
Abstract
Acute myocardial infarction (AMI) is associated with high morbidity and mortality worldwide. Although early reperfusion is the most effective strategy to salvage ischemic myocardium, reperfusion injury can develop with the restoration of blood flow. Therefore, it is important to identify protection mechanisms and strategies for the heart after myocardial infarction. Recent studies have shown that multiple intracellular molecules and signaling pathways are involved in cardioprotection. Meanwhile, device-based cardioprotective modalities such as cardiac left ventricular unloading, hypothermia, coronary sinus intervention, supersaturated oxygen (SSO2), and remote ischemic conditioning (RIC) have become important areas of research. Herein, we review the molecular mechanisms of cardioprotection and cardioprotective modalities after ischemia–reperfusion injury (IRI) to identify potential approaches to reduce mortality and improve prognosis in patients with AMI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
At present, myocardial infarction still has a high incidence of a disease and mortality. The area of myocardial infarction is often closely related to ischemia–reperfusion injury. |
This review summarizes the mechanisms by which ischemia–reperfusion occurs and strategies to protect the heart by reducing defect reperfusion injury. |
This provides a new approach to reduce the size of myocardial infarction and reduce mortality. |
1 Introduction
Prolonged myocardial necrosis, which arises as a result of coronary artery occlusion and death of myocardial tissue, is associated with cardiogenic shock and risk of death. Acute myocardial infarction (AMI) causes more than 15% of all deaths worldwide each year, with a higher incidence in men than women [1]. Currently, the mainstay of treatment for AMI is early and effective reperfusion of the infarcted artery, such as thrombolytic therapy and percutaneous coronary intervention (PCI). However, timely reperfusion is often accompanied by non-negligible reperfusion injury.
Evidence shows that myocardial infarct size, as measured by cardiac magnetic resonance (CMR) imaging or technetium-99m sestamibi single-photon emission computed tomography (SPET) within 1 month after AMI, was strongly associated with all-cause mortality and hospitalization for heart failure at 1 year [2]. Therefore, scholars are focusing their attention on cardioprotection. Interestingly, a variety of molecules and signaling pathways in organisms may be involved in cardioprotection, such as NLRP3, miRNAs, and extracellular vesicles (EVs). These molecules and signaling pathways are worth exploring. In addition, extensive research has been conducted into adjuvant treatment modalities for the heart after myocardial infarction. Cardioprotective modalities such as left ventricular unloading, myocardial cooling, supersaturated oxygen (SSO2), and stimulation of the vagus nerve are all being explored. In this review, we address the mechanisms of ischemia–reperfusion injury (IRI) development and cardioprotective strategies.
2 Molecular Mechanisms of Cardioprotection
Many molecules and signaling pathways are involved in the process of cardioprotection. Figure 1 lists three signaling pathways involved in cardioprotection, including the reperfusion injury salvage kinase (RISK) pathway, the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, and the NO/cGMP/PKG pathway. All of these pathways converge to the mitochondria, acting through the opening of the mitochondrial ATP-sensitive potassium channel (mKATP) and the mitochondrial permeability transition pore (mPTP). Reviewed below are important molecules involved in cardioprotection [3,4,5].

The main protection mechanisms in the IR injury. There are three major pathways : a the reperfusion injury salvage kinase (RISK) pathway, including phosphatidylinositol 3-kinase/Akt (PI3K/Akt), extracellular signal-regulated kinase (ERK1/2), and the downstream target glycogen synthase kinase-3 beta (GSK3β); b the NO/cGMP/PKG pathway, where NO stands for nitric oxide, cGMP for cyclic guanosine monophosphate, and PKG for protein kinase G; and c the survivor activating factor enhancement (SAFE) pathway, including the tumor necrosis factor alpha (TNFα) and the transcription factor transducer and activator of transcription-3 (STAT3). All of these pathways converge to the mitochondria, acting through the opening of the mitochondrial ATP-sensitive potassium channel (mKATP) and the mitochondrial permeability transition pore (mPTP).
2.1 Cytosolic Molecules: SUMOyation
Small ubiquitin-like modifier (SUMO) is a 10-kDa polypeptide. SUMO1–3 are ubiquitously expressed, whereas SUMO4 is limited to lymph node, kidney, and spleen [6]. The role of SUMO1 in cardiac function was tested in myocardium-specific SUMO1-expressing transgenic mice, and the results showed that SUMO1 overexpression inhibited pressure overload-induced cardiac hypertrophy and dysfunction, suggesting a cardioprotective role for SUMO1 [7]. SUMOylation is a lysine-targeted post-translational modification (PTM) [8, 9]. It is becoming increasingly apparent that many SUMO chemical molecules are localized in the extranuclear compartment and that they are involved in the regulation of protein functions, including intracellular trafficking, apoptosis, protein stability, and enzyme activity [10,11,12].
2.1.1 NF-κB
Nuclear factor (NF)-κB is a master regulator of cell survival and inflammation and plays an important role in various cardiac pathogenesis including ischemic preconditioning [13]. When cells are stimulated by agonists such as tumor necrosis factor (TNF), NF-κB dissociates from inhibitor of κB (IκB) and translocates into the nucleus and transactivates proinflammatory genes [14]. SUMOylation has been reported to be involved in different levels of NF-κB regulation. IκBα can be modified by SUMO1 to protect it from ubiquitination and degradation, thereby limiting NF-κB activation [15, 16]. IκB kinase, an important regulatory kinase complex that regulates NF-κB signaling, consists of two kinases (IKKα and IKKβ) and a regulatory subunit, nuclear factor-κB essential modulator (NEMO). It has been reported that depletion of NEMO in cardiomyocytes promotes apoptosis and subsequent cardiac dysfunction via inhibiting the expression of antioxidant genes such as superoxide dismutase 2 and ferritin heavy chain [17, 18]. In summary, activation of NEMO/IKKγ–NF-κB signaling can be cardioprotective by inhibiting apoptosis.
2.1.2 Protein Kinase C (PKC)
PKC contains multiple putative SUMOylation sites. Inactive PKCα is SUMOylated at the Lys465 site which can be de-SUMOylated by sentrin-specific peptidase1 (SENP1). It was shown that PKCα activation by calcium was achieved only after de-SUMOylation by SENP1 [19]. This suggests that SUMOylation can play an inhibitory role in PKCα kinase function. It is well known that PKCα negatively regulates cardiomyocyte contraction and that three specific extracellular PKCα substrates play a role in this regard [20]. First, PKCα phosphorylates inhibitor 1 (I-1) at Ser67, which upregulates protein phosphatase 1 activity, leading to more phosphatidylcholine (PLN) dephosphorylation and reduced sarcoplasmic reticulum (SR) Ca2+ ATPase (SERCA2) pump activity [21]. Second, PKCα activation increases G protein-coupled receptor kinase 2 (GRK2) phosphorylation and activity and impairs β-agonist-stimulated ventricular function via abolishing cyclase activity [22]. Lastly, PKCα can phosphorylate cardiac troponin I (cTnI), cTnT, titin, and myosin binding protein C, the effect of which is to decrease the Ca2+ sensitivity and contractility of cardiomyocytes [23,24,25,26]. In conclusion, inhibition of PKCα kinase activity by SUMOylation can be cardioprotective.
2.1.3 Adenosine Monophosphate-Activated Protein Kinase (AMPK) and Ubiquitin–Proteasome System (UPS)
AMPK is a stress-activated kinase, which can orchestrate the cellular response to a variety of stresses in the heart by regulating metabolism, protein synthesis, degradation, autophagy, and apoptosis. AMPK is a complex of three subunits: a catalytic subunit (α) containing a serine-threonine kinase domain (KD) with a Thr172 phosphorylation site, which is the target of liver kinase B1 (LKB1), and calcium–calmodulin-activated protein kinase kinase-β (CAMKKβ) and two regulatory subunits (β and γ). Most studies have shown that endogenous AMPK activation is protective against cardiac insults, including ischemia/reperfusion and pressure overload [27,28,29,30]. Rubio et al. reported that AMPK SUMOylation (SUMO2) stimulates AMPK activation and inhibits its ubiquitin-dependent degradation [31]. In addition, Yeh’s group recently found that LKB1 K178 SUMO1 modification promotes LKB1 binding to AMPKα SIM and accelerates AMPK activation [32]. These data suggest that the LKB1–AMPK SUMOylation can be cardioprotective. Robbins and his colleagues showed that depletion of Ubc9, the SUMO E2 conjugating enzyme, in cardiomyocytes caused accumulation of protein aggregates inside these cells and impaired cardiac function [33]. In addition, they found that Ubc9-mediated SUMOylation increased autophagy, thereby reducing protein aggregate formation, fibrosis, and hypertrophy, while improving cardiac function and survival [34]. In conclusion, most of the cardiac extranuclear SUMOylation events are cardioprotective against cardiac damage.
2.2 Mitochondrial Molecules
Mitochondria are essential organelles for the proper functioning of cardiac muscle cells and are known as the “energy processing plant.” In addition, mitochondria are involved in many important processes such as cell differentiation, information transfer, and apoptosis. Impaired mitochondrial function is an important cause of IRI [35,36,37,38]. Therefore, mitochondria can be an important research target for cardioprotective mechanisms. C. Penna et al. demonstrated that direct targeting of mitochondria with diazoxide activates the RISK pathway via a redox signaling, favors discretemitochondrial protein S-nitrosylation, and decreases signals of death [39]. Furthermore, by comparing exercised (5 days/week for 5 weeks) and sedentary Wistar rats, Doria Boulghobra et al. found that exercise training increases nitric oxide (NO) bioavailability in mitochondria, results in SNO of key proteins involved in the mitochondrial response to stress, and modulates Ca2+-dependent mPTP opening and reactive oxygen species (ROS) production in conditions that mimic ischemia–reperfusion [40]. All of these can demonstrate that mitochondria are involved in the process of IRI and that they can be targeted for cardioprotection after IRI.
2.3 NLRP3
The nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) are able to identify various damage-associated molecular patterns (DAMPs) and inflammatory factors, such as those released during ischemia–reperfusion. In particular, NLRP3 has been widely investigated in the setting of cardiac ischemia–reperfusion. The very low levels of NLRP3 expression in the healthy heart remain unchanged up to 3 h after reperfusion, while a significant increase has been detected by 6 h [41, 42]. The NLRP3 inflammasome is involved in many important processes, such as the efflux of cellular potassium, the release of cathepsin from damaged lysosomes, metabolic and mitochondrial dysfunction, Ca2þ-induced calpain activation resulting in the release of caspase-1 from actin, and impaired autophagy/mitophagy [43, 44]. The most important thing is that deletion of NLRP3 protein inhibits the ischemic preconditioning in an NLRP3 inflammasome-independent manner through an IL-6/STAT3-dependent mechanism [45]. Several experimental results indicate that the activation of NLRP3 inflammasome after reperfusion may concur with the progression of cardiac IRI, promoting cardiomyocyte loss by inducing pyroptosis, and favoring adverse remodeling by inducing the release of interleukin (IL)-1b and IL-18. However, reduced infarct size and improved cardiac function have been observed after inhibition or deficiency of one of the components of the NLRP3 inflammasome or using several NLRP3 inhibitors and gene silencing [46,47,48,49]. Interestingly, the fact that ischemic preconditioning (IPreC) was ineffective in NLRP3-deficient mice suggests that NLRP3 may play a double role in the ischemia–reperfusion heart, being able to trigger protective signaling and participating in IPreC, but also able to concur with the progression of cardiac injury and promoting cardiomyocyte loss after ischemia–reperfusion [50]. Furthermore, more about the inflammasome-independent role of NLRP3 and inflammasome protective signaling in AMI is described in detail in another review article [51, 52]. In conclusion, most of the studies proved that NLRP3 has a cardioprotective effect on the heart, and some of them suggested that its effect is two-sided. With in-depth research, scholars are becoming more aware of the complexity of the mechanism, which requires further experiments to confirm.
2.4 MicroRNA
Numerous studies have shown that microRNAs may regulate cytoprotective mechanisms and exert cardioprotection against ischemia–reperfusion [53]. Multiple microRNAs are described as key regulators of cardiocytoprotection and improvement of cardiac function after AMI. Among these microRNAs, miR-1 antagomir delivery exerted a significant protective effect on heart function, decreasing cardiomyocyte apoptosis and alleviating myocardial fibrosis and remodeling in the mouse model of MI. A study has shown that miR-1 antagomir decreased 19s proteasome, 20S proteasome, and ubiquitin ligase E3 levels, which play a pivotal role in the selective recognition and degradation of oxidized proteins. Meanwhile, both miR-21 and miR-146a showed a protective role against hypoxia-induced myocardial apoptosis and inflammation in the context of IRI [54,55,56,57]. In addition, miR-125b has been studied well, and knockdown of miR-125b-5p after transfection of its inhibitor results in enhanced post-AMI mortality and left ventricular dysfunction in a mouse model of AMI [58]. In summary, microRNAs have been proven to be involved in the mechanism of IR injury and cardioprotection, and mimics or antagomirs of certain microRNAs may serve as potential multitarget drugs for several cardiovascular pathologies.
2.5 mitochondrial-Derived Vesicles (MDVs) and EVs
Mitochondria are highly dynamic organelles; their function is crucial for maintaining cellular homeostasis. Mitochondrial dysfunction and dysregulation of mitochondrial remodeling processes play critical roles in the pathogenesis of cardiovascular disease [59]. Recently, mitochondrial-derived vesicles (MDVs) have been identified as a novel mechanism in mitochondrial quality control [60]. In cardiomyocytes, the formation of MDVs is a physiological process that is accelerated by oxidative stress [61]. MDVs emerge as an essential quality control mechanism in response to mitochondrial stress, in addition to mitophagy and fission/fusion processes that regulate mitochondrial turnover. MDVs may play a role in the cellular response to hypoxia. MDV formation in rat cardiomyocytes increased initially under hypoxic conditions, but decreased with prolonged hypoxia [62]. Doxorubicin, a chemotherapeutic agent known for its cardiotoxic potential [63], increased the generation of TOMM20+/PDH− and TOMM20−/PDH+ MDVs in rat cardiomyocytes. Doxorubicin-treated C57BL/6 mice exhibited a more than two-fold increase in cardiac MDV budding compared with control mice. MDV formation in the heart was observed under basal conditions and can be regarded as a physiologic process induced in response to cellular stressors, such as oxidative stress, which further modulates MDV cargo loading. Unfortunately, the role of MDVs in human cardiomyocytes or heart tissue, as well as the vascular system, is still unknown [64].
EVs are membrane-bound particles secreted from cells that carry biomolecules, such as proteins, nucleic acids, and lipids [65, 66]. Several studies have found mitochondrial content in EVs, establishing a link between EVs and MDVs. Interestingly, EVs may aid in mitochondrial uptake. Mitochondria-containing EVs improved bioenergetics in hypoxia-injured cardiomyocytes and left ventricular function in a mouse model of myocardial infarction [67]. This was not observed after injection of isolated naked mitochondria, strengthening the idea that vesicular delivery increases mitochondrial uptake. The therapeutic approach of transferring functional mitochondria to damaged cells or tissue, also known as mitochondrial transplantation, is gaining increasing attention [64]. Large animal and clinical trials should be conducted in the future to assess the safety and efficacy of EV-mediated mitochondrial transfer.
3 Cardiac Protection Strategies
3.1 Left Ventricular Unloading of the Heart
Burhoff defines acute left ventricular (LV) unloading as any manipulative, therapeutic, and interventional approach aimed at reducing total LV mechanical power. Cardiac LV unloading in AMI is aimed at preserving the myocardium, avoiding adverse ventricular remodeling and resulting heart failure [68, 69]. Limiting the size of myocardial infarction by LV unloading was initially used in patients with cardiogenic shock [70,71,72], or in high-risk patients with PCI and impaired LV function [73, 74].
Impella is a percutaneous interventional microaxial flow pump based on the Archimedean spiral principle, where impeller rotation assists the heart in generating blood flow, increasing cardiac output, peripheral tissue perfusion and coronary blood flow, and prolonging patient survival. Several animal studies have shown that initiating LV unloading via the Impella device reduces myocardial oxygen consumption during the ischemic and reperfusion phases and decreases infarct size [75,76,77]. Initial research by Meyns et al. in 2003 demonstrated that LV unloading using Impella-reduced myocardial oxygen consumption resulted in correlated infarct size (IS) reduction in a sheep model of AMI [78]. Some studies have demonstrated that the use of the Impella in patients undergoing high-risk coronary interventions can reduce LV end-diastolic pressure, elevate blood pressure, and maintain effective coronary perfusion pressure [79]. In the DTU-STEMI pilot trial (Table 1), patients with AMI were randomized into two groups: one group underwent intervention immediately after Impella implantation, and the other group underwent intervention 30 min after reduction of cardiac LV load with Impella. Interestingly, the study concluded that delayed reperfusion therapy after 30 min of LV load reduction significantly reduced myocardial infarct size in a subset of patients [80]. This demonstrates the safety and feasibility of Impella implantation in humans, and explores a new option for reperfusion therapy in patients with AMI, which needs to be validated in large-scale trials. However, using Impella devices for LV unloading starting prior to revascularization and delaying revascularization requires a femoral 14F access, which is invasive. Meanwhile, the mechanisms by which LV unloading affects the size of myocardial infarction are still unclear. It has been suggested that activation of the RISK pathway may be involved by maintaining the integrity of myocardial energy and mitochondrial function [81], which warrants further research.
3.2 Cryocardial Cooling
TTM (targeted temperature management) has been used to reduce systemic low-flow and no-flow reperfusion injury after cardiac arrest [82,83,84,85,86]. In the ischemic heart, TTM has shown infarct size reduction in several preclinical models with a variety of animal species, ischemia durations, cooling methods, cooling durations, magnitudes of cooling, and timings of cooling initiation [87, 88]. A study in rabbits identified a linear relationship between the myocardial infarct area and temperature when heart rate was controlled and body temperature was maintained at 35–42 °C. Moreover, a reduction in myocardial infarct area could only be achieved when cooling began during the ischemic phase, with no benefit of hypothermia following the initiation of reperfusion [89]. Clinical trials thus far have demonstrated that hypothermic myocardial cooling is beneficial only in patients cooled to < 35 °C and in those with anterior myocardial wall involvement [90]. The COOL-AMI EU trial showed a 40% reduction in myocardial infarct size 4–6 days after myocardial infarction in patients with STEMI using the ZOLL Proteus cooling system to achieve a mean temperature of 33 °C before and after PCI, compared with the PCI-only group [91]. Intracoronary cooling has been proposed as a promising method for improving IRI, which has been demonstrated in a porcine model of myocardial ischemia–reperfusion [92, 93]. The EURO-ICE trial of intracoronary cooling in patients with reperfusion after myocardial ischemia is ongoing [94]. Although the induction of hypothermia is associated with improvements in IRI, it may promote platelet activation, delay reperfusion, and cause hemodynamic instability when large amounts of cold fluid are infused.
3.3 Coronary Sinus Intervention
Increased coronary sinus pressure can induce a retrograde perfusion gradient in the ischemic myocardium and improve myocardial perfusion. Therefore, myocardial ischemia can be improved by regulating coronary sinus pressure. Existing methods of coronary sinus intervention include the retroperfusion technique, retroinfusion, and coronary sinus obstruction techniques [95]. The retroperfusion technique involves active pumping of blood into the coronary sinus, which has been shown to improve myocardial metabolism and reduce myocardial ischemia in animal models [96]. The reperfusion technique involves pumping substances into the coronary sinus, such as blood or other substances. Obstruction of the coronary sinus induces collateral blood flow into the area lacking perfusion. The diversion of blood flow improves subendocardial ischemia, which has been demonstrated in a canine model with anterior descending branch occlusion complicated by coronary sinus occlusion [97]. The most commonly used method of coronary sinus intervention is pressure-controlled intermittent coronary sinus occlusion (PiCSO). A coronary sinus pressure transducer is placed at the coronary sinus orifice, and a gradual increase in coronary sinus pressure can be observed during each cardiac cycle. The balloon is inflated until a pressure plateau (approximately 70 mmHg) is reached and then the balloon is deflated, allowing redistribution of coronary blood flow to the ischemic zone [98]. PiCSO has been shown to reduce cardiomyocyte edema in dogs during AMI and to accelerate the removal of harmful molecules [99]. Khattab et al. showed that PiCSO improved coronary perfusion pressure and improved myocardial oxygen consumption in a porcine closed-chest infarction model [100]. A clinical trial of PiCSO also demonstrated a reduction in myocardial infarct size in patients with STEMI in the anterior approach, which demonstrates the safety of PiCSO as an adjunctive therapy. The first randomized controlled trial of PiCSO is underway in patients with AMI (PiCSO-AMI-I; NCT03625869), which will determine the effect of PiCSO on myocardial infarction by CMR on day 5. Another randomized controlled trial (PiCSO-AMI-II) is ongoing in Europe and North America. The trial enrolled 300 patients with anterior STEMI presenting within 12 h of symptom onset. The primary efficacy endpoint was infarct size measured by CMR at 5 days. The primary safety endpoint was the performance target for device- and procedure-related adverse events at 30 days. Major adverse cardiac events and heart failure endpoints will be captured in the acute phase and up to 3 years.
3.4 Supersaturated Oxygen
The rationale for investigating the benefit of increasing plasma oxygen tension during myocardial ischemia has been provided by experiments that revealed both increase in coronary blood flow and ischemia alleviation during hyperoxia [101, 102]. A 1950s animal study of coronary sinus obstruction showed that the mortality rate in animals with coronary occlusion decreased with increased coronary sinus pressure [103]. Chardrack et al. ligated the coronary arteries of 162 dogs and the survival rate of the dogs was 52.5% at 1 atmosphere and rose to 77.8% at 4 atmosphere [104]. Several animal studies have demonstrated that hyperbaric oxygen improves IRI, reduces myocardial infarct size, and improves survival [105, 106]. Subsequently, researchers found that water carries ten times more oxygen than blood, and the application of water–oxygen reperfusion in animal experiments improved LV function and reduced infarct size [107]. Multiple studies have since demonstrated the benefits of SSO2. In 2002, a US–Italian multicenter study of 29 post-PCI patients with acute infarction treated with hyperbaric oxygen using the TherOx® device for 60–90 min demonstrated the safety of SSO2 [108]. In 2007, the AMI HOT trial demonstrated the benefits of SSO2 for adjunctive treatment of patients with anterior myocardial infarction within 6 h of symptom onset [109]. The 2019 IC-HOT trial demonstrated the feasibility and safety of transfusion of SSO via the left main coronary artery [110]. Starting in 2021, the TherOx® device has been routinely used as an adjunctive therapy for myocardial infarction at the Hannover Medical School [111]. In the same year, the ISO-SHOCK trial, which evaluated the administration of SSO2 into the coronary arteries of patients with acute myocardial infarction and cardiogenic shock, was initiated with an anticipated completion date of 2025.
3.5 Stimulation of the Vagus Nerve
The vagus nerve is the main parasympathetic component of the body and is vital for maintaining physiological activity. The main effects of the vagus nerve on the heart include lowering heart rate, slowing atrioventricular conduction, and decreasing myocardial contractility [112]. IRI can activate the sympathetic nerve, further complementing and aggravating myocardial injury. Activation of the vagus nerve by electrical stimulation, pressure, or chemical reflexes, which further reduces heart rate and increases coronary blood flow through NO-dependent mechanisms, may improve myocardial ischemia [113]. In a rat model of AMI, stimulation of the vagus nerve had protective effects on the heart, likely due to the activity of glucagon-like peptide 1 [114]. Stimulation of the vagus nerve also reduced the production of mitochondrial ROS in pigs, significantly reducing the size (about 59%) of myocardial infarction and the probability of ventricular fibrillation [115]. In a randomized controlled trial, patients undergoing PCI for STEMI were divided into two groups; the low-level tragus stimulation (LLTS) combined with sham group, and a LLTS alone group, where stimulation was performed 2 h after reperfusion. The results showed a decrease in cardiac biomarkers and an improvement in left ventricular ejection fraction in the LLTS alone group [116]. Vagus nerve stimulation is often accompanied by side effects such as nausea, vomiting, dizziness, and hoarseness. To improve the specificity of vagus nerve stimulation for cardioprotection, many selective vagus nerve stimulation (sVNS) models have been developed, such as fiber selective stimulation, spatially selective stimulation, and kilohertz electrical stimulation [117]. According to the current study, several mechanisms have been related to cardioprotection through efferent vagal stimulation, including improved mitochondrial function, attenuation of ROS formation, antiapoptotic cardiomyocyte signaling, and reduction of systemic and local inflammatory responses [118]. It is useful to continue to research the mechanisms by which the vagus nerve protects the cardiovascular system.
3.6 Remote Ischemia Regulation
Davidson et al. proposed a mechanistic basis for cardioprotection based on the mode of protection, time of application, cellular targets and intracellular targets. It can be divided into ischemic regulation, pharmacological cardioprotection, and physical intervention. Among them, ischemic modulation can be further divided into preischemic injury modulation, postischemic injury modulation, and RIC. As of now, RIC seems to be the most promising method for cardiac repair. RIC is the application of reperfusion to the vascular bed, corresponding tissues, and organs after a short period of ischemia. These ischemic reperfusions allow tissues and organs far from the point of application to resist damage caused by reperfusion after prolonged ischemia. Loukogeorgakis et al. investigated the neuromodulatory process of distal ischemic preconditioning of endothelial cells (RIPC). The early phase of RIPC is 4 h, followed by a maintenance phase that lasts for at least 48 h after 24 h of RIPC stimulation [117]. Their study found that the use of the autonomic ganglion blocker trimethaphan (trimethaphan 16 mg/min intravenous infusion) reduced both the early and maintenance phases of RIPC, suggesting that protection in both phases is dependent on preservation of autonomic function [119,120,121,122]. The exact mechanism of cardioprotection by RIC is not yet fully understood. In particular, it has been suggested that RIC involves the activation of three different pathways: the humoral, the neuronal, and the systemic pathway. Studies suggest that RIC may activate defense mechanisms in the cardiovascular system through humoral pathways and neuronal pathways at distal stimulation sites. They depend on the anatomical site (renal, mesenteric, or skeletal muscle) in which the ischemia–reperfusion stimulus has been applied. For instance, adenosine and erythropoietin are involved in RIC induced by renal IR, while bradykinin, cannabinoids, opioids, and CGRP, or adenosine, NO, opioids, noradrenaline, ROS, apolipoprotein-A-I, GLP- 1, stromal cell-derived factor-1a, prostanoids, IL-10, glycine, exosomes, and microRNAs can account for RIC after mesenteric or skeletal muscle ischemia–reperfusion, respectively. RIC reduces the platelet activation process of IRI and attenuates endothelial injury and inflammatory processes [123,124,125,126]. EVs have been proposed as a source of RIC signaling that initiates the protective effect of ischemia–reperfusion [127]. The specific mechanisms underlying these effects must be further explored.
3.7 C-Reactive Protein Monocollection
C-reactive protein (CRP) is a non-specific marker of inflammation and is one of the key players in the body’s natural immune barrier. CRP is associated with inflammation, infection, malignancy, and cardiovascular disease. CRP is also a mediator of myocardial infarction, which may promote localized tissue damage [128]. Elevated CRP predicts second-year heart failure, as well as cardiovascular mortality in patients with STEMI [129]. Therefore, lowering CRP levels is a potential method by which to reduce reperfusion injury [130]. The CAMI-1 clinical trial of 83 patients with STEMI showed that single extraction reduced CRP levels and did not correlate with final infarct size (FIS) or LV function [131]. A randomized controlled trial (NCT04939805) using CRP monocollection as an adjunctive treatment for patients with STEMI is currently underway in Austria and Germany, with results expected in the coming years.
4 Conclusion
Stenosis and occlusion of coronary arteries can lead to myocardial ischemia and infarction. Early unblocking of coronary arteries and restoration of blood flow reconstruction can reduce the infarcted area of the myocardium and improve prognosis. However, animal tests have demonstrated that almost half of the infarct size can be attributed to IRI [132, 133]. As a result, many scholars have focused their attention on cardioprotective mechanisms and strategies. Current studies suggest that SUMOylation of multiple substances in the cytoplasm and mitochondria can have cardioprotective effects. Several members of the miRNA family play important roles in cardioprotection. NLRP3 regulates a variety of signaling pathways. MDVs and EVs may be involved in mitochondrial translocation. All of these could be targets for cardioprotection.
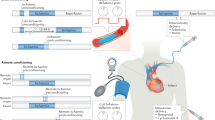
Myocardial infarct size partially determines the prognosis of patients with myocardial infarction, although regular administration of antiplatelet agents as well as statins after PCI does not greatly reduce the incidence of heart failure and recurrent myocardial infarction. Therefore, mitigating the potential for myocardial injury as a result of postischemic reperfusion and reducing the size of myocardial infarction, have become important research areas of focus. Recently, cardiac-assisted protection methods have shown potential for improving prognosis in patients with myocardial infarction. LV unloading, myocardial cooling, coronary sinus intervention, SSO2, stimulation of the vagus nerve, RIC, and CRP monocollection are effective cardioprotection methods that have been identified in recent years (Fig. 2). Most of these methods may prolong the time in the catheterization chamber, except for CRP monophoresis. However, only SSO2 has been shown to reduce myocardial infarct size in patients with antegrade STEMI. In addition to this, some studies have found that low doses of carbon monoxide can maintain the stability of the mitochondrial membrane potential and may have cardioprotective effects [134].

Cardioprotective strategies after ischemia–-reperfusion injury.
Unfortunately, although cardioprotective strategies are effective in reducing myocardial infarct size in some populations, they have low benefit in metabolic syndrome populations. Metabolic syndrome is known to be an important risk factor for myocardial ischemia, such as diabetes and obesity. Studies suggest that the diabetic heart is resistant to cardioprotective strategies, although clinical evidence is lacking [135]. Meanwhile, data suggest that susceptibility to the IR damage is increased and cardioprotection effectiveness is reduced in obesity [136]. In addition, being old has been identified to be an independent determinant of the extension of AMI and the outcome of cardioprotective strategies after an ischemia–reperfusion episode [137]. There have also been studies showing that the estrogen status of females modulates the susceptibility of the heart to IRI. Similarly, reduction in testosterone levels in older men worsens cardiovascular outcomes [138,139,140]. Future research on cardioprotection could focus on metabolic syndrome, older adults, and sex.
Most of the cardioprotective strategies discussed in this review have been validated in animal studies, and large-scale clinical trials are needed to confirm the results reported so far. Meanwhile, more research is needed to understand the mechanisms of cardioprotection and associated signaling pathways, which can provide new approaches for the prevention of IRI.
References
Elamin ABA, Forsat K, Senok SS, Goswami N. Vagus nerve stimulation and its cardioprotective abilities: a systematic review. J Clin Med. 2023. https://doi.org/10.3390/jcm12051717.
Stone GW, Selker HP, Thiele H, et al. Relationship between infarct size and outcomes following primary PCI: Patient-level analysis from 10 randomized trials. J Am Coll Cardiol. 2016;67(14):1674–83. https://doi.org/10.1016/j.jacc.2016.01.069.
Pagliaro P, Penna C. Redox signalling and cardioprotection: translatability and mechanism. Br J Pharmacol. 2015;172(8):1974–95. https://doi.org/10.1111/bph.12975.
Penna C, Alloatti G, Crisafulli A. Mechanisms involved in cardioprotection induced by physical exercise. Antioxid Redox Signal. 2020;32(15):1115–34. https://doi.org/10.1089/ars.2019.8009.
Penna C, Comità S, Tullio F, et al. Challenges facing the clinical translation of cardioprotection: 35 years after the discovery of ischemic preconditioning. Vascul Pharmacol. 2022;144: 106995. https://doi.org/10.1016/j.vph.2022.106995.
Gao C, Huang W, Kanasaki K, et al. The role of ubiquitination and sumoylation in diabetic nephropathy. Biomed Res Int. 2014;2014: 160692. https://doi.org/10.1155/2014/160692.
Kho C, Lee A, Jeong D, et al. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–5. https://doi.org/10.1038/nature10407.
Hay RT. SUMO: a history of modification. Mol Cell. 2005;18:1–12. https://doi.org/10.1016/j.molcel.2005.03.012.
Guo B, Yang SH, Witty J, Sharrocks AD. Signalling pathways and the regulation of SUMO modification. Biochem Soc Trans. 2007;35:1414–8. https://doi.org/10.1042/BST0351414.
Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8:947–56. https://doi.org/10.1038/nrm2293.
Chang E, Heo KS, Woo CH, Lee H, Le NT, Thomas TN, Fujiwara K, Abe J. MK2 SUMOylation regulates actin filament remodeling and subsequent migration in endothelial cells by inhibiting MK2 kinase and HSP27 phosphorylation. Blood. 2011;117:2527–37. https://doi.org/10.1182/blood-2010-08-302281.
Le NT, Martin JF, Fujiwara K, Abe JI. Sub-cellular localization specific SUMOylation in the heart. Biochim Biophys Acta Mol Basis Dis. 2017;1863(8):2041–55. https://doi.org/10.1016/j.bbadis.2017.01.018.
Jancso G, Lantos J, Borsiczky B, Szanto Z, Roth E. Dynamism of NF-kappaB and AP-1 activation in the signal transduction of ischaemic myocardial preconditioning. Eur Surg Res. 2004;36:129–35. https://doi.org/10.1159/000077253.
Xiao L, Liu Y, Wang N. New paradigms in inflammatory signaling in vascular endothelial cells. Am J Physiol Heart Circ Physiol. 2014;306:H317–25. https://doi.org/10.1152/ajpheart.00182.2013.
Desterro JM, Rodriguez MS, Hay RT. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol Cell. 1998;2:233–9. https://doi.org/10.1016/s1097-2765(00)80133-1.
Culver C, Sundqvist A, Mudie S, Melvin A, Xirodimas D, Rocha S. Mechanism of hypoxia-induced NF-kappaB. Mol Cell Biol. 2010;30:4901–21. https://doi.org/10.1128/MCB.00409-10.
Ramana KV, Friedrich B, Srivastava S, Bhatnagar A, Srivastava SK. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes. 2004;53:2910–20. https://doi.org/10.2337/diabetes.53.11.2910.
Mohan S, Konopinski R, Yan B, Centonze VE, Natarajan M. High glucose-induced IKK-Hsp-90 interaction contributes to endothelial dysfunction. Am J Physiol Cell Physiol. 2009;296:C182–92. https://doi.org/10.1152/ajpcell.00575.2007.
Sun H, Lu L, Zuo Y, Wang Y, Jiao Y, Zeng WZ, Huang C, Zhu MX, Zamponi GW, Zhou T, Xu TL, Cheng J, Li Y. Kainate receptor activation induces glycine receptor endocytosis through PKC deSUMOylation. Nat Commun. 2014;5:4980. https://doi.org/10.1038/ncomms5980.
Liu Q, Molkentin JD. Protein kinase Cα as a heart failure therapeutic target. J Mol Cell Cardiol. 2011;51:474–8. https://doi.org/10.1016/j.yjmcc.2010.10.004.
Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10(3):248–54. https://doi.org/10.1038/nm1000.
Malhotra R, D’Souza KM, Staron ML, Birukov KG, Bodi I, Akhter SA. G alpha(q)-mediated activation of GRK2 by mechanical stretch in cardiac myocytes: the role of protein kinase C. J Biol Chem. 2010;285:13748–60. https://doi.org/10.1074/jbc.M110.109272.
Belin RJ, Sumandea MP, Allen EJ, Schoenfelt K, Wang H, Solaro RJ, de Tombe PP. Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ Res. 2007;101:195–204. https://doi.org/10.1161/CIRCRESAHA.107.148288.
Sumandea MP, Pyle WG, Kobayashi T, de Tombe PP, Solaro RJ. Identification of a functionally critical protein kinase C phosphorylation residue of cardiac troponin T. J Biol Chem. 2003;278:35135–44. https://doi.org/10.1074/jbc.M306325200.
Kooij V, Boontje N, Zaremba R, Jaquet K, dos Remedios C, Stienen GJ, van der Velden J. Protein kinase C alpha and epsilon phosphorylation of troponin and myosin binding protein C reduce Ca2+ sensitivity in human myocardium. Basic Res Cardiol. 2010;105:289–300. https://doi.org/10.1007/s00395-009-0053-z.
Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. PKC phosphorylation of titin’s PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res. 2009;105:631–8. https://doi.org/10.1161/CIRCRESAHA.109.198465.
Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res. 2012;111:800–14. https://doi.org/10.1161/CIRCRESAHA.111.255505.
Russell RR 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. https://doi.org/10.1172/JCI19297.
Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, Lefer DJ. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57:696–705. https://doi.org/10.2337/db07-1098.
Xing Y, Musi N, Fujii N, Zou L, Luptak I, Hirshman MF, Goodyear LJ, Tian R. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J Biol Chem. 2003;278:28372–7. https://doi.org/10.1074/jbc.M303521200.
Rubio T, Vernia S, Sanz P. Sumoylation of AMPKbeta2 subunit enhances AMP-activated protein kinase activity. Mol Biol Cell. 2013;24(1801–1811):S1801-1804. https://doi.org/10.1091/mbc.E12-11-0806.
Ritho J, Arold ST, Yeh ET. A critical SUMO1 modification of LKB1 regulates AMPK activity during energy stress. Cell Rep. 2015;12:734–42. https://doi.org/10.1016/j.celrep.2015.07.002.
Gupta MK, Gulick J, Liu R, Wang X, Molkentin JD, Robbins J. Sumo E2 enzyme UBC9 is required for efficient protein quality control in cardiomyocytes. Circ Res. 2014;115:721–9. https://doi.org/10.1161/CIRCRESAHA.115.304760.
Gupta MK, McLendon PM, Gulick J, James J, Khalili K, Robbins J. UBC9-mediated sumoylation favorably impacts cardiac function in compromised hearts. Circ Res. 2016;118:1894–905. https://doi.org/10.1161/CIRCRESAHA.115.308268.
Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: united at reperfusion. Pharmacol Ther. 2007;116(2):173–91. https://doi.org/10.1016/j.pharmthera.2007.06.005.
Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res. 2003;60(3):617–25. https://doi.org/10.1016/j.cardiores.2003.09.025.
Piot C, Croisille P, Staat P, Thibault H, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359(5):473–81. https://doi.org/10.1056/NEJMoa071142.
Verma S, Fedak PW, Weisel RD, et al. Fundamentals of reperfusion injury for the clinical cardiologist. Circulation. 2002;105(20):2332–6. https://doi.org/10.1161/01.cir.0000016602.96363.36.
Penna C, Perrelli MG, Tullio F, Angotti C, Camporeale A, Poli V, Pagliaro P. Diazoxide postconditioning induces mitochondrial protein S-nitrosylation and a redox-sensitive mitochondrial phosphorylation/translocation of RISK elements: no role for SAFE. Basic Res Cardiol. 2013;108(5):371. https://doi.org/10.1007/s00395-013-0371-z.
Boulghobra D, Dubois M, Alpha-Bazin B, Coste F, Olmos M, Gayrard S, Bornard I, Meyer G, Gaillard JC, Armengaud J, Reboul C. Increased protein S-nitrosylation in mitochondria: a key mechanism of exercise-induced cardioprotection. Basic Res Cardiol. 2021;116(1):66. https://doi.org/10.1007/s00395-021-00906-3.
Jong WM, Leemans JC, Weber NC, Juffermans NP, Schultz MJ, Hollmann MW, Zuurbier CJ. Nlrp3 plays no role in acute cardiac infarction due to low cardiac expression. Int J Cardiol. 2014;177:41–3. https://doi.org/10.1016/j.ijcard.2014.09.148.
Toldo S, Marchetti C, Mauro AG, Chojnacki J, Mezzaroma E, Carbone S, Zhang S, Van Tassell B, Salloum FN, Abbate A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia- reperfusion in the mouse. Int J Cardiol. 2016;209:215–20. https://doi.org/10.1016/j.ijcard.2016.02.043.
Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17:588–606. https://doi.org/10.1038/nrd.2018.97.
Zhang Y, Rong H, Zhang FX, Wu K, Mu L, Meng J, Xiao B, Zamponi GW, Shi Y. A membrane potential- and calpain-dependent reversal of caspase-1 inhibition regulates canonical NLRP3 inflammasome. Cell Rep. 2018;24:2356–69. https://doi.org/10.1016/j.celrep.2018.07.098. (e5).
Zuurbier CJ, Jong WMC, Eerbeek O, et al. Deletion of the innateimmune NLRP3 receptor abolishes cardiac ischemic preconditioningand is associated with decreased IL-6/STAT3 signaling. PLoS ONE. 2012;7: e40643. https://doi.org/10.1371/journal.pone.0040643.
Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF, Abbate A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci USA. 2011;108:19725–30. https://doi.org/10.1073/pnas.1108586108.
Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. https://doi.org/10.1161/CIRCULATIONAHA.110.982777.
Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao C, Xin C, Zhu D, Li Y, Yan W, Xiong L, Gao E, Wang H, Tao L. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res Cardiol. 2014. https://doi.org/10.1007/s00395-014-0415-z.
Mastrocola R, Penna C, Tullio F, Femminò S, Nigro D, Chiazza F, Serpe L, Collotta D, Alloatti G, Cocco M, Bertinaria M, Pagliaro P, Aragno M, Collino M. Pharmacological inhibition of NLRP3 inflammasome attenuates myocardial ischemia/reperfusion injury by activation of RISK and mitochondrial pathways. Oxid Med Cell Longev. 2016. https://doi.org/10.1155/2016/5271251.
Penna C, Comità S, Tullio F, Alloatti G, Pagliaro P. Challenges facing the clinical translation of cardioprotection: 35 years after the discovery of ischemic preconditioning. Vascul Pharmacol. 2022;144: 106995. https://doi.org/10.1016/j.vph.2022.106995.
Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF, Abbate A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. 2011;108(49):19725–30. https://doi.org/10.1073/pnas.1108586108.
Mauro AG, Bonaventura A, Mezzaroma E, Quader M, Toldo S. NLRP3 Inflammasome in acute myocardial infarction. J Cardiovasc Pharmacol. 2019;74(3):175–87. https://doi.org/10.1097/FJC.0000000000000717.
Makkos A, Ágg B, Petrovich B, Varga ZV, Görbe A, Ferdinandy P. Systematic review and network analysis of microRNAs involved in cardioprotection against myocardial ischemia/reperfusion injury and infarction: involvement of redox signalling. Free Radic Biol Med. 2021;20(172):237–51. https://doi.org/10.1016/j.freeradbiomed.2021.04.034.
Cheng Y, Zhu P, Yang J, Liu X, Dong S, Wang X, Chun B, Zhuang J, Zhang C. Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via anti-apoptosis through its target PDCD4. Cardiovasc Res. 2010;87(3):431–9. https://doi.org/10.1093/cvr/cvq082.
Wang X, Ha T, Liu L, Zou J, Zhang X, Kalbfleisch J, Gao X, Williams D, Li C. Increased expression of microRNA-146a decreases myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2013;97(3):432–42. https://doi.org/10.1093/cvr/cvs356.
Qiao S, Olson JM, Paterson M, Yan Y, Zaja I, Liu Y, Riess ML, Kersten JR, Liang M, Warltier DC, Bosnjak ZJ, Ge ZD. MicroRNA-21 mediates isoflurane-induced cardioprotection against ischemia-reperfusion injury via Akt/nitric oxide synthase/mitochondrial permeability transition pore pathway. Anesthesiology. 2015;123(4):786–98. https://doi.org/10.1097/ALN.0000000000000807.
Ma N, Bai J, Zhang W, Luo H, Zhang X, Liu D, Qiao C. Trimetazidine protects against cardiac ischemia/reperfusion injury via effects on cardiac miRNA-21 expression, Akt and the Bcl-2/Bax pathway. Mol Med Rep. 2016;14(5):4216–22. https://doi.org/10.3892/mmr.2016.5773.
Bayoumi AS, Park KM, Wang Y, Teoh JP, Aonuma T, Tang Y, Su H, Weintraub NL, Kim IM. A carvedilol-responsive microRNA, miR-125b-5p protects the heart from acute myocardial infarction by repressing pro-apoptotic bak1 and klf13 in cardiomyocytes. J Mol Cell Cardiol. 2018;114:72–82. https://doi.org/10.1016/j.yjmcc.2017.11.003.
Nguyen BY, Ruiz-Velasco A, Bui T, Collins L, Wang X, Liu W. Mitochondrial function in the heart: the insight into mechanisms and therapeutic potentials. Br J Pharmacol. 2019;176(22):4302–18. https://doi.org/10.1111/bph.14431.
Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33(19):2142–56. https://doi.org/10.15252/embj.201488104.
Cadete VJ, Deschênes S, Cuillerier A, Brisebois F, Sugiura A, Vincent A, Turnbull D, Picard M, McBride HM, Burelle Y. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J Physiol. 2016;594(18):5343–62. https://doi.org/10.1113/JP272703.
Li B, Zhao H, Wu Y, Zhu Y, Zhang J, Yang G, Yan Q, Li J, Li T, Liu L. Mitochondrial-derived vesicles protect cardiomyocytes against hypoxic damage. Front Cell Dev Biol. 2020;17(8):214. https://doi.org/10.3389/fcell.2020.00214.
Zhao L, Zhang B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci Rep. 2017;16(7):44735. https://doi.org/10.1038/srep44735.
Heyn J, Heuschkel MA, Goettsch C. Mitochondrial-derived vesicles-link to extracellular vesicles and implications in cardiovascular disease. Int J Mol Sci. 2023;24(3):2637. https://doi.org/10.3390/ijms24032637.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213–28. https://doi.org/10.1038/nrm.2017.125.
Cocozza F, Grisard E, Martin-Jaular L, Mathieu M, Théry C. SnapShot: extracellular vesicles. Cell. 2020;182(1):262-262.e1. https://doi.org/10.1016/j.cell.2020.04.054.
Ikeda G, Santoso MR, Tada Y, Li AM, Vaskova E, Jung JH, O’Brien C, Egan E, Ye J, Yang PC. Mitochondria-rich extracellular vesicles from autologous stem cell-derived cardiomyocytes restore energetics of ischemic myocardium. J Am Coll Cardiol. 2021;77(8):1073–88. https://doi.org/10.1016/j.jacc.2020.12.060.
Uriel N, Sayer G, Annamalai S, Kapur NK, Burkhoff D. Mechanical unloading in heart failure. J Am Coll Cardiol. 2018;72(5):569–80. https://doi.org/10.1016/j.jacc.2018.05.038.
Burkhoff D, Sayer G, Doshi D, Uriel N. Hemodynamics of mechanical circulatory support. J Am Coll Cardiol. 2015;66(23):2663–74. https://doi.org/10.1016/j.jacc.2015.10.017.
Basir MB, Schreiber TL, Grines CL, et al. Effect of early initiation of mechanical circulatory support on survival in cardiogenic shock. Am J Cardiol. 2017;119(6):845–51. https://doi.org/10.1016/j.amjcard.2016.11.037.
O’Neill WW, Schreiber T, Wohns DH, et al. The current use of Impella 2.5 in acute myocardial infarction complicated by cardiogenic shock: results from the USpella Registry. J Interv Cardiol. 2014;27(1):1–11. https://doi.org/10.1111/joic.12080.
Schäfer A, Werner N, Burkhoff D, et al. Influence of timing and predicted risk on mortality in Impella-treated infarct-related cardiogenic shock patients. Front Cardiovasc Med. 2020;7:74. https://doi.org/10.3389/fcvm.2020.00074.
O’Neill WW, Kleiman NS, Moses J, et al. A prospective, randomized clinical trial of hemodynamic support with Impella 2.5 versus intra-aortic balloon pump in patients undergoing high-risk percutaneous coronary intervention: the PROTECT II study. Circulation. 2012;126(14):1717–27. https://doi.org/10.1161/CIRCULATIONAHA.112.098194.
Baumann S, Werner N, Ibrahim K, et al. Indication and short-term clinical outcomes of high-risk percutaneous coronary intervention with microaxial Impella® pump: results from the German Impella® registry. Clin Res Cardiol. 2018;107(8):653–7. https://doi.org/10.1007/s00392-018-1230-6.
Meyns B, Stolinski J, Leunens V, Verbeken E, Flameng W. Left ventricular support by catheter- mounted axial flow pump reduces infarct size. J Am Coll Cardiol. 2003;41:1087–95. https://doi.org/10.1016/s0735-1097(03)000846.
Meyns B, Stolinski J, Leunens V, Verbeken E, Flameng W. Left ventricular support by catheter-mounted axial flow pump reduces infarct size. J Am Coll Cardiol. 2003;41(7):1087–95. https://doi.org/10.1016/s0735-1097(03)00084-6.
Saku K, Kakino T, Arimura T, et al. Left ventricular mechanical unloading by total support of impella in myocardial infarction reduces infarct size, preserves left ventricular function, and prevents subsequent heart failure in dogs. Circ Heart Fail. 2018;11(5): e004397. https://doi.org/10.1161/CIRCHEARTFAILURE.117.004397.
Saku K, Kakino T, Arimura T, et al. Total mechanical unloading minimizes metabolic demand of left ventricle and dramatically reduces infarct size in myocardial infarction. PLoS ONE. 2016;11(4): e0152911. https://doi.org/10.1371/journal.pone.0152911.
Alqarqaz M, Basir M, Alaswad K, O’Neill W. Effects of impella on coronary perfusion in patients with critical coronary artery stenosis. Circ Cardiovasc Interv. 2018;11(4): e005870. https://doi.org/10.1161/CIRCINTERVENTIONS.117.005870.
Kapur NK, Alkhouli MA, DeMartini TJ, et al. Unloading the left ventricle before reperfusion in patients with anterior ST-segment-elevation myocardial infarction. Circulation. 2019;139(3):337–46. https://doi.org/10.1161/CIRCULATIONAHA.118.038269.
Rossello X, Yellon DM. The RISK pathway and beyond. Basic Res Cardiol. 2018;113(1):2. https://doi.org/10.1007/s00395-017-0662-x.
Schäfer A, Bauersachs J, Akin M. Therapeutic hypothermia following cardiac arrest after the TTM2 trial—more questions raised than answered. Curr Probl Cardiol. 2023;48(3): 101046. https://doi.org/10.1016/j.cpcardiol.2021.101046.
Akin M, et al. Mortality in patients with out-of-hospital cardiac arrest undergoing a standardized protocol including therapeutic hypothermia and routine coronary angiography: experience from the HACORE registry. JACC Cardiovasc Interv. 2018;11(18):1811–20. https://doi.org/10.1016/j.jcin.2018.06.022.
Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346(8):557–63. https://doi.org/10.1056/NEJMoa003289.
Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346(8):549–56. https://doi.org/10.1056/NEJMoa012689.
Lascarrou JB, Merdji H, Le Gouge A, et al. Targeted temperature management for cardiac arrest with nonshockable rhythm. N Engl J Med. 2019;381(24):2327–37. https://doi.org/10.1056/NEJMoa1906661.
Bergmann SR, Angelakos ET, Torres JC. Salutary effects of moderate hypothermia on the circulatory and myocardial consequences of acute coronary occlusion in dogs. Cryobiology. 1985;22:555–68. https://doi.org/10.1016/0011-2240(85)90032-x.58.
Otake H, Shite J, Paredes OL, Shinke T, Yoshikawa R, Tanino Y, Watanabe S, Ozawa T, Matsumoto D, Ogasawara D, et al. Catheter-based transcoronary myocardial hypothermia attenuates arrhythmia and myocardial necrosis in pigs with acute myocardial infarction. J Am Coll Cardiol. 2007;49:250–60. https://doi.org/10.1016/j.jacc.2006.06.080].
Kohlhauer M, Berdeaux A, Ghaleh B, Tissier R. Therapeutic hypothermia to protect the heart against acute myocardial infarction. Arch Cardiovasc Dis. 2016;109(12):716–22. https://doi.org/10.1016/j.acvd.2016.05.005.
Erlinge D, Götberg M, Grines C, et al. A pooled analysis of the effect of endovascular cooling on infarct size in patients with ST-elevation myocardial infarction. EuroIntervention. 2013;8(12):1435–40. https://doi.org/10.4244/EIJV8I12A217.
Noc M, Erlinge D, Neskovic AN, et al. COOL AMI EU pilot trial: a multicentre, prospective, randomised controlled trial to assess cooling as an adjunctive therapy to percutaneous intervention in patients with acute myocardial infarction. EuroIntervention. 2017;13(5):e531–9. https://doi.org/10.4244/EIJ-D-17-00279.
Kim H, Lee J, Song W, et al. Feasibility and safety of regional myocardial hypothermia during myocardial ischemia and infarction in pigs. Coron Artery Dis. 2005;16(2):125–9. https://doi.org/10.1097/00019501-200503000-00008.
Otake H, Shite J, Paredes OL, et al. Catheter-based transcoronary myocardial hypothermia attenuates arrhythmia and myocardial necrosis in pigs with acute myocardial infarction. J Am Coll Cardiol. 2007;49(2):250–60. https://doi.org/10.1016/j.jacc.2006.06.080.
El Farissi M, Keulards DCJ, van’t Veer M, et al. Selective intracoronary hypothermia in patients with ST-elevation myocardial infarction. Rationale and design of the EURO-ICE trial. EuroIntervention. 2021;16(17):1444–6. https://doi.org/10.4244/EIJ-D-19-00471.
Meerbaum S, Lang TW, Osher JV, et al. Diastolic retroperfusion of acutely ischemic myocardium. Am J Cardiol. 1976;37(4):588–98. https://doi.org/10.1016/0002-9149(76)90400-8.
Romeo FJ, Mazurek R, Sakata T, et al. Device-based approaches targeting cardioprotection in myocardial infarction: the expanding armamentarium of innovative strategies. J Am Heart Assoc. 2022;11(23): e026474. https://doi.org/10.1161/JAHA.122.026474.
Ido A, Hasebe N, Matsuhashi H, Kikuchi K. Coronary sinus occlusion enhances coronary collateral flow and reduces subendocardial ischemia. Am J Physiol Heart Circ Physiol. 2001;280(3):H1361–7. https://doi.org/10.1152/ajpheart.2001.280.3.H1361.
Egred M, Bagnall A, Spyridopoulos I, et al. Effect of Pressure-controlled intermittent Coronary Sinus Occlusion (PiCSO) on infarct size in anterior STEMI: PiCSO in ACS study. Int J Cardiol Heart Vasc. 2020;28: 100526. https://doi.org/10.1016/j.ijcha.2020.100526.
Mohl W, Gangl C, Jusić A, Aschacher T, De Jonge M, Rattay F. PICSO: from myocardial salvage to tissue regeneration. Cardiovasc Revasc Med. 2015;16(1):36–46. https://doi.org/10.1016/j.carrev.2014.12.004.
Stone GW, Martin JL, de Boer MJ. Effect of supersaturated oxygen delivery on infarct size after percutaneous coronary intervention in acute myocardial infarction. Circ Cardiovasc Interv. 2009;2(5):366–75. https://doi.org/10.1161/CIRCINTERVENTIONS.108.840066.
Cason BA, Wisneski JA, Neese RA, Stanley WC, Hickey RF, Shnier CB, Gertz EW. Effects of high arterial oxygen tension on function, blood flow distribution, and metabolism in ischemic myocardium. Circulation. 1992;85:828–38. https://doi.org/10.1161/01.cir.85.2.828/.
Ribeiro LG, Louie EK, Davis MA, Maroko PR. Augmentation of col-lateral blood flow to the ischaemic myocardiumby oxygen inhalation following experimental coronary artery occlusion. Cardiovasc Res. 1979;13:160–6. https://doi.org/10.1093/cvr/13.3.160.
Kloner RA. Current state of clinical translation of cardioprotective agents for acute myocardial infarction. Circ Res. 2013;113(4):451–63. https://doi.org/10.1161/CIRCRESAHA.112.300627.
Chardack WM, Gage AA, Federico AJ, Cusick JK, Matsumoto PJ, Lanphier EH. Reduction by hyperbaric oxygenation of the mortality from ventricular fibrillation following coronary artery ligation. Circ Res. 1964;15:497–502. https://doi.org/10.1161/01.res.15.6.497.
Peter RH, Rau RW, Whalen RE, Entman ML, McIntosh HD. Effects of hyperbaric oxygenation on coronary artery occlusion in pigs. Circ Res. 1966;18(1):89–96. https://doi.org/10.1161/01.res.18.1.89.
Sterling DL, Thornton JD, Swafford A, et al. Hyperbaric oxygen limits infarct size in ischemic rabbit myocardium in vivo. Circulation. 1993;88(4 Pt 1):1931–6. https://doi.org/10.1161/01.cir.88.4.1931.
Bartorelli AL. Hyperoxemic perfusion for treatment of reperfusion microvascular ischemia in patients with myocardial infarction. Am J Cardiovasc Drugs. 2003;3(4):253–63. https://doi.org/10.2165/00129784-200303040-00004.
Dixon SR, Bartorelli AL, Marcovitz PA, et al. Initial experience with hyperoxemic reperfusion after primary angioplasty for acute myocardial infarction: results of a pilot study utilizing intracoronary aqueous oxygen therapy. J Am Coll Cardiol. 2002;39(3):387–92. https://doi.org/10.1016/s0735-1097(01)01771-5.
O’Neill WW, Martin JL, Dixon SR, et al. Acute Myocardial Infarction with Hyperoxemic Therapy (AMIHOT): a prospective, randomized trial of intracoronary hyperoxemic reperfusion after percutaneous coronary intervention. J Am Coll Cardiol. 2007;50(5):397–405. https://doi.org/10.1016/j.jacc.2007.01.099.
David SW, Khan ZA, Patel NC, et al. Evaluation of intracoronary hyperoxemic oxygen therapy in acute anterior myocardial infarction: the IC-HOT study. Catheter Cardiovasc Interv. 2019;93(5):882–90. https://doi.org/10.1002/ccd.27905.
Schäfer A, Akin M, Diekmann J, König T. Intracoronary application of super-saturated oxygen to reduce infarct size following myocardial infarction. J Clin Med. 2022. https://doi.org/10.3390/jcm11061509.
Capilupi MJ, Kerath SM, Becker LB. Vagus nerve stimulation and the cardiovascular system. Cold Spring Harb Perspect Med. 2020. https://doi.org/10.1101/cshperspect.a034173.
Chen M, Li X, Yang H, Tang J, Zhou S. Hype or hope: vagus nerve stimulation against acute myocardial ischemia-reperfusion injury. Trends Cardiovasc Med. 2020;30(8):481–8. https://doi.org/10.1016/j.tcm.2019.10.011.
Basalay MV, Li X, Yang H, Tang J, Zhou S. Glucagon-like peptide-1 (GLP-1) mediates cardioprotection by remote ischaemic conditioning. Cardiovasc Res. 2016;112(3):669–76. https://doi.org/10.1016/j.tcm.2019.10.011.
Shinlapawittayatorn K, Chinda K, Palee S, et al. Vagus nerve stimulation initiated late during ischemia, but not reperfusion, exerts cardioprotection via amelioration of cardiac mitochondrial dysfunction. Heart Rhythm. 2014;11(12):2278–87. https://doi.org/10.1016/j.hrthm.2014.08.001.
Yu L, Huang B, Po SS, et al. Low-level tragus stimulation for the treatment of ischemia and reperfusion injury in patients with ST-Segment elevation myocardial infarction: a proof-of-concept study. JACC Cardiovasc Interv. 2017;10(15):1511–20. https://doi.org/10.1016/j.jcin.2017.04.036.
Fitchett A, Mastitskaya S, Aristovich K. Selective Neuromodulation of the Vagus Nerve. Front Neurosci. 2021;15: 685872. https://doi.org/10.3389/fnins.2021.685872.
Chen M, Li X, Yang H, Tang J, Zhou S. Hype or hope: vagus nerve stimulation against acute myocardial ischemia- reperfusion injury. Trends Cardiovasc Med. 2020;30:481488. https://doi.org/10.1016/j.tcm.2019.10.011.
Loukogeorgakis SP, Panagiotidou AT, Broadhead MW, Donald A, Deanfield JE, MacAllister RJ. Remote ischemic preconditioning provides early and late protection against endothelial ischemia-reperfusion injury in humans: role of the autonomic nervous system. J Am Coll Cardiol. 2005;46(3):450–6. https://doi.org/10.1016/j.jacc.2005.04.044.
Chen Y, Shin YK, Bassham DC. YKT6 is a core constituent of membrane fusion machineries at the Arabidopsis trans-Golgi network. J Mol Biol. 2005;350(1):92–101. https://doi.org/10.1016/j.jmb.2005.04.061.
Weinbrenner C, Schulze F, Sárváry L, Strasser RH. Remote preconditioning by infrarenal aortic occlusion is operative via delta1-opioid receptors and free radicals in vivo in the rat heart. Cardiovasc Res. 2004;61(3):591–9. https://doi.org/10.1016/j.cardiores.2003.10.008.
Khanna G, Diwan V, Singh M, Singh N, Jaggi AS. Reduction of ischemic, pharmacological and remote preconditioning effects by an antioxidant N-acetyl cysteine pretreatment in isolated rat heart. Yakugaku Zasshi. 2008;128(3):469–77. https://doi.org/10.1248/yakushi.128.469.
Shahid M, Tauseef M, Sharma KK, Fahim M. Brief femoral artery ischaemia provides protection against myocardial ischaemia-reperfusion injury in rats: the possible mechanisms. Exp Physiol. 2008;93(8):954–68. https://doi.org/10.1113/expphysiol.2007.041442.
Pedersen CM, Cruden NL, Schmidt MR, et al. Remote ischemic preconditioning prevents systemic platelet activation associated with ischemia-reperfusion injury in humans. J Thromb Haemost. 2011;9(2):404–7. https://doi.org/10.1111/j.1538-7836.2010.04142.x.
Kharbanda RK, Mortensen UM, White PA, et al. Transient limb ischemia induces remote ischemic preconditioning in vivo. Circulation. 2002;106(23):2881–3. https://doi.org/10.1161/01.cir.0000043806.51912.9b.
Konstantinov IE, Arab S, Kharbanda RK, et al. The remote ischemic preconditioning stimulus modifies inflammatory gene expression in humans. Physiol Genomics. 2004;19(1):143–50. https://doi.org/10.1152/physiolgenomics.00046.2004.
Shimizu M, Saxena P, Konstantinov IE, et al. Remote ischemic preconditioning decreases adhesion and selectively modifies functional responses of human neutrophils. J Surg Res. 2010;158(1):155–61. https://doi.org/10.1016/j.jss.2008.08.010.
Barani B, Rajasingh S, Rajasingh J. Exosomes: outlook for future cell-free cardiovascular disease therapy. Adv Exp Med Biol. 2017;998:285–307. https://doi.org/10.1007/978-981-10-4397-0_19.
Lagrand WK, Niessen HW, Wolbink GJ, et al. C-reactive protein colocalizes with complement in human hearts during acute myocardial infarction. Circulation. 1997;95(1):97–103. https://doi.org/10.1161/01.cir.95.1.97.
Stumpf C, Sheriff A, Zimmermann S, et al. C-reactive protein levels predict systolic heart failure and outcome in patients with first ST-elevation myocardial infarction treated with coronary angioplasty. Arch Med Sci. 2017;13(5):1086–93. https://doi.org/10.5114/aoms.2017.69327.
Sheriff A, Schindler R, Vogt B, et al. Selective apheresis of C-reactive protein: a new therapeutic option in myocardial infarction? J Clin Apher. 2015;30(1):15–21. https://doi.org/10.1002/jca.21344.
Ries W, Torzewski J, Heigl F, et al. C-reactive protein apheresis as anti-inflammatory therapy in acute myocardial infarction: results of the CAMI-1 study. Front Cardiovasc Med. 2021;8: 591714. https://doi.org/10.3389/fcvm.2021.591714.
Hausenloy DJ, Yellon DM. Time to take myocardial reperfusion injury seriously. N Engl J Med. 2008;359(5):518–20. https://doi.org/10.1056/NEJMe0803746.
Shemarova I, Nesterov V, Emelyanova L, Korotkov S. Mitochondrial mechanisms by which gasotransmitters (H(2)S, NO and CO) protect cardiovascular system against hypoxia. Front Biosci (Schol Ed). 2021;13(2):105–30. https://doi.org/10.52586/S556.
Penna C, Andreadou I, Aragno M, Beauloye C, Bertrand L, Lazou A, Falcão-Pires I, Bell R, Zuurbier CJ, Pagliaro P, Hausenloy DJ. Effect of hyperglycaemia and diabetes on acute myocardial ischaemia-reperfusion injury and cardioprotection by ischaemic conditioning protocols. Br J Pharmacol. 2020;177(23):5312–35. https://doi.org/10.1111/bph.14993.
Femminò S, Pagliaro P, Penna C. Obesity and cardioprotection. Curr Med Chem. 2020;27(2):230–9. https://doi.org/10.2174/0929867326666190325094453.
Briston T, Selwood DL, Szabadkai G, Duchen MR. Mitochondrial permeability transition: a molecular lesion with multiple drugtargets. Trends Pharmacol Sci. 2019;40:50–70. https://doi.org/10.1016/j.tips.2018.11.004].
Korzick DH, Lancaster TS. Age-related differences in cardiacischemia-reperfusion injury: effects of estrogen deficiency. Pflügers Arch. 2013;465:669–85. https://doi.org/10.1007/s00424-013-1255-7).
Ruiz-Meana M, Boengler K, Garcia-Dorado D, Hausenloy DJ, Kaambre T, Kararigas G, Perrino C, Schulz R, Ytrehus K. Ageing, sex, and cardioprotection. Br J Pharmacol. 2020;177(23):5270–86. https://doi.org/10.1111/bph.14951.
Arvidsson I, Eriksson E. Postoperative TENS pain relief after knee surgery: objective evaluation. Orthopedics. 1986;9(10):1346–51. https://doi.org/10.3928/0147-7447-19861001-06.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by Zhejiang Provincial Natural Science Foundation of China (grant no. LGF21H020006), Pioneer Innovation Team of Jiaxing Institute of Atherosclerotic Diseases (Grant no. XFCX—DMYH), The Key Medicine Disciplines Co‐Construction Project of Jiaxing Municipal (Grant no. 2019‐ss‐xxgbx), Program of the First Hospital of Jiaxing (grant no. 2021-YA-011), Jiaxing Key Laboratory of Arteriosclerotic Diseases (grant no. 2020-dmzdsys).
Conflicts of Interests
Honghong Zhang, Huilin Hu, Changlin Zhai, Lele Jing, and Hongen Tian have no relevant financial or non-financial interests to disclose.
Authors’ Contributions
The first draft of the manuscript was written by all authors. All authors read and approved the final manuscript.
Ethics Approval
Not applicable.
Informed Consent
Not applicable.
Data Availability
Not applicable.
Compliance with Ethical Standards
Not applicable.
Consent for Publication
Not applicable.
Code Availability
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Zhang, H., Hu, H., Zhai, C. et al. Cardioprotective Strategies After Ischemia–Reperfusion Injury. Am J Cardiovasc Drugs 24, 5–18 (2024). https://doi.org/10.1007/s40256-023-00614-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40256-023-00614-4