
Abstract
The economic evaluation of medical technology has evolved as a key element in supporting health budget allocation decisions. Among suppliers of innovation, the medical device industry is one of the most dynamic fields of medical progress with thousands of new products marketed every year. Accordingly, the broad variety of technologies covered by the umbrella term ‘medical devices’ have come under increasing scrutiny regarding their cost effectiveness. In the process, a number of device-specific factors have become apparent, each of which can complicate a thorough economic evaluation and limit its informative value. Some of these factors relate to specific characteristics of device functioning. Examples of such factors include the fact that most technologies require, or form part of, a procedure and that many devices have multiple indications or purposes. Others in turn reflect external conditions and are more general in character, such as the regulatory framework that a medical device manufacturer faces prior to market approval and the structure of the medical device industry. Drawing on the available literature, these complicating factors and their practical implications are discussed and used as a basis to elaborate on the emerging challenges for the economic evaluation of medical devices.
Similar content being viewed by others


• Those conducting economic evaluations of medical devices face a variety of challenges stemming from external regulatory circumstances and product-specific characteristics
• The current regulatory framework for medical devices, characteristics of the medical device industry and market and device-related constraints to certain aspects of clinical trial design all limit the availability of high-quality evidence at the time of reimbursement decisions
• Issues such as device reusability, procedural integration and temporal changes in determinants of medical device performance challenge the calculation, validity and generalizability of the cost effectiveness of a medical device
• Possible solutions to the challenges encountered in economic evaluations of medical devices include changes in legal regulations, the use of different health technology (re-)assessment methods at different points in time, awareness of the various determinants of device effectiveness and resource use and innovative reimbursement strategies
1 Introduction
Daily clinical practice relies heavily on a vast array of medical devices. For the two biggest medical device markets, the US and Europe [1], the total number of registered products has been estimated to exceed 200,000 [2, 3]. In an environment of increasing demand for information on the economic value of healthcare innovation, there has been growing interest in the assessment of medical devices. A number of factors related to the development, functionality and use of devices have been found to complicate the evaluation of clinical and economic value. The goal of our study was to analyse these factors, provide a summary of practical challenges that researchers face when conducting economic evaluations of medical devices, and examine some ways to address these challenges.
A logical starting point in this paper is the answer to a seemingly basic question, namely what is a medical device? Regulatory documents published by the US Food and Drug Administration (FDA) [4], the European Commission (EC) [5] and the Global Harmonization Task ForceFootnote 1 (GHTF) [6] provide guidance here. From these directives, two central, recurring statements stand out and serve as a working definition for this paper. Specifically, a device will be considered a ‘medical device’ if: (1) it is used for the prevention, diagnosis, monitoring or treatment of a certain health state; and (2) its primary mode of action is not based on a pharmacological, metabolic or immunological process. If a medical device is defined in this way, it becomes clear that apart from being numerous, medical devices comprise a very heterogeneous collection of technologies. The risk classification system proposed by the GHTF provides a basic structure [7]. Categories in this system range from class A (low-risk devices) to class D (high-risk devices) [7]. Table 1 shows a non-exhaustive list of medical devices for each category to illustrate the wide variety of technologies covered. Differences between the various technologies in several characteristics such as target indication, invasiveness, duration of use and passive or active mode of functioning are evident.
Given the general definition of medical devices provided, what makes economic evaluations of these technologies difficult? When investigating the cost effectiveness of a medical device, the researcher faces the “…basic tasks of any economic evaluation [namely] to identify, measure, value, and compare the costs and consequences of the alternatives being considered” [8]. Following this notion, we have identified a number of external factors and device-level factors from the available literature that can complicate these basic tasks. External factors originate from the regulatory and industry environment, whereas device-level factors emerge from the specific nature of medical device functioning. This paper first introduces these two categories and the challenges they entail regarding economic evaluations. This is followed by a discussion of their practical implications for the decision maker and possible solutions to these challenges.
2 Methods
After an initial exploration of secondary literature, we performed a qualitative, non-systematic review of primary data and secondary sources on the challenges that arise when performing economic evaluations of medical devices. Given the very broad spectrum of devices covered and the resulting difficulties in formulating a search strategy that could identify all publications relating to economic evaluations of all types of medical devices, a systematic approach was not deemed feasible.
3 Complicating Factors in the Economic Evaluation of Medical Devices
3.1 External Factors
The ability to perform a thorough economic evaluation of health technologies relies heavily on the evidence base that exists at that moment. The quality and timeliness of available data are essential considerations if an economic evaluation is to support reimbursement decisions about an innovative medical technology. In the case of medical devices, both aspects are hindered by a number of external restraints.
3.1.1 Regulatory Framework
The highest level of clinical evidence is derived from the structured review of randomized controlled trials (RCTs) [9]. Considering the limited availability of RCT data for the majority of medical devices, the current regulatory approval environment plays a decisive role [10, 11]. Both the American and especially the European regulatory bodies apply less stringent rules when it comes to pre-market clinical evaluation of medical devices compared with the standards for new pharmaceuticals [12]. For example, the FDA requires a single RCT demonstrating safety and effectiveness for high-risk devices only [13]. For the EU market, randomized study design is not mandatory for any medical device to obtain market approval [3, 13]. In addition, there is a gradient in evidence requirements relating to the device classification system presented in Table 1. Each class is associated with specific conditions regarding quality assurance, technical documentation and the provision of clinical data that must be addressed by manufacturers in the pre-market approval procedure. For technologies assigned to lower-risk classes, the effectiveness and safety requirements are less rigorous than for high-risk devices [3, 14]. Low-risk technologies, at the same time, constitute the vast majority of products licensed [15]. With thousands of new medical devices being marketed every year [15], the resulting evidence gap is therefore substantial. In summary, the regulatory framework does not require device manufacturers to undertake clinical studies as a universal prerequisite. In those cases where they are indicated due to the risk profile of a certain device, RCTs are the exception rather than the rule.
3.1.2 Industry Structure
The fragmented structure and the dynamic development processes in the medical device industry impede the realization of extensive clinical trials. The medical device industry mainly consists of small and medium-sized companies with a very limited portfolio [1, 10]. Their products typically serve small markets and often depend on venture capital until product launch [16]. For these manufacturers, the financial burden of conducting reasonably powered RCTs can be impossible to bear. Financing problems are particularly evident in cases where the clinical trial of a device in an established clinical setting entails laborious and costly organizational adaptations, for instance because of the need for additional training of clinical staff or the modification of facilities.
3.1.3 Short Product Life-Cycle
The practical relevance of clinical studies with a time-consuming recruitment process and long follow-up periods is further diminished by the short product life-cycle of many devices [17]. The time needed to develop the next generation of a device might outpace the time needed to properly evaluate the previous one, rendering the results of the evaluation irrelevant the moment they are known. Not surprisingly, determining the optimal timing for assessing fast-changing technologies has been acknowledged as an area of research in and of itself [18, 19].
3.1.4 Early Market Diffusion
It may seem self-evident that the optimal time to perform an initial economic evaluation of a device can only be prior to its widespread use. However, in the case of medical devices, rapid market diffusion, facilitated by the aforementioned minor regulatory prerequisites and early adoption by key opinion leaders, counteract this desire [18, 19]. Rapid market diffusion can even precede the demonstration of clinical safety. In a 5-year analysis of FDA device recalls due to a “…reasonable chance that [the products recalled] could cause serious health problems or death,” [20] it was observed that the majority of recalls had been related to devices that had not been subjected to rigorous clinical testing due to their classification by the FDA [21]. The possible conflict of interest arising for individual clinical investigators due to a close, business-like relationship with the medical device industry has also been criticized [22, 23]. Apart from incentives offered for testimonials, clinicians might feel generally inclined to adopt new technologies because of the associated prestige, a genuine interest in exploring new ways to treat patients, or the attractive financial rewards for performing device-related procedures. Eventually, premature clinical diffusion can lead to a situation where professional or patient impartiality towards the available technologies is compromised. If that is the case, there may be resistance to performing prospective comparative studies [19, 24].
In summary, four external factors that have a potential impact on economic evaluations of medical devices can be identified: (1) the regulatory environment for market approval, (2) the structure of the medical device industry, (3) the short product life-cycle, and (4) the early market diffusion of medical devices. One could argue that the current regulatory environment stands out in importance because of its potential to influence the other three factors.
The resulting implications for economic evaluations are evident. For one thing, there may be no opportunity to rely on a pre-market clinical trial as a means to obtain patient-level economic data (i.e., a ‘piggyback’ approach) [25]. More importantly, for economic evaluations based on a systematic review of available data, the evidence base might be insufficient, especially regarding RCT-level evidence. The overall impact of this problem is further compounded by the high volume of low-risk technologies being developed and the limited need to demonstrate clinical effectiveness and safety for market approval of these devices.
If the aim of an economic evaluation is to provide a quantitative underpinning for reimbursement decisions, this role can be questioned if the data used in the evaluation are qualitatively weak or if the evaluation has to be delayed until sufficient data are collected. Consequently, evidence limitations at the time of an initial reimbursement decision can be viewed as the most significant challenge for the economic evaluation of medical devices.
3.2 Device-Level Factors
Besides the external factors that complicate the economic evaluation of a medical device, there are other factors that are more device-specific. That is, the collection and analysis of input data used in the economic evaluation, as well as the generalizability of results, are hampered by several factors that vary depending on the functional nature of the device. These device-level factors are the focus of attention in this section.
3.2.1 Constraints to Study Design
Clinical studies of medical devices are subject to so-called ‘inherent constraints’ [10] to study design. These include the impossibility of (double-)blinding in trials of many devices and the controversial ethics of sham procedures as a way to enable a ‘placebo’ control and blinding of patients [10, 26]. While in certain instances the blinded assessment of health outcomes may be feasible, this will not always resolve all important threats to the internal validity of a study. Patient recruitment poses another critical challenge. With many medical devices already serving small target populations, it may be difficult to recruit sufficient numbers of patients for trials. This problem might be aggravated if invasive or otherwise unpleasant procedures are involved in device use. Moreover, the structural and procedural requirements of a medical device study might limit the number of eligible trial centres and curtail the size of the study population even further [10].
Even where well designed RCTs of medical devices are conducted, the study population is often small and the follow-up period is short [27]. These two properties limit the ability to detect rare but important adverse events; moreover, they also limit the ability to detect small but potentially important differences in effectiveness or costs between a medical device and its comparator. The limitations of small trials will be particularly problematic if there is a high degree of variability in effectiveness between individual patients. Data from observational studies are therefore a common necessity in economic evaluations of medical devices since they can close evidence gaps arising when experimental studies cannot easily be performed or the generalizability of results from these studies is limited [27, 28]. However, the use of observational studies comes at a price, since they are known to be more vulnerable to bias and confounding than RCTs. Inappropriate use of observational data can therefore result in severely biased estimates of clinical outcomes and cost effectiveness.
3.2.2 Procedural Integration
The inseparable link between the function of a medical device and the procedure that it forms a part of presents an important challenge in economic evaluations of medical devices. In contrast to pharmaceuticals, which have been described as ‘embodied technologies’ [29], most devices are embedded in a cascade of professional activities and associated technology. While endoscopes and coronary catheters are indispensable for minimally invasive surgery and percutaneous coronary interventions, respectively, they nevertheless comprise only two of the many factors that contribute to the clinical outcome and cost effectiveness of the respective overall procedure. This procedural integration has important implications when assessing the impact of a medical device on health outcomes and costs.
To begin with, the integration in a ‘compound’ intervention means that the clinical performance of a medical device is a function of several determinants. Ramsey et al [10]. state that the “…effectiveness [of a medical device] varies with: (1) the intrinsic net efficacy resulting from the function of the device (consider harms and benefits); (2) the skill of the medical professional that installs, operates and monitors the device; and (3) patient factors…”. One could add two other factors to complete this list: (4) the performance of required ancillary technology; and (5) the organizational setting. Consequently, the transferability of a cost-effectiveness estimate to a specific setting will depend on the degree of variability between healthcare settings in relevant input parameters [30]. The point can be made that this problem is not unique to medical devices since it is also found with pharmaceuticals. While it is true that they have some things in common (e.g., the importance of intrinsic efficacy and patient mix), it can be argued that variations in user proficiency and organizational setting are important additional considerations that are particularly present with medical devices. These determinants act as important ‘disrupting factors’ in the assessment of device-related effectiveness and their influence means that individual study results need to be interpreted with caution. For example, in a cost-effectiveness analysis of surgical robot-assisted cholecystectomy compared with conventional laparoscopic cholecystectomy, Heemskerk et al. [31] mention systematic variation in surgical experience between the two study groups and little overall experience with robotic surgery at their institution as limiting factors for the interpretation of study results. Obviously, the impact of these factors on the cost effectiveness of medical devices can challenge claims of generalizability even within healthcare systems or hospitals.
The cost calculations involved in an economic evaluation of a medical device may be more complex than that of a drug, particularly because of their aforementioned procedural integration. Costs can accrue from a number of related activities such as training of personnel or patients, the application and maintenance of the device and the need for complementary technology and shared facilities [10]. Accounting for all of the relevant cost items that are associated with medical device use can therefore be laborious and the choice of cost components might affect the decision between micro- and gross-costing methods [32]. What is more, every additional cost item represents yet another source of uncertainty to be considered when assessing the transferability of cost-effectiveness estimates between healthcare settings.
Other uncertainties in the economic evaluation of medical devices relate to how utilities are included in the evaluation. The generally observed focus on outcome assessment does not account for theoretically relevant process measures. Consideration of only intermediate or final outcomes, an attitude critically referred to as ‘narrow consequentialism’ [33], disregards the potential for (dis-)utility associated with the process of device use [33, 34]. It might be appropriate to assume that in most cases the administration of a drug will not have an important impact on the value that a patient gives to a medicine, since the patient would very likely be more concerned about its ultimate efficacy and safety. In contrast, the situation with medical devices may be different, since the ease, comfort of use or the unpleasantness of undergoing an associated invasive procedure might add to overall benefits and harms. This particularly applies to technologies such as hearing aids or limb prostheses that involve regular use. The available evidence dealing with the relevance of process utilities is limited and empirical results are mixed [33, 35]. Whether process utility indeed forms a measurable entity that can and should always be assessed separately from final health outcomes and costs without the risk of double-counting remains up for discussion and constitutes an interesting field of research.
The best examples of procedural integration can be found with diagnostic tests, which comprise an important subcategory of medical devices. Since a new diagnostic test will often be used in practice as one part of a diagnostic strategy, it may be necessary to compare two diagnostic strategies involving a combination of tests and not just two single tests if the effectiveness outcome is simply overall diagnostic accuracy. The evaluation would also have to consider what happens when there are equivocal results as well as the costs of confirmatory testing. However, one could also opt to base the assessment of effectiveness on health outcomes instead of diagnostic performance. After all, while parameters like diagnostic accuracy have often been employed as effectiveness outcomes in cost-effectiveness analyses, better diagnostic performance does not necessarily translate into improved health outcomes [36]. Shifting the focus from diagnostic accuracy to health outcomes requires consideration of treatment decisions made on the basis of the test results. In addition to considering the effectiveness of these treatments, one would also need to consider occurrences such as health loss caused by delayed treatment due to a false negative diagnosis or from unnecessary treatment following a false positive diagnosis. As a result, three stages in a diagnostic process need to be considered in an economic evaluation of a test: obtaining a test result, deciding on the next steps in diagnosis and therapy, and estimating the resulting health outcomes [37]. Since these three stages are rarely studied together in clinical trials, modelling and evidence synthesis play a critical role in economic evaluations of diagnostic devices.
3.2.3 Reusability
Some medical devices can be used repeatedly and for different health conditions. They can have one ‘intended use’ for a number of ‘indications of use’ [29]. Reusable technologies such as MRI scanners or robotic surgery devices serve a variety of target indications and exhibit different clinical effectiveness for each of these [38, 39]. Furthermore, reusable technologies often require large capital investments that need to be attributed to individual use units. Clinical effectiveness, use capacity and utilization rate of such technologies vary with indication, rendering the calculation of an overall cost-effectiveness ratio of these shared devices a difficult undertaking [29, 37].
Investments such as acquisition costs or adaptations of facilities have to be annuitized over an assumed device lifetime to calculate the equivalent annual cost [8]. In addition to the fixed costs of device use, the equivalent annual cost of initial investment needs to be attributed to individual use units (e.g., one CT scan performed) to enable cost-effectiveness calculations [8]. It is therefore necessary to estimate the lifetime and use capacity of a device. If fixed costs contribute substantially to overall costs, uncertainty about the lifetime and the actual degree of capacity utilization will translate into uncertainty surrounding the cost effectiveness of the device. If actual utilization exceeds the utilization predicted during the economic evaluation, the device may in truth be more cost effective than estimated and vice versa.
A distinction can be made between reusable and non-reusable medical devices regarding the impact of use volume on total budget and cost effectiveness. The overall utilization of non-reusable medical devices will primarily affect the total budget impact. In the case of reusable technologies, however, the overall utilization over the product lifetime will also affect their cost effectiveness. The consideration of a conservative and an optimistic scenario of utilization rate can help to determine a range of possible cost-effectiveness results.
The issue of multiple indications also needs special consideration in the case of reusable devices, as their overall cost effectiveness will be a composite measure of multiple cost-effectiveness ratios. Specifically, such a composite cost-effectiveness ratio would be a combination of the cost-effectiveness ratios and frequencies of all indications served [29]. In summary, upon deciding about the implementation of reusable devices, the challenge of attributing fixed costs and accounting for varying performance over indications must be met.
3.2.4 Dynamics of Effectiveness and Resource Use
The influence of the skill of the medical professional on the effectiveness of health technologies has attracted special interest in the past. Ramsay et al. [40] investigated the impact of individual and institutional learning curves and concluded that they are complicating factors for health technology assessment (HTA) studies. Besides baseline differences in experience and skill between clinicians, the aptitude in using a device is likely to change over time and at varying rates. This adds a dynamic element to the effectiveness of a medical device, reflecting the operator’s current position on a hypothetical learning curve. It is worth noting that learning effects can also apply to patients if device performance depends on their actions (e.g., the use of powered wheelchairs) [41]. The impact that individual and institutional learning can have on resource use has also been acknowledged [40, 42].
User skill, however, is not the only unstable factor in economic evaluations of medical devices. The ‘problem’ of learning is aggravated by the fact that devices in their early stages of development are often subject to continued modification and frequent product updates; this can also be seen even after their entry into clinical practice [43, 44]. The previously discussed early market diffusion and little field-testing typically mean that users often deal with an ‘unfinished’ product that relies on their feedback for further improvements. Frequent changes in medical device prices (or acquisition costs) pose yet another issue. Early market entry of competing technologies for devices, enabled by “building around the patent” [45], plus different pricing and procurement strategies than those seen in the pharmaceutical industry, result in a comparably short steady-state phase of medical device prices [29, 46]. Taken together, the dynamics of learning effects, product evolution and price instability contribute to a situation where the researcher has to estimate the effectiveness and cost effectiveness of a ‘moving target’ [43]. Viewed in that way, economic evaluations can have a limited shelf-life, just like the device being evaluated. One could therefore argue that re-evaluations at appropriate intervals (or suggested expiry dates) in the future be conducted [30]. Little research has been conducted on the determinants of cost effectiveness that change over time [40]. One study re-evaluated the cost effectiveness of a drug one decade after its initial assessment and found a more favourable estimate of cost effectiveness [47]. Changes in cost and outcome parameters over time are, without a doubt, not exclusive to medical devices. However, the number of varying factors and the possible occurrence of important changes within short periods of time make the generalizability of the results of economic evaluation particularly challenging for medical devices.
To summarize, four major device-level issues complicate the analysis of cost and effectiveness data of medical devices. These include: (1) inherent constraints to clinical study design, (2) the procedural integration of medical devices, (3) the reusability of medical devices, and (4) the dynamics of outcome and resource use parameters over time.
4 Summary and Possible Solutions
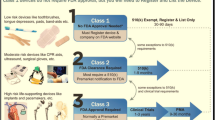
Based on an examination of external and device-level factors that complicate the assessment of health outcomes and resource use of medical devices, several challenges in undertaking an economic evaluation can be formulated. A summary of these challenges and their underlying factors is also provided in Fig. 1.

Challenges in undertaking economic evaluations of medical devices and their underlying factors
-
1.
Medical devices often have a fairly weak evidence base for an economic evaluation at the time of market entry. Consequently, when evaluating a new device, a balance has to be struck between data availability (and its quality) and the timeliness of an economic evaluation.
-
2.
The need to draw on data from observational and small or nonblinded experimental studies translates into greater parameter uncertainty and threatens both the validity and precision of cost-effectiveness calculations.
-
3.
Determinants such as user skill and organizational factors can confound the generalizability of economic evaluations due to variation in these determinants between healthcare settings.
-
4.
A sufficiently valid and precise cost analysis can be a complex – if not impossible – undertaking due to the number and shared character of relevant cost items associated with device use.
-
5.
The question about whether to include process utility in the outcome assessment of a medical device warrants consideration.
-
6.
In the case of reusable devices, the attribution of fixed costs and varying clinical effectiveness and utilization rates for different target indications have to be considered.
-
7.
Temporal changes in the determinants of device-related outcomes and costs mean that estimates of cost effectiveness can have a limited shelf-life.
What do these challenges imply for the decision maker and how can they be addressed? We have seen that in many cases the data on clinical performance and resource use of medical devices might euphemistically be described as imperfect. Under such circumstances, the decision maker has two options when it comes to reimbursing or implementing a new medical device: (1) to make an immediate decision based on the limited evidence available; or (2) to delay the decision until important evidence gaps have been filled.
It should be clear that an economic evaluation based on poor quality clinical studies may lead to an underestimate or overestimate of the true cost effectiveness of a device. However, the availability of good quality data will not be sufficient if the quantity of data on a device is too small (number of patients, measurements or clinical events), there will be substantial uncertainty about its cost effectiveness, which in turn could result in important uncertainty about whether or not to reimburse it. Two unfavourable scenarios are imaginable: an incorrect decision to reimburse the device and an incorrect decision to reject it. Both decisions will result in inefficient use of resources, and possibly health loss, through diversion of funds away from more cost-effective interventions. One way to assist decision makers would be to perform an expected value of perfect information (EVPI) analysis to decide whether it is worthwhile to collect certain additional evidence to reduce uncertainty about cost effectiveness; if generating additional data is deemed not to be worth the money, then a decision does not have to be delayed [48, 49]. However, it could be argued that this EVPI approach needs revision, since many believe that reimbursement decisions should be based on more than just cost effectiveness.
The generation of additional evidence will often require the medical device to enter clinical use. This could be managed by means of conditional coverage solutions such as risk-sharing agreements between payer and producer, outcome-based reimbursement schemes and volume or target population restrictions [50]. Regardless of the strategy chosen, the initial implementation and reimbursement decision can be made conditional on evidence development to enable the reassessment of effectiveness and cost effectiveness later on (coverage with evidence development, CED) [48, 50]. The decision about whether or not to continue reimbursement or change the terms of reimbursement (e.g., a lower price) would depend on the findings of that reassessment. However, the chance that conditional coverage of a device would ever be considered by payers is dependent on various factors relating to the disease indication, device, organization and stakeholder preferences in general. In these times of budget cuts, it might be safe to assume that the potential ability to reduce short-term costs or to improve health outcomes quickly would certainly improve the chance. There are, of course, other ways than conditional coverage to facilitate the assessment of a device. For example, both payers and industry are keen to appraise the value of devices as soon as possible in their development. For this reason, ‘early HTA’ techniques such as technology horizon scanning, early warning systems and iterative technology assessment (e.g., the IDEAL method [51, 52]) are increasingly being used in anticipation of early market diffusion and limited time for evidence development [53, 54]. In contrast to the reassessment of a device after market entry, they try to establish an early grip on the likely future performance of a technology, often long before implementation, and estimate its possible impact on healthcare. Originally applied by the industry to identify promising technologies worthy of further investment, the value of ‘early HTA’ for health policy making has been recognized as well. ‘Rapid HTA’ is yet another instrument to deal with the narrow time frame and evidence limitations associated with the economic evaluation of medical devices. Here, feasibility and timeliness of an assessment are given priority over comprehensiveness [55].
It is worth noting that all approaches to dealing with the external factors challenging device evaluation reflect the dilemma for decision makers that has been created by the current regulatory situation. The problem of granting access to new medical technologies despite uncertainty about their true clinical and economic value would ideally require a more radical solution, possibly using pharmaceutical legislation as a reference point. In light of recent high profile incidents such as defective Sprint Fidelis defibrillator leads and the Poly Implant Prothèse (PIP) breast implant scandal [56, 57], it appears more than questionable whether current device regulation can justifiably be sustained from a mere safety standpoint. With a history of “unfortunate events [having] catalysed the development of medicines regulation” [58], the case of thalidomide-related birth defects serving as the most prominent example, it remains to be seen whether the same will occur for medical devices.
In light of these considerations, one could argue that the challenges for economic evaluations caused by device-level factors are not as influential as the very prominent issue of too little clinical evidence. Nevertheless, a few points warrant extra consideration even though the solutions for several of them are rather straightforward.
Regarding the constraints to study design, an iterative approach to clinical assessment called the IDEAL method has been recommended for medical devices, which starts at an early point in product development and uses varying study designs [51, 52]. While such an approach is not suited to addressing the difficulties in designing and conducting experimental trials, it provides guidance on the appropriate method, size and setting for clinical studies at each stage of product development. This step-by-step approach can help to identify major sources of uncertainty about effectiveness early enough to enable a tailored approach for subsequent investigation.
As far as procedural integration is concerned, every effort should be made to identify and measure all important determinants of device effectiveness and resource use. Transparent and exhaustive reporting of the measurement methods can assist the later adjustment of variable determinants when transferring the cost-effectiveness results from one clinical setting to another.
For cost estimations, an up-front sensitivity analysis employing reasonable assumptions about the true values of different cost parameters would help to estimate the impact of these parameters on total costs and decide whether a more detailed assessment is worthwhile [42]. If deemed necessary, one could determine at a later time whether the values used in the evaluation differ substantially from those used in the original evaluation.
When it comes to addressing the temporal changes in input parameters that influence the cost effectiveness of a device, some parameters will be more challenging than others. While changes in product price can easily be corrected for if they are known, changes in user proficiency might be difficult to measure. Identification and comprehensive documentation of the values of dynamic input parameters at the time of assessment is an essential first step in resolving this problem. It is very important to distinguish baseline differences in user proficiency from the concept of learning effects. Whereas baseline differences in proficiency make it difficult to estimate the true level of device effectiveness across users, learning effects will affect an estimate of effectiveness (for both a single user and an organization) over time. If the other determinants are known, the impact of individual skills on device performance and its change over time can in principle be assessed and quantified by regression analysis [40]. The shape of the learning curve can also be approximated in such an analysis. The cost effectiveness of a medical device could then be determined for a particular user or set of users, both at the time of market launch and after reaching certain learning benchmarks (e.g., based on time since launch or on number of units used). Another way to address learning effects would be to repeatedly assess the outcomes achieved by the same users at certain points in time to estimate the shape of the learning curve and the temporal change in cost effectiveness [40]. The positive side effects of measuring user performance over time include the identification of mistake-prone device-related activities and the ability to make recommendations on the focus, optimal timing and duration of training.
5 Discussion
The challenges to be faced when conducting or interpreting economic evaluations of medical devices are manifold and it is clear that they translate into important caveats for HTA researchers and policy makers in healthcare. Some of the factors causing these challenges are simply unavoidable (e.g., procedural integration, reusability) and HTA researchers and others will have to develop the best ways to deal with them. Other factors, particularly the regulatory framework for medical devices, could be directly addressed by regulatory authorities, industry, and HTA methodologists; the failure to do so will mean that HTA researchers will have to find the best ways to address these challenges as well.
An important shortcoming of this work is rooted in the diversity of medical devices. As explained earlier, medical devices can be distinguished on the basis of a variety of attributes. As these attributes vary between devices, so does the impact of the challenges presented here. Certain challenges may even be completely irrelevant for some medical devices. While an examination spanning the entire domain of medical devices enabled a comprehensive analysis of issues relating to their economic evaluation, our findings cannot be generalized, since the properties of the device under investigation determine which challenges will have to be faced for an economic evaluation. If a device involves an invasive intervention requiring a high level of user proficiency and organizational adaptations, its economic evaluation will obviously face greater challenges than a non-invasive device with less user involvement. Similarly, devices in a late stage of product development will serve as a comparably ‘steady target’ for evaluation and will likely have a better evidence base than new innovations with little market and user experience.
Given the broad spectrum of devices that exist, a systematic review of the literature on economic evaluations of medical devices was deemed unfeasible since it would have required an examination of all economic evaluations of all medical devices. The factors and challenges discussed in our review should be explored in specific areas in healthcare where devices are used. Future research should focus on specific device classes or specific challenges and thus, by narrowing the scope, make a more systematic approach possible. A different approach would be to examine published systematic reviews of all economic evaluations of one specific medical device (e.g., coronary stents). This would effectively be a review of reviews, which would enable us to see how the challenges discussed in this paper could be amended or modified and, more importantly, to see what can be done to address these challenges to improve the quality of economic evaluations of specific medical devices.
It should be stated that several of the challenges discussed in this paper are not just relevant for evaluations of medical devices, but also relevant for assessments of drugs or medical procedures. However, since the aim of this work was to provide a comprehensive rather than exclusive analysis of device-related challenges, and since medical devices can combine properties of both drugs and procedures, we believe that certain overlaps have to be expected and accepted.
The list of challenges described here can serve as a reference for what should be considered when planning or interpreting an economic evaluation of a medical device. While some problems, such as the complexity of costing or the identification and measurement of determinants of a device’s effectiveness, may be resolvable in the assessment phase, many will not. We argue that the limitations to the availability and quality of evidence on the clinical performance of medical devices pose the most fundamental challenges for their economic evaluation. Indeed, the regulatory controls for medical devices to obtain market approval have repeatedly come under criticism, also with respect to their influence on the feasibility of HTA studies [12, 34, 59]. All in all, an economic evaluation that has properly addressed all methodological challenges cannot make up for all of the shortcomings in the underlying evidence base. At best, the results of such an evaluation could be used to clarify the aims and value of future research [48].
Thus, if lack of data constitutes the primary obstacle of an economic evaluation, the question arises as to whether medical devices that lack comprehensive clinical evidence should undergo an economic evaluation. Taylor and Iglesias [60] therefore argue that the reimbursement of medical devices and pharmaceuticals is subject to the same budget constraints and should therefore meet the same requirements for appraisal. However, as we have seen, the clinical evidence requirements that drugs and devices have to meet in order to obtain market approval are obviously not the same. If concerns about patient safety are not sufficient to legally force device manufacturers to provide clinical evidence of comparable quality, concerns about the cost effectiveness of their products might serve as an additional motivation in light of tightening healthcare budgets.
It has been stated that “no [health economic] study is likely to be completely free of flaws, so it is important that study users know how to detect them.” [61] This advice is of particular significance regarding the assessment of medical devices. An increased awareness of these challenges will help in detecting possible weaknesses of economic evaluations and enable the correct interpretation of their results. More importantly, greater awareness might serve as an impulse to formulate necessary changes at the regulatory level, to revisit methodological approaches to economic evaluations of medical devices and to optimize innovative reimbursement strategies in the future.
6 Conclusions
The economic evaluation of medical devices is aggravated by a variety of factors that stem partly from the regulatory and market environment and partly from functional device-specific characteristics. One central factor is the frequent lack of high-quality evidence on clinical performance, which severely hampers efforts to obtain valid estimates of incremental effectiveness and cost effectiveness. After all, if the clinical benefit and harm of a medical device are insufficiently understood, how much confidence can we place in statements about its cost effectiveness? Current medical device regulation can arguably be labelled as the principal origin of clinical evidence gaps. While device-related challenges could be addressed by rigorous documentation, regular reassessment and some methodological adaptations, the challenges caused by relatively few regulatory requirements would ideally be met at a different level. A very fundamental change, possibly using drug legislation as a paradigm, may be necessary to close the aforementioned evidence gaps, thereby enabling a comprehensive and valid assessment of each medical device’s benefit/risk balance and ultimately its cost effectiveness.
Notes
The Global Harmonization Task Force was formed by national authorities and industrial representatives from the EU, US, Canada, Australia and Japan, with the aim of introducing international uniformity concerning the regulatory approval procedures for new medical devices.
Reference
The European Commission. Medical devices competitiveness—facts and figures (online). http://ec.europa.eu/health/medical-devices/competitiveness/facts-figures/index_en.htm. Accessed 28 Aug 2012.
U.S. Food and Drug Administration. Medical device registration (online). http://www.accessdata.fda.gov/premarket/ftparea/Registration.zip. Accessed 28 Aug 2012.
Fraser AG, Daubert JC, Van de Werf F, et al. Clinical evaluation of cardiovascular devices: principles, problems, and proposals for European regulatory reform: report of a policy conference of the European Society of Cardiology. Eur Heart J. 2011;32(13):1673–86.
U.S. Food and Drug Administration. Is the product a medical device? (online). http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/ClassifyYourDevice/ucm051512.htm. Accessed 28 Aug 2012.
The European Commission. Directive 2007/47/EC of the European Parliament and of the Council of 5 September 2007 amending Council Directive 90/385/EEC on the approximation of the laws of the Member States relating to active implantable medical devices, Council Directive 93/42/EEC concerning medical devices and Directive 98/8/EC concerning the placing of biocidal products on the market.
GHTF Study Group 1 on behalf of the Global Harmonization Task Force. Information document concerning the definition of the term “Medical Device” (online). 2005 Jul. http://www.imdrf.org/docs/ghtf/final/sg1/technical-docs/ghtf-sg1-n29r16-2005-definition-medical-device-050520.pdf. Accessed 6 Dec 2012.
GHTF Study Group 1 on behalf of the Global Harmonization Task Force. Principles of Medical Devices Classification (online). 2006 Aug. http://www.imdrf.org/docs/ghtf/final/sg1/technical-docs/ghtf-sg1-n15-2006-guidance-classification-060627.pdf. Accessed 6 Dec 2012.
Drummond MF, Sculpher MJ, Torrance GW, et al. Methods for the economic evaluation of health care programmes. 3rd ed. Oxford: Oxford University Press; 2005.
Oxford Centre for Evidence Based Medicine (CEBM). Levels of evidence (online). 2009 Mar. http://www.cebm.net/index.aspx?o=1025. Accessed 28 Aug 2012.
Ramsey SD, Luce BR, Deyo R, et al. The limited state of technology assessment for medical devices: facing the issues. Am J Manag Care. 1998;4(Spec. No.):SP188–99.
Hartling L, McAlister FA, Rowe BH, et al. Challenges in systematic reviews of therapeutic devices and procedures. Ann Intern Med. 2005;142(12 Pt 2):1100–11.
Cohen D, Billingsley M. Europeans are left to their own devices. BMJ. 2011;342:d2748. doi:10.1136/bmj.d2748.
Hulstaert F, Neyt M, Vinck I, et al. The pre-market clinical evaluation of innovative high-risk medical devices. Health Services Research (HSR) Brussels; Belgian Health Care Knowledge Centre (KCE) 2011. KCE Report 158C. D/2011/10.273/31.
GHTF Study Group 5 on behalf of the Global Harmonization Task Force. Clinical Investigations (online). 2010 Feb. http://www.imdrf.org/docs/ghtf/final/sg5/technical-docs/ghtf-sg5-n3-clinical-investigations-100212.pdf. Accessed 6 Dec 2012.
Feigal DW, Gardner SN, McClellan M. Ensuring safe and effective medical devices. N Engl J Med. 2003;348(3):191–2.
Kaplan AV, Baim DS, Smith JJ, et al. Medical device development: from prototype to regulatory approval. Circulation. 2004;109(25):3068–72.
Siebert M, Clauss LC, Carlisle M, et al. Health technology assessment for medical devices in Europe: what must be considered. Int J Technol Assess Health Care. 2002;18(3):733–40.
Mowatt G, Bower DJ, Brebner JA, et al. When and how to assess fast-changing technologies: a comparative study of four generic technologies. Health Technol Assess. 1997;1(14):1–149.
Robert G, Stevens A, Gabbay J. ‘Early warning systems’ for identifying new healthcare technologies. Health Technol Assess. 1999;3(13):1–108.
U.S. Food and Drug Administration. Medical device recalls (online). http://www.fda.gov/MedicalDevices/Safety/ListofRecalls/default.htm. Accessed 28 Aug 2012.
Zuckerman DM, Brown P, Nissen SE. Medical device recalls and the FDA approval process. Arch Intern Med. 2011;171(11):1006–11.
Wilmshurst P. The regulation of medical devices. BMJ. 2011;342:d2822. doi:10.1136/bmj.d2822.
Cohen D. Out of joint: the story of the ASR. BMJ. 2011;342:d2905. doi:10.1136/bmj.d2905.
Lilford RJ, Jackson J. Equipoise and the ethics of randomization. J R Soc Med. 1995;88(10):552–9.
O’Brien B. Economic evaluation of pharmaceuticals: Frankenstein’s monster or vampire of trials? Med Care. 1996;34(12 Suppl.):DS99–108.
Macklin R. The ethical problems with sham surgery in clinical research. N Engl J Med. 1999;341(13):992–6.
Sorenson C, Tarricone R, Siebert M, et al. Applying health economics for policy decision making: do devices differ from drugs? Europace. 2011;13(Suppl. 2):ii54–8.
Black N. Why we need observational studies to evaluate the effectiveness of health care. BMJ. 1996;312(7040):1215–8.
Drummond M, Griffin A, Tarricone R. Economic evaluation for devices and drugs-same or different? Value Health. 2009;12(4):402–4.
Sculpher MJ, Pang FS, Manca A, et al. Generalisability in economic evaluation studies in healthcare: a review and case studies. Health Technol Assess. 2004;8(49):1–192.
Heemskerk J, van Dam R, van Gemert WG, et al. First results after introduction of the four-armed da Vinci Surgical System in fully robotic laparoscopic cholecystectomy. Dig Surg. 2005;22(6):426–31.
Raftery J. Costing in economic evaluation. BMJ. 2000;320(7249):1597.
Donaldson C, Shackley P. Does “process utility” exist? A case study of willingness to pay for laparoscopic cholecystectomy. Soc Sci Med. 1997;44(5):699–707.
Cookson R, Hutton J. Regulating the economic evaluation of pharmaceuticals and medical devices: a European perspective. Health Policy. 2003;63(2):167–78.
Birch S, Melnikow J, Kuppermann M. Conservative versus aggressive follow up of mildly abnormal Pap smears: testing for process utility. Health Econ. 2003;12(10):879–84.
Redekop WK. Cost-effectiveness analyses of diagnostic strategies: a literature survey using the NHS Economic Evaluation Database. Expert Rev Pharmacoecon Outcomes Res. 2006;6(1):41–8.
Sassi F, McKee M, Roberts JA. Economic evaluation of diagnostic technology: methodological challenges and viable solutions. Int J Technol Assess Health Care. 1997;13(4):613–30.
Kertesz A, Black SE, Nicholson L, et al. The sensitivity and specificity of MRI in stroke. Neurology. 1987;37(10):1580–5.
Maeso S, Reza M, Mayol JA, et al. Efficacy of the Da Vinci surgical system in abdominal surgery compared with that of laparoscopy: a systematic review and meta-analysis. Ann Surg. 2010;252(2):254–62.
Ramsay CR, Grant AM, Wallace SA, et al. Statistical assessment of the learning curves of health technologies. Health Technol Assess. 2001;5(12):1–79.
Sorrento GU, Archambault PS, Routhier F, et al. Assessment of Joystick control during the performance of powered wheelchair driving tasks. J Neuroeng Rehabil. 2011;8:31.
Brouwer W, Rutten F, Koopmanschap M. Costing in economic evaluations. In: Drummond M, McGuire A, editors. Economic evaluation in health care. Oxford: Oxford University Press; 2001. p. 68–93.
Fineberg HV. Technology assessment: motivation, capability and future directions. Med Care. 1985;23(5):663–71.
Raab GG, Parr DH. From medical invention to clinical practice: the reimbursement challenge facing new device procedures and technology. Part 1: Issues in medical device assessment. J Am Coll Radiol. 2006;3(9):694–702.
United States International Trade Commission. Medical Devices and equipment: competitive conditions affecting U.S. trade in Japan and other principal foreign markets (online). 2007 Mar. http://www.usitc.gov/publications/332/pub3909.pdf. Accessed 28 Aug 2012.
Brown A, Meenan BJ, Dixon D, et al. Application of the experience curve to price trends in medical devices: implications for product development and marketing strategies. J Med Mark. 2008;8(3):241–55.
Remák E, Hutton J, Jones M, et al. Changes in cost-effectiveness over time: the case of Epoetin Alfa for renal replacement therapy patients in the UK. Eur J Health Econ. 2003;4(2):115–21.
Sculpher M, Claxton K. Establishing the cost-effectiveness of new pharmaceuticals under conditions of uncertainty: when is there sufficient evidence? Value Health. 2005;8(4):433–46.
Claxton K, Sculpher M, Drummond M. A rational framework for decision making by the National Institute For Clinical Excellence (NICE). Lancet. 2002;360(9334):711–5.
Trueman P, Grainger DL, Downs KE. Coverage with evidence development: applications and issues. Int J Technol Assess Health Care. 2010;26(1):79–85.
McCulloch P. Wanted: an appropriate evaluation template. BMJ. 2011;342:d3540. doi:10.1136/bmj.d3540.
McCulloch P, Altman DG, Campbell WB. No surgical innovation without evaluation: the IDEAL recommendations. Lancet. 2009;374(9695):1105–12.
Wild C, Langer T. Emerging health technologies: informing and supporting health policy early. Health Policy. 2008;87(2):160–71.
Ijzerman MJ, Steuten LM. Early assessment of medical technologies to inform product development and market access: a review of methods and applications. Appl Health Econ Health Policy. 2011;9(5):331–47.
Watt A, Cameron A, Sturm L, et al. Rapid reviews versus full systematic reviews: an inventory of current methods and practice in health technology assessment. Int J Technol Assess Health Care. 2008;24(2):133–9.
Mehrotra AK, Knight BP, Smelley MP, et al. Medtronic Sprint Fidelis lead recall: determining the initial 5-year management cost to Medicare. Heart Rhythm. 2001;8(8):1192–7.
Berry MG, Stanek JJ. The PIP mammary prosthesis: a product recall study. J Plast Reconstr Aesthet Surg. 2012;65(6):697–704.
Rägo L, Santoso B. Drug regulation: history, present and future. In: van Boxtel CJ, Santoso B, Edwards IR, editors. Drug benefits and risks: international textbook of clinical pharmacology. revised 2nd ed. IOS Press and Uppsala Monitoring Centre; 2008. p. 65–77.
Freemantle N. Commentary: evaluating and regulating device therapy. BMJ. 2011;342:d2839. doi:10.1136/bmj.d2839.
Taylor RS, Iglesias CP. Assessing the clinical and cost-effectiveness of medical devices and drugs: are they that different? Value Health. 2009;12(4):404–6.
Drummond M, Sculpher M. Common methodological flaws in economic evaluations. Med Care. 2005;43(7 Suppl.):5–14.
Acknowledgments
The authors would like to thank Sarowar Golam and Anna Renner for their valuable comments on the concept, structure and content of an earlier version of this work. We are also grateful for the comments and recommendations made by the three anonymous reviewers and the editor, who helped to improve the quality of the paper.
Disclosures
No sources of funding were used to prepare this manuscript. The authors have no conflicts of interest that are directly relevant to the content of this article.
Author contributions
A.K. and W.K.R. contributed equally to the conceptual design of this work. A.K. identified relevant literature and provided an original draft. W.K.R. amended and finalized the original draft. A.K. and W.K.R. contributed equally to the revision of the paper after independent peer review. A.K. is guarantor of this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kirisits, A., Redekop, W.K. The Economic Evaluation of Medical Devices. Appl Health Econ Health Policy 11, 15–26 (2013). https://doi.org/10.1007/s40258-012-0006-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40258-012-0006-9