and all patients provided written informed consent.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Suh, CH., Yoo, D.H., Berrocal Kasay, A. et al. Long-Term Efficacy and Safety of Biosimilar CT-P10 Versus Innovator Rituximab in Rheumatoid Arthritis: 48-Week Results from a Randomized Phase III Trial. BioDrugs 33, 79–91 (2019). https://doi.org/10.1007/s40259-018-00331-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-018-00331-4