
Abstract
Because of the value of hydrogen as the future energy in no distant time, demand for efficient and scalable hydrogen production via electrochemical water splitting process has recently attracted considerable attention from industrial and scientific communities. Yet, several challenges associated with production remain to be addressed. One of the overriding challenges is the sluggish kinetics of oxygen evolution reaction (OER), which can have significant impact on the H2 production due to overpotential. To overcome this limitation, developing low-cost, robust and stable electrocatalysts very close to the same electrode activity as seen for iridium metal is crucial to solving the efficiency issue in the process. Therefore, timely review of progress in the field is vital to identify the electrocatalytic systems with the highest potential and, more importantly, to understand the factors which have positive contribution towards the electrocatalysts performance. We reviewed the progress made in the direction of designing binary and ternary alloys of transition metal-based electrocatalysts tuned with carbon materials. The review focuses more on the modulation of structural design and electronic conductivity that have been carried out by manipulating chemical compositions to moderate the surface adsorption free energies of the reaction intermediates, targeted to reduce overpotential. The strategic routes are discussed thoroughly with respect to the OER mechanisms and their derived-descriptors. However, numerous opportunities still remain open for exploration, particularly on the key challenge to obtain a route to unify electronic structure-activity and activity-multi-descriptor relationships for rational design of efficient electrocatalysts.
Similar content being viewed by others

Introduction
The generation of energy required to meet the increasing global demand should not compromise the environmental quality and sustainability of future life on the planet. It has been noted that the excessive use of non-renewable sources of mineral fuels in the last several decades caused a series of environmental pollution problems and especially the greenhouse effect and climatic changes. Inevitably, scientists and engineers initiated, many decades ago, the use of renewable energy sources which are environment friendly. Therefore, renewable energies have been identified as potential alternatives to fossil fuels that are associated with CO2 emissions. Sunlight is by far the most plentiful renewable energy resource, providing Earth with enough power to meet several hundred times of all humanity's needs [1]. However, it is both diffuse and intermittent and therefore, how best to harvest the energy and store it for times when the sun is not shining presents the challenge. Devices that use sunlight to split water into hydrogen and oxygen could be one solution to these problems, as hydrogen is an excellent energy carrier—a convenient form of energy. Hence, from sustainability point of view, a synergy between hydrogen and electricity and renewable energy sources is particularly gaining more interest. Thus, it worth investigating as the direct conversion of solar energy to hydrogen is considered the most sustainable energy production process without causing pollutions to the environment [2]. Accordingly, hydrogen production by water splitting process has emerged as a promising approach for converting huge amount of stored energy in sunlight to clean fuel called hydrogen fuel (H2). Hydrogen, as a sustainable energy carrier, not only has high energy conversion and storage efficiency, but it also emits no pollutants as its combustion process produces only water as a by-product. This limits unwanted releases into the environment and thereby can sustain earth’s hospitality.
Switching to low and ultimately no-carbon generation options of energy production, the history of past transitions can help us understand how the entire world moves towards climate-neutral energy transition. In the transition, there are clearly visible changes and more significant ones are still to come. The bell-ringing weather statistics of the growing levels of CO2 in the earth’s atmosphere (Fig. 1a), which causing rise in average global temperatures coupled with the projections of the data under different scenarios have led to suggested paths of actions [3]. As a result, a dominant trend in the change of energy source transition dynamics is the pursuance of different approaches in energy decarbonization from high-carbon energy source to zero-carbon energy option in the form of hydrogen (Fig. 1b). The impetus for this change comes from the deep impacts of human societies have had on the earth’s ecological environment during the past decades. This is in addition to the forecasts about what will happen in the future if stay without transformative action within the next decades. Accordingly, more and more countries are seeking ways towards zero-emissions in their energy sector, which is the central focus that pulls the attention of the scientific communities in today’s energy research—the need for decarbonization in global energy landscape. As a result, the development of water oxidation through electrochemical splitting process using electrolytic cells for hydrogen production from renewable sources has become a global motive towards a future sustainable energy package.

Copyright of World Carbon Budget, 2017, b a diagram depicting the evolution and transition of fuels in terms of H:C ratio. Reproduced with permission from ref. [5]. Copyright of Springer Open, 2021
a CO2 emission from fossil fuel combustion around the world. Reproduced with the authors’ permission from ref. [4].
Generally, the overall electrochemical water splitting process is encompasses of two half-cell reactions, namely hydrogen evolution reaction (HER) and oxygen evolution reaction (OER). In the process, water is reduced and oxidized to form H2 and O2 at the cathode and anode electrodes, respectively. Although the two half reactions are required to proceed simultaneously, but the cathodic reaction (HER) moves faster kinetically than the OER process. The latter is characterized with high overpotential due to its sluggish kinetics compared to the former with very low overpotentials [6]. Therefore, it is the OER barrier critically limiting the process in practical applications extensively [7, 8]. Consequently, electrocatalysis has a crucial role to play in H2O splitting process. Because the OER half electrochemical reaction demands highly effective electrocatalysts to minimize its inherent overpotentials toward efficient hydrogen production. As a result, electrocatalysis can play a crucial role in breaking the kinetic energy barrier of OER process that limiting the efficiency of the electrochemical reactions of water oxidation. Hence, the role of carbon-tunable alloys of transition metals in enhancing performance of electrocatalytic hydrogen production through favorable modulation of surface adsorption energy of OER intermediates is the focal point of this review.
In attempts to meet the demands of highly effective electrocatalysts for water splitting process, design and development of electrocatalysts to suit reaction conditions of the water electrolysis is part of the challenges associated with the process, particularly the half-reaction producing O2 from water oxidation. On the operating conditions, the water splitting process is either perform under acidic or alkaline condition. Often times, one condition has some advantages over other condition, such as high proton conductivity and lower gas permeability. Or sometimes one condition is characterized with high energy efficiency and fast hydrogen production kinetics [9]. For example, the operating condition requirement(s) of acidic medium for water splitting process restricts the functionality of OER electrocatalysts to generally noble metal and noble metal oxide electrocatalysts. This requirement necessitates high cost for the process arising from costs of electrochemical cell electrodes [10]. In comparison, conducting the reaction in alkaline medium broadens the selection decision between the non-noble metal and non-noble metal oxide electrocatalysts. This implies that electrocatalyst materials under different operating conditions meet one perfectly while are poor in the other. This has considerably engaged community of material scientists, particularly the surface scientists and engineers in development of new multi-operational electrocatalysts with suitable electronic and physical properties to offer superior performance with low-cost economic practicability [11,12,13,14,15,16]. Consequently, design of optimum OER electrocatalysts to fit in different reaction media and operating conditions with economic practicability, high activity, and excellent stability for electrolytic water splitting process still remains very challenging.
Therefore, this review has chosen to work on alloys of transition metals (mainly metals not of their other forms such as oxides or sulfides, phosphides, nitrides, carbides, borides, etc.) with respect to modulation of surface adsorption energy of OER intermediates for the overall water splitting process. However, it has been practically established that the alloys suffered instability and other morphological deficiencies caused by aggregation during synthesis [17, 18]. These identified limitations have opened windows for integration of conductive carbon supports with the alloys of transition metals (TMs). Consequently, there remain numerous avenues to discuss detailed routes to modulate surface adsorption energy of the materials in terms of structural and electronic properties. This also including morphology that synergistically has effects between various compositional components. Because these are variant factors that facilitate the adsorption/desorption ability towards the key reaction intermediates or regulate charge transfer during water electrocatalysis. Other factors including multifunctional active sites and improved electrical conductivity, porosity, and surface area architectural design are also used to overcome diffusion and mass transport of ions and produced gases and their relationship with OER activity and stability in both acidic and/or alkaline medium. These are design objectives towards one fundamental aim, which is to reduce energy consumption or overpotential. In particular, identification of the key contributions of surface adsorption free energies of the carbon-tunable alloys of TMs for the reaction intermediates in enhancing overall water splitting process have been reportedly achieved, but are dispersed and characterized with heterogeneity. Thus, guidelines or routes for designing electrocatalysts towards achieving that have not been fully established and therefore largely lacking. This implies that there is an obvious gap required to be bridged between the two disconnects. Hence, more efforts are required to be devoted to this point to establish the inherent and coherent trends in the electrocatalytic ability of carbon-tunable alloys of TMs electrocatalysts in OER process. These are what constitute the focus of the present review. The idea of the focus stems from the fact that, in addition to the electronic conductivity of noble metal-free nanoparticles, carbon matrix serves as a conducting medium that quickens the electron and charge transfer. Besides, carbon enhances adsorption and as well provides protective layer that enhances phase stability and prevention of aggregation of noble metal-free nanoparticles during synthesis. Moreover, the flexibility of carbon-containing electrocatalysts offer the feasibility to manipulate material structural design and electronic conductivity modulations via (1) constructing unique architectural surfaces that expose a large density of surface active sites, (2) integrating the noble metal-free nanoparticles with conducting carbon materials accelerates charge transfer and mobility of electrons and ions, thereby limits the kinetic reaction barriers of the OER electrochemical process, (3) building nanostructured architecture of the noble metal-free nanoparticles over high-conducting carbon materials to tuning electronic structure and optimize the thermodynamic OER intermediates adsorption/desorption on the surfaces of electrocatalysts, (4) capping the surface of carbon matrix with different surface dopants or functional groups not only disable the spontaneous surface oxidation of noble metal-free nanoparticles but also results in increase in charge carrier density of the target nanocomposite, which leads to increase in electrode–electrolyte interaction and enhance surface charge capacitance of the prepared material, (5) building architecture of the target electrode with an enormous surface area and varied hole sizes (porosity that controls diffusion) on which the oxygen evolution reaction occurs seamlessly, as large bubbles of oxygen escape easily through the big holes in the carbon matrix. As well, the structural architecture of carbon-based electrocatalyst nanocomposite prevents wetting of electrode surface—a common problem that makes electrodes less efficient. Also, opportunities to further manipulate carbon-tunable alloys of noble metal-free nanoparticles remains open for more exploration. The introduction of hetero-species rich with lone pair of electrons into bulk carbon matrix to enhance electrocatalysts performance with multifunctional surface sites/groups of such as N, and/or –NH2 that will play important roles in electron-transfer reactions. This in effect could further offer enhancement of carbon-based electrocatalysts performance activity due to reduced O-containing functionality and increased N-containing terminal nucleophilic sites instead of electrophiles.
In this review, the discussion centered on the performance index such as surface adsorption free energies of reaction intermediates used to evaluate the carbon-tunable alloys of TMs electrocatalysts activity, stability, and efficiency in water splitting process. Finally, future research prospects in the design of carbon-tunable alloys of TMs for OER towards effective and efficient hydrogen production are also discussed in the perspective of mathematical approaches comprised of high-throughput calculations and statistics under the machine learning process.
Fundamentals of electrocatalytic reactions of water splitting process
Electrolysis of water is nowadays considered as an essential and clean way to produce hydrogen, aiming to address global energy crisis and long-term energy-causing environmental pollution. Considering the fact that hydrogen could be believed to be an everlasting and promising energy resource owing to global water volume estimated to be around 1.4 × 109 km3 [19, 20]. Therefore, the process can easily be integrated with renewable energy sources such as solar, particularly that water is widely accepted as the most interesting source of sustainable hydrogen production [20].
The overall electrochemical water splitting process can be simply presented as in Eq. (1):
The electrolytic reaction appears simple, but this production method of hydrogen through electrochemical reactions taking place between two electrodes are more complicated than the described simple reaction. The reaction process involves multiple reaction steps. The multiplicity of the process is described by electrons captured or released by electrolytic ions at the surface of electrodes, resulting in a multiphasic gas–liquid–solid transition occurring within the overall process. During the multiphasic switch, water is continually split into hydrogen and oxygen (O2) through two crucial multi-proton/electron combined half-cell reactions, which are the cathodic hydrogen evolution reaction (HER) and anodic oxygen evolution reaction (OER). Therefore, in the quest to achieve an efficient water oxidation/splitting process, a clear and thorough understanding of both HER and OER mechanisms in different pH environment is crucial. This is also an important part considered in designing efficient and effective electrocatalysts. Also, it is undoubtedly an important factor that determines the acceptance of future large-scale application of the technology to satisfy the global renewable and clean energy demand, including free environment from pollution liabilities caused by the conventional hydrocarbon energy sources.
Oxygen evolution reaction (OER)
The electrochemical reaction process of oxygen evolution reaction (OER) is a four-electron transfer process occurring at the anodic electrode, which is regardless of whether the reaction is conducted under acidic or alkaline or neutral condition. The overall reactions for O2 evolution in different electrolytic solutions are as follows presented in Eq. (2 and 3).
It is the four-electron and/or four-electron–proton transfer process in alkaline and acidic reaction environment, respectively that always make the kinetics of OER proceeds sluggishly. This goes with attendant consequence of high overpotential caused by complex reaction mechanism characterized by more possible reaction pathways compared with HER. Owing to the sluggish nature of such electrochemical reaction kinetics, the reaction mechanisms and reaction pathways of OER are found to be more complicated than that of HER. The OER mechanism involves four-electron transfer process that produces multiple reaction intermediates, such as OH*, O*, and OOH* regardless of whether the reaction is carried out in acidic or alkaline/neutral electrolytic solutions [21]. It is worth mentioning that OER fundamentally occurs through the two main mechanisms, namely adsorbate evolution mechanism (AEM) and lattice oxygen mediated mechanism (LOM).
In the AEM reaction pathway for O2 molecules evolution, normally the proton–electron transfer process occurs both in acidic and alkaline reaction medium, as the sequence of reaction steps shown below and proven [22, 23]. The AEM reaction pathway proceeds through the HO*, O*, HOO*, and O*2 intermediate species.
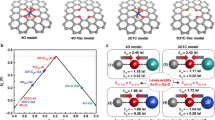
In acidic electrolytic solution:
In neutral or alkaline electrolytic solution:
where in the above scheme of reaction mechanism of OER, * symbolizes the active site on the surface of electrocatalysts and HO*, O*, HOO*, and O2* are designated intermediates absorbed at the active site of the electrocatalysts. From the above reaction steps whether in acidic or neutral/alkaline, the key important intermediate in the entire reaction procession is HO* intermediate. Therefore, the activity performance of an OER electrocatalyst can be primarily measured by its ability of adsorbing HO* over the surface and then producing adsorbed HOO* freely (see Eqs. (4), (5), (6) and (9), (10), and (11) for acidic and neutral/alkaline medium, respectively). For this reason, an in-depth understanding of the adsorption strength of these reaction intermediates over the surface of anodic electrode catalyst is vital for the overall OER electrocatalyst activity performance. This is owing that Gibbs free energy of adsorption is a key parameter governing the electrochemical reaction overpotential. In other words, it is therefore right to say that the value of the difference between (∆GO* − ∆GHO*) has a pronounced influence on the activity of an OER electrocatalyst. For example, it has been reported that the adsorption behavior of transition metal (TM) electrocatalysts and carbon (in the form of assembly of Fe, Ni, Co–N-doped graphene and CNTs) is directly connected to their electronic structure governed by the adsorption energies of O* and HO* intermediates [24,25,26,27,28,29]. Consequently, the difference between ∆GO* and ∆GHO* has traditionally been utilized as a universal descriptor to predict the OER activity of anodic electrode catalysts. This is represented by the Sabatier’s volcano-shaped relationship that has been commonly employed to explain the OER activity trends of the metal oxide electrocatalysts applied in both acidic and alkaline electrolytic solutions. On the account of the volcano-shaped curve, the best electrocatalysts reported so far are IrO2 and RuO2 in terms of their lowest theoretical overpotential for OER. These electrocatalysts are found worthy because of their optimum adsorption strength over the surface of the noble-metal oxide electrocatalysts, which are neither too strong nor too weak (Fig. 2a) [27, 30]. From the volcano-curve, the surface active site with the highest efficiency of electrocatalytic activity is the point at the tip of the volcano curve, which implies that the strength of the binding or adsorption energies of M–*OH → M–*O and M–*O → M–*OOH is similar, resulting in the lowest overpotential [31]. Similarly, by an ideal diagram of free energy of the different reaction steps presented in Fig. 2b, there will be no overpotential required to drive OER to proceed to the formation of reaction product if the free energy gap for each of the elementary steps presented above would remain the same at 1.23 eV [26]. However, that is an ideal situation which is impractical to be achieved. In a different approach, the overpotential of the electrochemical process of OER is determined by the rate-determining step of the reaction that is derived from the adsorption energies of the M–OH, M–OOH and M–O, as shown in Fig. 2c. The explanation of the plot indicates that the adsorption or binding energies of the M–OH and M–OOH maintained a constant difference of 3.2 eV (∆GHOO* − ∆GHO*). This occurred because both HOO* and HO* intermediates are adsorbed over the metal active site of electrocatalyst via the same adsorption pattern with an oxygen atom through a single bond because of similar symmetry [32].

Copyright of Wiley–VCH, 2011
a The volcano curve for OER activities for various metal oxide surfaces, b illustration of Gibbs free energy plot for an ideal OER electrocatalyst under potential U = 0 and U = 1.23 V, c correlative relationship between the adsorption energies of HOO* and HO* on a series of oxide of OER electrocatalysts. Reproduced with the permission of ref. [27].
It has been reported that the OER electrochemical reaction pathways are pH-dependent as well and therefore, the transfer of electron process occurs both in acidic and alkaline reaction environment [23]. The reaction pathways are presented in Fig. 3a. To achieve effective hydrogen production through water splitting process, a thorough understanding of the electrochemical OER mechanism is essentially required. Beginning from the conventional understanding of the OER mechanism over metal electrocatalyst surface, the reaction is characterized by involvement of four concerted proton–electron transfer processes with participation of the metal surface active site (M). This is what leads to production of O2 molecules from H2O molecules both in acidic and alkaline reaction media (Fig. 3a). The reaction mechanism is generally known as adsorbate evolution mechanism (AEM). In the illustration, the reaction pathways of acidic OER is presented in blue line. Generally, with the participation of electrocatalyst surface active site, the commonest reaction intermediates involved are M–OH, M–O, and M–OOH [24]. In an alkaline reaction environment which characterized with red line, the electrochemical OER takes different pathways. Initially, M–OH forms through an electron oxidation of OH─ ion adsorbed over the surface active site of the electrocatalyst. This is followed by conversion of M–OH into M–O after a couple of proton–electron transfer process. Thereafter, M–O undergoes two different reaction pathways to produce O2 molecules. In the first possible pathway, M–O reacts with OH− to form M–OOH intermediate, which subsequently produce O2 molecules along with the regeneration of the surface active site of the electrocatalyst via deprotonation of the intermediate (M–OOH). The other reaction route illustrated with the green color involves the direct combination of the two M–O species, which leads to evolution of O2 molecules and regeneration of the surface active site of the electrocatalyst. However, as the direct combination of the two M–O species is considered to have a high activation barrier [28, 33], it is therefore less likely to occur. Thus, another reaction route with complete elimination of the direct coupling of the M–O species in the scheme of the reaction mechanism has also been proposed (Fig. 3b) [33]. Besides, in situations where electrochemical reaction involved the lattice oxygen oxidation participation in the scheme of OER mechanism, the release of oxygen at the anodic electrode cannot be explained using the traditional OER mechanism [28]. Because the traditional mechanism with concerted proton–electron transfer process has failed to support explanation of the pH-dependent kinetics of OER processes, which was conducted recently with fabricated composite oxides of La, Sr, and Co; Pr, Ba, and Co; and Sr and Co. This was fundamentally detected in a study where the observed oxygen evolution or release was as a result of the involvement of lattice oxygen oxidation of different oxygen atoms, which were labelled as 34O2 and 36O2 in the OER process for La0.5Sr0.5CoO3–δ, Pr0.5Ba0.5CoO3–δ, and SrCoO3–δ. In addition, it was also found that their OER kinetics were pH-dependent on the RHE scale (Fig. 3d, e) [28]. The figures illustrate that all the measurements made with respect to pH were found to increase with increasing pH for La0.5Sr0.5CoO3–δ, Pr0.5Ba0.5CoO3–δ, and SrCoO3–δ, but LaCoO3 was not pH responsive. This is to say that pH controls the availability of lattice oxygen for kinetics function and further indicated that the electrocatalysts only function well with their surface mostly covered by lattice oxygen in a close interval of high pH values ranged between 12.5 and 14. In addition, the result further suggested that at high pH oxides are prone to formation of oxygen vacancy when their electronic band structures are highly hybridized [28]. Moreover, having pH sensitivity, the electrocatalysts masked the AEM reaction route and made the OER mechanism responded mainly to LOM. This result is quite a challenge to the most widespread explanation and understanding of the OER mechanism based on metal ions as surface active sites where the classical four proton–electron transfer steps takes place. On the other hand, the result contributed in providing new insights into the understanding of the OER mechanism, which at the same time, established new and/or alternative OER mechanistic pathways through which some of the overpotential-causing intermediates are completely avoided.

Reproduced with permission from ref. [24]. Copyright of the Royal Society of Chemistry, 2017; b, c Schematic graphical presentations of the commonly known oxygen evolution reaction mechanisms, namely, AEM and LOM, respectively. Reproduced with the permission from ref. [33]. Copyright of Royal Society of Chemistry, 2020, d, e pH-dependent OER activity on the RHE scale, the d plot represents a curve of CV measurements from O2-saturated 0.03 M KOH (pH12.5) to 1 M KOH (pH 14) recorded at 10 mV s−1 and the e plot portrays specific OER activity (current normalized by oxide BET surface area) at 1.55 V against RHE after iR correction as a function of pH. Reproduced with the permission of ref. [28]. Copyright of Nature, 2017
a The mechanistic pathways of oxygen evolution reaction on electrocatalyst surfaces represented by blue and red lines for acidic and alkaline reaction media, respectively.
Although much earlier than this investigation, it was previously reported and shown that La0.8Sr0.2CoO3–δ and La0.6Sr0.4CoO3–δ exhibited pH-dependent OER activity on the RHE scale [34,35,36,37]. Recently, Shi et al. [38] was also further observed and reconfirmed a noticeable pH-dependent OER kinetics with strontium cobaltite. All these results provided new landscape for fine tuning and structural adjustment of electrocatalyst towards the pH-dependent OER activity through the stress effect of lattice oxygen in the architectural structure of the electrocatalysts. This quietly suggests that lattice of an oxide electrocatalyst is useful for oxidation in the scheme of OER mechanism, acting as a reservoir for oxygen, storing and releasing it for reactions at the electrocatalyst surface under alkaline conditions. In this part of the efforts, lattice oxygen mediated mechanism (LOM) was introduced and its utilization has progressed significantly in the description of OER mechanism in the recent years [28, 33,34,35, 38]. However, the usefulness of AEM has not been discarded and thus is still relevant in the nonparticipation of lattice oxygen oxidation and lack of pH-dependent OER kinetics. This is particularly observed for LaCoO3 where the OER kinetics over the surface of the LaCoO3, which was mainly explained by the conventional concerted proton–electron transfer mechanism on the cobalt surface active site [32, 39].
In LOM of OER, the reaction mechanism proceeds with lattice oxygen-participation on the basis of exchange of lattice oxygen species in oxides of OER electrocatalysts, which involved an active strategic role of surface oxygen vacancies [40,41,42]. In the LOM-type reaction mechanism of OER, the reaction mechanism proceeds on the surface of two adjacent metal sites, which is quite different from the scheme of reaction steps in the traditional AEM (Fig. 3b, c) [35]. In this case, the reaction mechanism begins with an initiation of deprotonation of the two HO* on the adjacent metal sites, resulting in the generation of two metal-oxo species (Fig. 3c). In the following step, the two adjacent metal-oxo species directly couple to form the twosome O–O bound molecule rather than combining with H2O or HO* to produce HOO*, which eventually bypass the thermodynamic limitations of HO* and HOO* as it occurs in AEM. Thus, the OER kinetics of LOM must be faster than that of AEM because of the absence of concerted proton–electron transfer process. This is how O2 is formed and released, resulting in emptiness of the two unoccupied metal centers that are made to be available for subsequent occupation by OH─ for the next chain of oxidative reaction. Because LOM is independent on the limitation of HOO* intermediate in the electrocatalytic cycle, the overpotential problem characterized to OER is minimized by scaling the reactive relation between HO* and HOO*. Consequently, the LOM-type reaction mechanism of OER consumes less energy compared to AEM route. The underlying chemical physics of LOM explains that the Fermi level is lowered into oxygen 2p band induced by the strong overlap of metal 3d with oxygen 2p (Fig. 4a) [41], through which O2 molecule via direct coupling of metal cation and lattice anion is formed. This process occurs faster with less OER kinetics barrier compared to the reaction pathway through the concerted proton–electron transfer process of AEM. This was earlier proven where DFT studies have pointed out that trivalent ions such as La3+ was reportedly substituted with Sr2+ ion on the A-site of the perovskite structure, which resulted in move of the Fermi level much nearer to the computed O 2p-band center that was accompanied by a narrowed energy gap between the metal 3d and O 2p-band centers (Fig. 4b) [43,44,45]. In effect, it makes the metal center often at higher valence state and is responsible for promoting the generation of more electrophilic oxygen atoms that increases the interaction between metal and oxygen [46]. In a different study, it was further proved that as the Fermi level shifts down and becomes closer to the O 2p state from La CoO3 to Sr CoO3, the antibonding state below the Fermi level showed oxygen character, an indication of mutual covalency between metal and oxygen in form of metal–oxygen bond (Fig. 4c) [47]. In this context, it has been reported that enhancing the hybridization overlap between the metal 3d and the oxygen 2p orbitals can trigger and initiate the lattice oxidative OER activity. This can be achieved by changing the redox sites from the electronic structure of metal ions to the lattice oxygen. This is what makes and explains the oxidation of lattice oxygen in the perovskites which is thermodynamically favorable when the Fermi level of O 2p states in the electronic structure of perovskite lies above the redox potential or energy of the O2/H2O couple, as presented in Fig. 4c [28, 48]. However, it is important to note that the factor responsible for the observed rapid kinetics of OER mediated by LOM is that O p-band centre was near enough to Fermi level [44]. This could be an exposed surface site purposely designed to cause structural adjustment in the materials (constructible Sr–Co-based oxides in alkaline OER) and perhaps initiated through an abridged pathway for creation of spin triplet O2 form. Because that the oxygen species on the surface of electrocatalyst are highly dependent on the pH of electrolyte [49], which could be a pathway for structural degradation in oxide electrocatalysts to unstable electrocatalytic activity during long-term electrochemical reaction cyclic rounds. Overall, despite the existing difference the two mechanistic pathways for OER, the oxidative phenomena occurring in the process of an OER can still be explained by both the AEM and LOM theories. Because, for example, the excessive oxidation of surface active sites of metal-based electrocatalysts for the AEM and participation of lattice oxygen for the LOM generally lead to material instability used for electrocatalysis [48]. However, it is also essential to note that the amorphous phase of metal oxides mostly exhibit better electrocatalytic activity compared to the same well-crystalline materials. Because the lattice oxygen can participate in the electrocatalytic oxidative reaction easily in the former than in the latter one [50].

Copyright of Macmillan Publisher Limited, 2016; b, c the electronical presentation of structure of Co-containing perovskite oxide, b the electronic difference between the Co 3d-band center and O 2p-band center against the O 2p-band center relative to the Fermi level for stoichiometric ACoO3 perovskite; c Schematic rigid band diagrams of LaCoO3 and SrCoO3 combined with the position of the O2/H2O redox couple at pH 14 (4OH− → O2 + 2H2O + 4e−) was 1.23 V against RHE. Reproduced with permission from ref. [28]. Copyright of Nature, 2017
a Schematic illustration of band structure of perovskite oxides to demonstrate the difference redox process at the energy of M d-band decreased with respect to the O 2p-band. Reproduced with the permission from ref. [35].
Therefore, as in the case of AEM, OER activity performance of an electrocatalyst is directly linked to the kinetics of the transfer of electrons and protons involved in the electrochemical reaction. Accordingly, the reaction rate is determined also by the number of surface active sites. Therefore, it is important to prepare electrocatalyst with sufficient surface area full of active sites to enable facile protons/electrons and other mass transport. On the contrary, in the case of LOM, OER activity performance is determined by full participation of lattice oxygen and thus, composition modification of an electrocatalyst through doping may expose or increase more active reaction sites by creation of more lattice oxygen species. This could possibly induce and confer moderate surface adsorption strength on oxygen intermediates due to surface defects, vacancies and edges that can enhance the inherent activity of OER electrocatalysts.
pH-dependent LOM and its implication on overpotential of OER
It is worth showing how OER enhancement by LOM is pH-dependent. As earlier mentioned, the evolution of O2 molecules from the oxygen species on the surface of electrocatalyst is highly influenced by the pH of electrolyte. This was proven via 18O-isotopic labelling experiment [28]. Specifically, in alkaline electrolytic solution, the LOM occurrence has been connected with the observation of pH-dependent OER kinetics on the RHE scale, which can be deduced from Eq. (14), as applied by Yang et al. [51].
where i is the OER current, θ is the surface coverage of the adsorbed OH or OOH intermediates, \({C}_{\mathrm{OH}}\) is the concentration of OH ions, ∆G is the reaction free energy, R is the universal gas constants and T is the temperature during the measurement.
From the equation above, it is very clear to understand that altering the pH to increase kinetics of activity can result in the modification of the exponential term by changing the energy of the adsorbed intermediates or increase the pre-exponential term by increasing the surface coverage or the OH− concentration, leading to increase of OER kinetics. This has previously been confirmed [28, 32, 38, 41, 52, 53], where increase of pH from 12.5 to 14 led to an improved OER activity. Accordingly, these studies have proven pH-dependence of the OER kinetics and their electrochemical reactions passed through LOM pathway. The reported OER kinetic currents were increased progressively with change of pH values and as a result, in all the experiments conducted, the highest OER kinetic current corresponded to pH 14.
Furthermore, the strong metal–oxygen covalence is also playing role in reducing overpotential problem of OER. This is primarily induced and triggered by lattice-oxygen oxidation during OER process. Between the two OER mechanisms, the conventional AEM that takes the reaction route through the four concerted proton–electron transfer steps with multiple adsorbed intermediates is much restricted always with a minimum theoretical overpotential of ~ 0.37 V [32, 54, 55]. On the contrary, the recently accepted LOM relies on the participation of a lattice-oxygen oxidation and thus, is a non-concerted proton–electron transfer process. Thus, it enables the mechanism to bypass the earlier mentioned limitation for AEM and thereby potentially offers better OER activity with low overpotential [52, 55,56,57], as depicted in Fig. 5. The figure displays the free energy diagram under potential U = 0 and 1.23 V of Sr3(Co0.8Fe0.1Nb0.1)2O7–δ (Ruddleden-popper oxide). This was used for OER through both AEM and LOM where the theoretical thermodynamic overpotential (η) of the oxide electrocatalyst based on LOM recorded the least value of 0.22 V (Fig. 5b). This was found to be significantly more than 3 times lower than the value of 0.71 V (Fig. 5a) attained by AEM [48]. Similar results were previously reported with state-of-the-art oxides full of activated oxygen sites, such as Zn0.2Co0.8OOH (0.270) [55] and La0.5Ba0.25Sr0.25CoO2.9δF0.1 (0.256) for OER [58]. This supports the potential of LOM as an efficient and energy profitable route for electrocatalyzing the anodic reaction involving lattice oxygen sites for OER characterized with a problem of overpotential. As such, the mechanism can proceed via reaction pathway with low theoretical thermodynamic overpotential as compared to AEM. This means that the lattice oxygen mechanism possesses a lower reaction energy barrier compared to AEM.

Copyright of Wiley, 2020
a, b The AEM and LOM with their corresponding free energy of oxygen evolution reaction steps under potential U = 0 and 1.23 V, respectively, of which LOM and AEM showed varied overpotential values. The LOM showing low overpotential of OER compared to AEM reaction pathway of the same anodic electrochemical process. Reproduced with the permission from ref. [48].
Integration of transition metal alloys and conductive carbon materials for OER process
Transition metal alloys
Several studies conducted in the past several years produced a significant number of transition metal compounds including their alloys, which were primarily designed and developed as efficient OER electrocatalysts. Because that transition metals possess several unique physical and chemical properties with flexibility of fine-tuning such properties into desired structures electronically and crystal form as well as creation of defect sites for the surface adsorption of intermediates, oxygen vacancy/excess and excellent mass transport ability. In addition, these materials are mated into alloys to contain enough surface active sites for an excellent electrocatalytic activity for OER in the cause of water splitting process. It is with such uniqueness, transition metal alloys are considered as ideal model materials for major electrocatalytic studies with the prime objective to establish correlative relationship between their physico-chemical properties and the corresponding electrocatalytic activities in OER process. In this context, the discussion here will basically dwell on structure–properties–electrocatalytic activity relationship in terms of reactants and intermediates surface interactions for OER process. Because structure provides a framework for arrangement, rearrangement and strategic placement of key surface active sites.
On the basis of the OER volcano curve (Fig. 2a), the electrocatalysts are divided into two sides of the curve with those on the left side of the curve have a strong ability to bind with oxygen, signifying the potential-determining step of the HOO* intermediates propagation. However, on the right-hand side of the curve are metal oxides with low oxygen-binding strength representing the potential-determining step of the HO* intermediates deprotonation step. Nevertheless, to aid ways of achieving a high OER electrochemical activity, an optimum oxygen adsorption strength of the same value of (∆GO* − ∆GHO*) = 1.6 eV is required [17], which corresponds to the arrowhead point of the OER volcano curve summit. From the curve, it is very clear that transition metal oxides are also part of the OER electrocatalysts, particularly Co, Ni, and Mn and are on the left-hand side of curve, which translates their possession of strong oxygen-binding behavior that can negatively affect the rate-determining step of the HOO* intermediates propagation. Therefore, to tailor an optimum oxygen adsorption energy of the OER intermediates over the surface of transition metal alloyed electrocatalysts along with their electrical conductivity, several efforts have been exercised including alloying through compositional manipulation towards morphological restructuring for enhanced performance in OER process.
Overall, alloying approach allowed development of varieties of hybrid structures of transition metal-based materials with synergetic effect acceptable to both HER and OER processes [17]. Alloying among the transition metals is a strategy of downshifting the d-band centers of combined metals to match the Fermi level. In addition, the interface between two or three blended transition metals could result in the formed alloyed metals to retain the suitable hydrogen (H) and hydroxyl (HO) adsorption energies, leading to an optimum Gibbs free energy of the HO* and HOO* intermediates’ state. Thus, granting a superior OER in the overall water splitting technological process. This is achievable, particularly in transition metal atoms that hold large number of unpaired d-band electrons and unfilled d-orbitals. Therefore, according to Brewer–Engel theory the unpaired d-band electrons are susceptible to initiate chemisorption bonds with hydrogen atoms [59], which could easily facilitate HER in the overall water splitting process. Similarly, in the case of hydroxyl, delocalization, and resonance of electrons in the alloys cause electron deficiencies in some part of the bulk system of the alloy and enables them to adsorb OH− and OOH− for activation or defects provide surface active sites for activation of OH− and granting an enhanced OER activity performance. Alloying binary or ternary composition of transition metals offer more possibilities in improving electrocatalytic performance of their electrocatalysts through tuning metals composition and proportions [60, 61]. On the other hand, bare alloyed transition metal-based electrocatalysts suffer from shortage of low conductivity due to aggregation normally caused by lack of self-supporting platform and thus causing low surface area. This results in poor mass transport and stability in acidic and alkaline electrolytic solutions [60,61,62]. These are some of the challenges, which to date, have greatly obstructed the actual and full utilization of unsupported transition metal alloyed electrocatalysts for broad applications in large scale production of hydrogen. To address these challenges, addition of a highly conductive materials such as carbon has become necessary.
Transition between alloys of transition metal and carbon-tunable alloys of transition metal
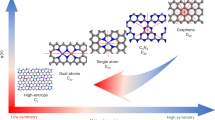
As generally known, conductive carbon supports such as graphite, carbon nanotubes, and graphene possessed excellent stability and conductivity. It has been reported that integrating carbon nanomaterials with transition metal into hybrids or alloys addressed the above mentioned challenges encountered in the utilization of bare transition metal alloyed materials in electrocatalysis [62,63,64,65,66]. In addition, it has been identified that the mode of adsorption of integrated forms of transition metal alloys and carbon supports is directly linked to their electronic structure [67]. Therefore, supporting transition metal alloys on conductive carbon phase openly tune to extend the surface utilization and stability of the alloyed phase, as well as enhancing the electrical conductivity. This is closely related to the creation of cooperation resulted from interfacial interaction between carbon and alloy of metal particle components. As shown in Fig. 6, the carbon phase provides conductive network that enhances electrical connectivity between metal alloyed phase and the electrode, which as a result could grant full utilization of the alloyed electrocatalyst for OER processes. Also, in the hybrid, carbon phase brings about enhancement of surface active sites exposition that accelerates transport of charged carriers and intermediates, as well as electron transfer that always occurs at the and across hetero-interface of transition metal alloyed particles and carbon phase. Thus, the electronic and structural hybridization between carbon phase and transition metal alloyed particles can regulate their adsorption behavior towards OER reactants and intermediates, which in turn can greatly enhance the electrochemical activity of water splitting process for hydrogen production.

Copyright of Wiley–VCH, 2017
Imagery modelling of the role of carbon in the carbon-tunable alloys of transition metal electrocatalyst for simultaneous hydrogen evolution and oxygen evolution reactions. Adapted with the permission from ref. [42].
In comparison with the corresponding counterparts with a single metal part, carbon-tunable alloys are more promising owing it to their demonstration of highly impressed activity for the OER process in water splitting process compared to single or unalloyed systems (see Table 1). The promotion in their activity resulted due to promoted synergy between carbon nanostructure and the transition metal alloyed nanoparticles [68,69,70]. Hybridization between the transition metal alloyed atomic orbitals and carbon nanostructure creates unique interface that exposes and augment surface area as well as active sites. Such effect promotes both adsorption and transportation of reactants and intermediates for OER process and in return, stimulate high electrocatalytic water splitting performance [71,72,73]. On the material protection, the integration of transition metal alloys with carbon materials reduces aggregation and oxidation of the active components of the formed material through supporting and coating, respectively by the carbon materials. As a result, this improved the activity life-span of the material with superior long-lasting stability [74,75,76,77,78,79,80,81,82,83,84]. This was further confirmed with the alloys of FeCo supported on GRO designed for OER process. The unique structure of the carbon in the materials promoted electron transfer, mass transport ability and stability of the material [79, 84]. In addition, it was also reported that carbon support restricted aggregation and oxidation of Mo nanoparticles and consequently, the electrocatalyst exhibited a low overpotential and excellent long-lasting stability with a negligible current density loss at 20 mAcm−2 within a period of 10 h [74].
Furthermore, in OER process, carbon framework also plays vital role in the electrochemical reaction. The conductive carbon nanomaterials framework possess electronic chemical structure comprised of both sp2 and sp3 hybridizations, which is in addition to rich pool of oxygen-containing surface functional groups and defects, allows the carbon structure to efficiently improve the electron transfer process and superior surface contact with electrolytes [78]. It is such physico-chemical properties of conductive carbon supports coupled with their intimate electronic interaction with transition metal alloys that offers an excellent avenue for efficient mass transfer process to occur on the surface of the combined materials. This eventually leads to optimization of the surface adsorption free energies of the reaction intermediates of the overall water splitting process, particularly for the universal activity performance descriptors, i.e., (∆GO* − ∆GHO*) for OER process. It is therefore worthwhile to show a series of electrocatalytic performance of carbon-tunable alloys of TMs relative to material design for water oxidation electrocatalysis on the scale of descriptors of Gibbs free energy of adsorption for O* and HO*(∆GO* − ∆GHO*) intermediates in the overall water splitting process.
Designing efficient OER electrocatalysts based on AEM and LOM as descriptors of activity performance
Knowing that the renewable hydrogen fuel production via electrocatalytic water splitting process is always accompanied with the evolution of oxygen that takes more energy conception and consume a lot of it due to its sluggishness in nature kinetically and therefore, in-depth understanding on how the materials that electrocatalyze the OER function is essential for the development of efficient water splitting process relative to energy consumption. It is broadly believed that OER proceeds via two distinct mechanisms, namely, adsorbate evolution mechanism (AEM) and lattice oxygen mediated mechanism (LOM). In this part, the discussion will largely focus on the utilization of the difference between ∆GO* and ∆GHO* (∆GO* − ∆GHO*) employed as a universal key descriptor for the prediction of OER activity performance and LOM that is fundamentally involves redox reactions of lattice oxygen. This type of reaction mechanism is different from the conventional OER schemes of reaction particularly that the metal–oxygen covalency is its powerful descriptor of performance for OER activity.
4.1 Design of efficient OER electrocatalysts based on (∆G O* − ∆G HO*) as descriptor of activity performance through AEM
The OER in the electrocatalytic water splitting process is generally entailed to occur on metal surface that proceeds through four concerted proton–electron transfer steps, which are understood taking place on ionic metal centers [26], harvesting O2 molecules through a pH-independent activity on the reversible hydrogen electrode scale [101]. With this type of mechanism, high OER activity can be regulated to occur through optimization of adsorption strength of the reaction intermediates (HO*, O*, HO*, and O2*) on the surfaces of metal-based electrocatalysts that should be neither too strong nor too weak. This has been supported with experimental results that the OER activities on metal surfaces is directly associated with electrocatalysts electronic structural parameters such as estimated eg occupancy of surface transition metal ions [102]. It is important to know that though HO*, O*, HOO*, and O2* are designated intermediates absorbed on the active sites of the electrocatalysts. No matter whether reaction is taking place in acidic or neutral/alkaline, the key important intermediate in the entire reaction procession of OER is HO* intermediate and therefore, the activity performance of OER an electrocatalyst is primarily determined by the ability of adsorbing HO*over its surface and then producing adsorbed HOO* freely.
Although this traditional mechanism that focused on the redox surface chemistry of metal ion has been strongly challenged by the following observatory facts, viz, (1) that some highly active oxides exhibited pH-dependent OER activity characteristic on the RHE scale [37, 38, 103], implying that non-concerted proton–electron transfers participated in the electrocatalytic process of OER [104]; (2) that the bulk oxide electronic structure of perovskites was reportedly participated in electrocatalyzing OER activities. This was evidently proved by enhanced OER activities correlated with increased oxygen 2p-band centers close to Fermi level [44, 45] and metal–oxygen hybridization [103, 105], indicating the role of bulk electronic structure in the OER kinetics [42, 46, 106, 107]; (3) that the mass spectrometry measurements of oxides such as NiCo2O4 [108], IrO2 [109], RuO2 [110, 111], and Co–Pi [112] all used for OER provided evidence of the evolved oxygen emerged not only from the water molecules but also from the oxides [112,113,114]. Therefore, the mechanism is still relevant in the discussion of activity descriptor of OER. In OER, the difference between ∆GO* and ∆GHO* (∆G*O − ∆G*OH) is employed as a universal descriptor to predict their OER activity [115, 116]. For this reason, establishing a relationship between the adsorption free energies of intermediates and overpotential of electrocatalysts is key to rational design of electrocatalysts [98, 117]. Thus, to work within the frame of an ideal electrocatalyst for OER, it is required for the adsorption strength of the key intermediates (HO*, HOO*, and O*) to be neither too strong nor too weak.
In accordance with the above context, metallic constituents of Fe, Co, Ni, FeCo, and CoNi, as well as FeNi alloys were fully encapsulated within the single graphene layer shell to avoid oxidation of the alloyed nanoparticles when exposed to oxygen in an air environment [98]. The characterization results confirmed that the corresponding alloys were effectively encapsulated in the single layer graphene shell and tested for OER in 1 M NaOH electrolytic solution. As presented in Fig. 7a, several linear sweep voltammograms (LVSs) were conducted to measure the OER activity performance of all the prepared electrocatalysts along with the reference materials, i.e., CNTs, few-layer-graphene nanosheets and IrO2. The polarization curve disclosed that all the transition metals and their corresponding alloys electrocatalysts outperformed with greater OER activity than pristine CNTs and graphene nanosheets. In the trend of overpotential, FeNi@NC possessed the best OER activity compared to all other electrocatalysts. More interestingly, the onset potential of the best electrocatalyst (FeNi@NC) was only 1.44 V vs RHE, and the potentials at 10 and 20 mA cm−2 were 1.51 and 1.54 V vs RHE, respectively (Fig. 7a), which was even better than the commercially based IrO2—the universally acceptable OER reference electrocatalyst used to check quality and validate activity performance of any other developed OER electrocatalyst. The Tafel slope of the FeNi@NC was 70 mV dec−1 that is closer to the value of 63 mV dec−1 of IrO2 (Fig. 7b), signifying that the FeNi@NC worked in a similar ability closer to IrO2 by driving the OER process at low overpotential. In addition, the electrochemical stability of FeNi@NC was quite superb, as the polarization curve of FeNi@NC after 10,000 cycles remained almost the same as in its initial test (Fig. 7c). Moreover, it was further indicated that the potential values of FeNi@NC recorded at different current densities of 40 and 100 mA cm−2 before and after tests were only slightly increased (Fig. 7d), which was obviously even superior to the electrochemical durability of the commercially based IrO2.

Copyright of the Royal Society of Chemistry, 2016
Electrocatalytic OER performance test of transition metal elements and their corresponding graphene encapsulated alloys (M@NC). a Comparison of OER polarization plots between M@NCs and the two of CNTs and IrO2 with the same mass loading, b Tafel comparison plots betwenn FeNi@NC and IrO2, c Durability test of FeNi@NC in comparison with IrO2 in alkaline electrolytic solution, d comparison of potential changes in current densities at 40 and 100 mA cm−2 for FeNi@NC and IrO2 during the durability test. Reproduced with the permission from ref. [98].
Although the difference in activity of all the developed electrocatalysts was fundamentally associated to the difference in the metal types, but to gain in-depth understandings on the nature of structural configuration of the graphene shell encapsulated transition metal alloys (M@NCs) associated to the OER activity performance, DFT calculations were also employed. As presented in Fig. 8, the OER activity performance of the prepared electrocatalysts were evaluated on the basis of the surface adsorption free energies of the reaction intermediates, i.e., HO*, HOO*, and O*, as there is strong correlation between the adsorption free energies of reaction intermediates and the overpotentials that inform about energy consumption of an electrochemical process [98, 117]. From the DFT analysis, the research group found that the FeNi@C involved in the electrochemical interaction as exactly as in the generally accepted OER steps, as shown the Fig. 7a. As a result, the free energy of various intermediates for the OER electrocatalyzed by FeNi@C at the different constant potentials were analyzed and found that the free energies for all the steps involved, an overpotential downhill of about 0.49 V compared with the equilibrium potential is required to drive the OER process over FeNi@C electrocatalyst (Fig. 8b). This was controlled by the surface adsorption free energies of the reaction intermediates HO*, HOO*, and O* over the graphene surface. This was evidently shown in Fig. 8c, where a good scaling relationship between the adsorption free energies of HO* and HOO* was established, implying that the two intermediates were adsorbed over graphene surface with the same type of bond. The result is consistent with the wide range of oxides including rutile, perovskite, spinel, and rocksalt and thus, was considered as a universal scaling relation analogous to other results reported in the literature [27]. Accordingly, the overpotential for the OER process over the FeNi@C was measured by the difference between the adsorption free energy of O* and HO*, denoted by ∆G(O*) − ∆G(HO*) and employed as the activity descriptor for OER process. The calculated overpotential of the process displayed a volcano shape, as presented in Fig. 8d. At the end, the calculated overpotential was compared with the experimental values and quite agreed with each other (Fig. 8e), which means that the theoretical model employed to describe the activity performance of the prepared electrocatalysts by the use of ∆GO* − ∆GHO* was quite efficient in the prediction and thus is a promising route to electrocatalysts performance evaluation for OER process. Hence, the development here to learn is that the structural electronic properties of graphene shell was evidently the responsible factor that altered the electrons transfer ability of the transition metal alloys nanoparticles and further tuned the adsorption energies of the reaction intermediates on the graphene surface. This was promoted by wide electronegativity difference Fe and Ni that enabled allowable flow of charges between the two metal. Consequently, the OER activity performance descriptor (∆GO* − ∆GHO*) can be adjusted to the optimum value by changing the metal type from either of the Fe, Ni, and Co and their corresponding alloys as it occurred experimentally. Overall, the activity descriptor can serve as an efficient way towards a rational design and development of highly efficient and low-cost OER electrocatalyst, as well as an energy saving strategy for this the most energy consuming part of the water splitting process.

Copyright of Royal Chemical Society, 2016
The theoretical interpretation of OER over transition metal elements of Fe, Co, and Ni and their corresponding alloys encapsulated in graphene shell labelled as M@Cs (a) an imagery models of the OER steps over the transition metal alloys encapsulated in graphene shell, in which the graphitic carbon shells encapsulated the atoms of Fe and Ni represented with red and green colors, respectively while the C with gray color, b free energy profile for the OER over FeNi@C under potential (U = 0), equilibrium potential for oxygen evolution (U = 1.23 V), and minimum potential (U = 1.72 V) where all steps run downhill, c linear relation between the free energy of HO* (∆G(HO*), and ∆G(HOO*) intermediates on different electrocatalysts, d the calculated negative overpotential (ηcal) against the universal descriptor ∆G(O*) − ∆G(HO*) on different electrocatalysts. The data of oxides were cited from ref. [27], e calculated overpotential ((ηcal) vs the experimental overpotential ((ηexp) over M@C electrocatalysts. All experimental overpotential ((ηexp) values were recorded at 2 mA cm−2. Reproduced with the permission from ref. [98].
Contrary to the HER, OER is kinetically sluggish and as a result consumes a lot of energy, which remains as an effective resistance to the overall electrocatalytic water splitting process and its large scale application. However, with the tuning of surface Gibbs free energy from HO* to O* intermediates that are believed as the rate-determining descriptor for the theoretically intrinsic activity of OER electrocatalysts [52, 81], such reaction challenge can be overcome. In recent years, it has been widely believed that non-noble-metal-based electrocatalysts have attracted research attention incredibly, as they have shown much higher OER than the commercially and reference-based electrocatalysts of Ir/C and RuO2 in alkaline solution [81]. Among the class of non-noble-metal-based electrocatalysts, transition elements because of their low cost and long-term stability under low oxidation condition due to their special 3d electronic configurations [118], are considered as the most attractive in the class. However, because of the exhibition of poor electrical conductivity among some of them, low specific surface area and poor intrinsic activities, their OER overpotential are still found to be greater than 200 mV in alkaline media [24, 118]. Likewise, under acidic condition, the noble-metal-based materials, particularly Ir-based materials have still remained the reference and benchmark OER electrocatalysts [119]. Integration of carbon matrix with alloys of TMs can enhance the chemisorption characteristics of oxygen-containing intermediates to promote the kinetics of OER activity. Therefore, combining alloys of TMs and a substrate with strong electronegativity is one of the most favorable strategies for constructing high-activity OER electrocatalysts. To prove such, a novel alloy containing CoP nanoparticles embedded in N-doped carbon nanotube hollow polyhedron (NCNHP) was fabricated as a bifunctional electrocatalyst for the overall water splitting process [120]. In this research, the DFT calculation shown that transfer of electrons from NCNHP to Co improved the electronic state of the metal’s d-orbital to be nearby the Fermi level, which was responsible in increasing the adsorption strength of the generated overall intermediates of OER and thus, enhanced the overall electrocatalytic water splitting activity (Fig. 9a–d). Fundamentally, the overall process of OER over the electrocatalyst was facilitated though deoxygenation of H2O molecules and dehydrogenation of OH− ions by enhanced chemisorption of HOO*, and O* over the anodic electrode electrocatalyst of Co embedded in NCNHP. Accordingly, the electrocatalyst was able to facilitate synergistic chemisorption of all the reaction intermediates on its surface, which eventually led an enhanced OER activities seamlessly (Fig. 9a–d). Therefore, tuning surface structure of an electrocatalyst with sufficient surface area that enables facile electron and other masses transfer efficiently and alter the interaction between metal and carbon framework can facilitate optimum surface adsorption free energies of OER intermediates particularly HO* and O* species. Therefore, can be employed as activity descriptor of an enhanced OER process.

Copyright of American Chemical Society, 2017
a, b the comparison of polarization plots of different electrocatalysts for hydrogen and oxygen evolution reactions, respectively, c the water splitting polarization curve of the CoP/NCNHP||CoP/NCNHP electrodes, d the calculated free energy diagram of hydrogen evolution reaction over the electrocatalyst from the overall water splitting process where 1 M KOH was used as an electrolytic solution. Reproduced with the permission from ref. [120].
In recent years, transition metal alloyed nanoparticles tuned with heteroatom-doped carbon supports through the pyrolysis of metal–organic framework (MOFs) has become a hot topic for the development of highly efficient electrocatalysts. In this preparatory process, in the presence of carbon or reducing agents sourced externally, transition metals such as Fe, Co, and Ni ions around the organic framework in presence of doped-carbon can be reduced in situ to metal or alloyed nanoparticles encapsulated in a heteroatom-doped carbon frame. Transition metal alloys including FeNi [95, 99], FeCo [93, 95, 121, 122], and CoNi [123, 124] have been synthesized from MOFs by pyrolysis for their applications in the area of electrocatalysis. Recently, Zhang et al. reported FeCo bimetallic N-doped porous carbons (FeCo–C/N) sourced from the calcination of ZIFs [121]. Of all the electrocatalysts derived from MOFs exhibited excellent OER activity performance owing to their distinct structural, electronic and compositional features. For example, ultrathin CoNi-MOF nanosheet arrays (CoNi-MOFNA), which was highly active OER electrocatalyst [125], as with the electrocatalyst an extreme low overpotential of 215 mV at a current density of 10 mA cm−2 was achieved. In addition, its mass activity was 14 times that of commercially based RuO2. Moreover, its current density of CoNi-MOFNA was not significantly deteriorated even after 300 h of uninterrupted electrolytic process. The outstanding electrocatalytic activity performance of all these MOF-derived transition metal alloys was ascribed to improved transfer of charge from the core metal alloyed nanoparticles to the carbon nanosheet layers [125]. In addition, the carbon support prevented the aggregation of the core metal alloy nanoparticles during the OER process. This not only contributed to the creation of more active sites, but also ensured that sufficient used of the active sites. At the same time, the porous nature of the MOF-derived transition metal alloys facilitated easy mass transport that was responsible for observed accelerated OER reaction kinetics. This implies that the high electro-conductivity observed was promoted by intimate contact between core metal alloys and their corresponding graphitic carbon structure that ensured fast electron transport due to shortened distance of diffusion by the carbon electronic and porosity architecture. This further encouraged fabrication of not only other binary alloys but also the ternary alloy such as FeCoNi encapsulated with nanocarbon on RGO. In brief, MOF-derived transition metal alloys encapsulated in carbon nanostructure exhibited prospect in the application of OER process. Because of compositional optimization and structural functionalization, the MOF-derived transition metal alloy possessed electron conductivity that made the reaction intermediates formation route to follow through the low conversion kinetics. Consequently, such allowed the reaction to proceed with remarkable OER electrocatalytic behavior (Fig. 10a). In comparison, although MOF-derived electrocatalysts synthetic mechanism and characterization are still in the initial or early stage, however, Fig. 10b shows that with appropriate compositional and structural design, high OER activity with overpotential of no more than 300 mV at 10 mA cm−2 is practicably achievable [112]. Therefore, more wide scale and thorough mechanistic studies are superficially and desirably in need to guide the future rational fabrication of MOF-derived transition metal alloyed electrocatalysts with definite structure fit for electrocatalytic and long-lasting OER activity. Specifically, DFT calculation studies on the surface adsorption energies of the OER intermediates is highly required. Because it is an outcome of the concept of the thermodynamic-limiting step that describes the OER kinetics in either acidic or alkaline medium. In fact, a link between surface electro-adsorption energies of OER intermediates, particularly the HO* and O*, and OER electrocatalytic activity can provide an opportunity route to exploit more of electronic and structural engineering of surface of MOF-derived electrocatalysts in the future designs and development.

Copyright of Wiley–VCH, 2021
An illustrative comparison of activity performance evaluated for different OER electrocatalysts conducted 1 M KOH, a metal organic framework (MOF)-based and d metal organic framework (MOF)-derived electrocatalysts. Reproduced with permission of ref. [126].
Strategies for regulating electronic structure for optimization of the values of ∆G O* − ∆G HO*
Based on what has been presented, it is possible to design and develop an excellent active electrocatalyst of OER process by modulating the electronic structure to optimize the value of free energy of surface adsorption of OER intermediates. This means that optimizing the free energy of adsorption (∆G) of an electrocatalyst can fundamentally accelerate the reaction kinetics of the concerted four-proton coupled reaction in the course of the OER process. On the other hand, although it has been argued that in some catalytic reactions associated with surface adsorption of hydrogen neither electron transport ability nor surface area of a catalytic material exert influence on the reaction kinetics, but electrocatalytic OER process are parts of the essential requirements for outstanding activity. This is therefore to some extent, fine tuning the free energy (∆G) of adsorption of electrocatalyst is most often regarded as an essential route to enhance electrocatalytic activity [126, 127]. However, improvement of transfer of electrons and surface area could lower the consumption of energy (overpotential) of an electrocatalytic process and consequently, cannot be left unattended.
Strategies of tuning free energy of adsorption (∆G) of electrocatalysts
The strategies mainly include
-
i.
Elemental doping of carbon support To enhance activity performance of OER process, elemental doping of hetero-atom is one of the routes to improve the OER activity of carbon-tunable alloys of transition metal electrocatalysts because the electronic structure and adsorption energy of the intermediates in the course of the OER process could be remarkably enhanced [24, 128]. By considering the volcano curve, designing multi-constituent alloy with optimized surface chemistry is an ideal route to produce not even for OER but bifunctional materials for the overall water splitting process. In this regard, it was demonstrated by Kang et al. [100] that a synthesized nanoporous NiMnFeMo alloy (np-NiMnFeMo) showed an ultrahigh electrocatalytic activity with a large current density of 1000 mA cm−2 at 570 mV for OER. However, the N-doped graphene-decorated NiCo alloy combining with mesoporous NiCoMoO nanosheet grown on 3D NF and produced (NiCo@C-NiCoMo/NF) displayed an outstanding electrocatalytic activity and needed a small voltage of 1.90 V at 1000 mA cm−2 and maintain structural stability for 43 h under 6.0 M KOH electrolyte at 60 °C [129].
Despite the debate surrounding the influence of doping element in the transition metal based OER electrocatalysts, the reintroduction of the mechanism of elemental doping effect [130, 131] could help in strengthen the idea in the direction of rational design of carbon-tunable alloys of transition metal electrocatalysts. It was reported that a synergetic effect between V and Fe was achieved, whereby the doping of V was substantially reduced the adsorption free energies ∆Gs of the reaction intermediates (HO*, O*, and HOO*) that ordinarily led to evolution of O2 efficiently. The approach resulted in a low overpotential in the OER process (Fig. 11a). In addition, to further unveil the mechanism of OER process facilitated by elemental doped-transition metal-based electrocatalysis, the DFT calculation assisted in revealing information on the variation of OH adsorption energy (∆GHO*) connected to the compositional changes in FeCoW electrocatalyst (Fig. 11b). From the calculation, it was noted that Fe-doped Co electrocatalyst increased the adsorption energy of OH (∆GHO*), whereas the W-dopant decreased the value of the relatively higher ∆GHO* of the undoped CoOOH. Thereafter, however, such abnormality was delimited by codoping W and Fe into CoOOH and as a result, optimum ∆GHO* was achieved. Consequently, the term ∆GO* − ∆GHO* was considered as the electrocatalytic activity descriptor of the OER process (Fig. 11c). Therefore, with optimum ∆GO* − ∆GHO*, the electronic optimization of an electrocatalyst can be achieved for favorable OER process in term of low energy consumption caused by rapid reaction kinetics.
-
ii.
Interface It has been reported that hybridizing the electrocatalytically active phase with conductive support is not only enhancing the transfer of charges between the two hybridized components of the formed electrocatalyst, but also enhances the activity of the active site [133]. In this context, it has been found that an electrocatalyst support changes the adsorption energies of reaction intermediates by disrupting the electronic structure of the active sites or direct participation in the electrochemical reaction during the OER process [134,135,136,137,138,139,140,141]. With respect to this, conductive carbon materials have been proven providing excellent foundation for supporting more active phase such as transition metal alloys in the case of OER. However, it is unfavorable to use carbon materials in the absence of electrocatalytic active phases such as metal alloys, as are invariably contain defects and/or surface functional groups that expose carbon to suffer from carbon oxidation under the high potential of OER process. This to some extent leads to its electrochemical instability [142]. Based on the analysis of oxygen-to-carbon ratios upon electrooxidation [143], it was evidently confirmed the oxidation of carbon which reportedly proceeded through several reactions including the formation of phenols, hydroxyls, carbonyls/quinones, and carboxylic acids. In a similar study, TEM imaging authenticated the corrosion of the surface of carbon materials, which was resulted in through chemical attack [144]. The chemical attack was probably due to the formation of some active OER intermediates, particularly the OH-radicals. This is quite detrimental to the stability of the carbon materials and one should always take account of structural stability due to corrosion in the course of OER process in alkaline media. However, a contrary result was observed when carbon was mixed the oxide of perovskite. The carbon component was protected from corrosion by the oxide. This gave hope that carbon combined with metal oxides could be electronically a very promising OER electrocatalysts. Moreover, an electrochemical oxidation of carbon xerogels with different physico-chemical features was assessed and a contrary observation was reported [145]. The presence of oxygen functionalities on the surface, and in particular of phenolic groups, hinders the electro-oxidation of carbon by creating stable oxygen groups (carbonyl/quinone) that no longer oxidize to CO2. On the other hand, presence of wider pores and carboxylic groups contributed to the carbon-electrolyte interface, which favored the electro-oxidation by water. These results can help to rationalize strategies to ameliorate carbon electrooxidation in electrodes based on amorphous structures. On the combination of carbon with transition metals, carbon supports mainly contribute in carbon-tunable alloys of transition metal electrocatalysts as interconnected electron pathway, platform for excellent dispersion of active phases, hierarchical porosity meant for free mass transfer, accessible route to active sites, strongly couple interfaces, and regulation of active phases [146]. Thus, hybridization between transition metal alloyed phase and multi-contributor carbon support creates interface and will no doubt result in enhanced performances of electrocatalytic OER activity.

Copyright of Wiley–VCH, 2017 and the American Association of Advancement of Science, 2016, respectively
a Graphical presentation of Gibbs free energy changes in respect to different surface active sites in V-doped FeOOH, b fine tuning the adsorption energies of OER intermediates by changing the compositional amounts of constituents via alloying of Fe with W (c) illustration of OER activity variation among the Fe, Co, oxyhydroxides and W, Fe-codoped CoOOH, CoW, and WOx derived from calculated DFT + U. Reproduced with the permission from Refs. [130] and [132] for a–c, respectively.
The interface between the metal core center and the support causes re-arrangement of electrons originating from the support and anchored metal atoms, which as a result the direction of charge transfer is controlled and driven by the Fermi level difference between the metal active center and the support [147, 148]. Interestingly, interface creates direct and effective contact between electrocatalytic active sites and reactants/intermediates, which in due course drives the electrocatalytic reaction rapidly at the interface. This was recently confirmed, as shown in Fig. 12a, the FeNi alloys in FeNi/C exhibited a low current density of 0.18 A mg-1 at 1.55 V. Comparatively, the OER activity performance was extremely improved by the FeNi alloy nanoparticles in FeNi–Mo2C/C with the mass current density increased to 1.89 A mg−1 at the 1.55 V [149]. This was initiated by improved interface with increased active sites. In addition, since the mass current density is calculated from multiplying intrinsic activity by population of active site per unit mass of electrocatalyst, the inherent activity was then assessed by dividing the mass current density by the population of active sites of each of the two different electrocatalysts. After normalization, the active site population ratio between FeNi–Mo2C/C versus FeNi/C was found to be 5.41, meaning that the inherent activity of FeNi alloys in FeNi–Mo2C/C was greater compared to FeNi/C (Fig. 12b, c). All these was then translated in terms of reaction mechanistic paths and surface adsorption free energies of the OER intermediates (HO*, O*, and HOO*). Initially, the adsorption of O* was hampered by high adsorption free energy at Ni sites (3.87 eV), but with the incorporation of Mo2C/C the binding free energy was reduced and that benefitted the OER activity accompanied with reduced overpotential (Fig. 12d). Similarly, it has also been reported that transition metal alloys encapsulated in N-graphene has been discovered to be promising hybrid electrocatalysts for OER process [60, 66, 72, 95, 130]. The observed electrocatalytic OER process with transition metal alloys encapsulated in N-graphene was attributed to enhanced transfer of electrons at the interface between the metal core particles and graphene shell, which was the main advantage for the improved OER activity. Moreover, by modulating the chemical compositional proportion of the transition metal core components in company with thickness adjustment of the graphene shell layers, the electrocatalytic activity can be further tuned to enhance the OER activity [150].
-
iii.
Modification of electrocatalyst electron transport ability In an attempt to alter an intrinsic adsorption free energies of OER intermediates (∆G) to optimum level, electron transport ability is a key factor required to be achieved for accelerated OER process. The OER active electrocatalysts, such transition metal alloys, or hydroxides and oxyhydroxides, suffer from unsatisfied electron transport abilities caused by (1) their inherent and relatively low electronic conductivity and (2) their limited contact areas between their electrocatalysts and electrode due to nanosize effects. To halt such energy loss between electrode and active sites of their electrocatalysts where the OER reaction occurs, the electronic conductivity and mobility through interface between electrode and their electrocatalysts need to be optimized. One of the possible ways to effectively address this issue including an approach of combining electrocatalysts with conducting support [151,152,153,154,155,156]. This involves the use of stable carbon-based materials such as graphitic carbon, graphene and carbon nanotubes, all of which exhibited activity wonderfully in OER process (Fig. 13a). In carbon-tunable alloys of transition metal electrocatalysts, carbon is mainly considered as an electronic conductivity trigger and electron transporter due to its limited participation in the overall electrochemical OER activity. A carbon-based material enhances facial contact between the electrocatalyst active sites and electrode, as well as allows easy transport of electrons, OH− reactants, intermediates and diffusion of produced O2 molecules (Fig. 13b).

Copyright of Elsevier, 2021
a Comparison of calculated mass current density of FeNi alloys in FeNi/C and FeNi–Mo2C/C; b illustration of inherent OER activities of FeNi alloys in FeNi/C and FeNi–Mo2C/C; c imagery model of surface-oxidized atomic structure models of FeNi alloys in FeNi/C in the left and FeNi-Mo2C/C in the right side; d a comparison of the calculated binding free-energy diagram of OER processes for FeNi/C and FeNi-Mo2C/C, in which the insets are the atomic models where red, blue, pink, yellow, gray, purple and black represent O, intermediates, H, Fe, Ni, Mo, and C, respectively. Reproduced with the permission from ref. [149].

Reproduced with permission from ref. [157] and [158]. Copyright of Wiley–VCH, 2021 and 2018 for a, b, respectively
a Schematic presentation of different classes of advanced materials use as support of active phase of OER electrocatalysts, b Schematic illustration of the required features for OER electrocatalysts.
Recently, a group of researchers fabricated Ni2P/NPC electrocatalyst for OER process. The group was able to critically examine the role of carbon support from the energetic and electronic perspective by the application of DFT calculations. From the result, it was found that Ni2P/NPC had a more significant contribution at the Fermi level compared to the other type without the carbon support, i.e., Ni2P. This suggested that higher electrical conductivity of Ni2P/NPC was benefitted from the elevated electron transfer caused by carbon and thereby enhanced the OER process (Fig. 14a) [159]. Obviously, the transfer of electrons from supported Ni2P nanoparticles to NPC support substrate was largely responsible in enriching the electronic orbital distribution closer to the EF. The integration of the support substrate shifted the d-center upward from the Fermi level (Fig. 14b), which resulted in the d-center to increase from − 2.12 to − 1.78 eV for Ni2P and Ni2P/NPC, respectively and in effect increased the binding ability of OH−. It is remarkable to note that the shift of d-center was responsible in decreasing adsorption free energy of OH─ and thus quickly facilitated the activation of HO* on the surface of the electrocatalyst which enhanced the kinetics of OER process. The observation made in this work is similar to previous study [160]. In addition, the integration of NPC support substrate also caused great difference in the electronic structure of Ni2P, more especially for the Ni 3-d orbitals. This was evidently ascertained with charge density analysis, as presented in Fig. 14c, d. Figure 14c, d clearly show the strong coupling between Ni2P and NPC substrate that was responsible for efficient charge transfer, as well as difference in accumulation of electrons at NPC nanosheets, respectively. On the other hand, the support substrate also exerted support effect on the Ni, which as a result caused the Ni site in Ni2P/NPC became more positively charged (Fig. 14f) than that of the Ni in Ni2P (Fig. 14e). The positively charged Ni site in Ni2P/NPC was essentially responsible in facilitating the adsorption of HO−by electrostatic force. Consequently, the Ni site acted as the optimum electrocatalytic active site due to decreased surface adsorption free energy barrier (1.44 eV) for HO* generation compared to P site (2.74 eV). This was caused by the contribution of carbon support substrates, which was attributed to the variation in the ability to transfer electrons and that led to the dissimilarity in the values of adsorption free energy of ∆GHO* [161, 162]. However, as presented in Fig. 14h, g, the formation of O* over Ni2P was energetically favorable than in Ni2P/NPC, which as a result the OER was remarkably limited by the observed energy gap. Overall, despite that the generation of HOO* was viewed as the potential rate-limiting step due to lesser surface adsorption free energy barrier (Fig. 14h, g), carbon exerted its support effect and triggered Ni2P/NPC to decrease the energy barrier for the formation of HO* and HOO* that favored the superior OER performance.

Copyright of Elsevier, 2021
a Density of state of Ni2P and Ni2P/N,PC; b partial density of state of Ni2P and Ni2P/N,PC; c variant charge densities of the Ni2P supported by N and P-doped graphene, where yellow and cyan areas represent positive and negative charge, respectively, while in the composition of the electrocatalyst, the pink, blue, gray, and pink balls represent P, N, C, and Ni, respectively; d the planar average electron density difference between Ni2P supported by N and P-doped graphene; e molecular imagery models of Ni2P and f Ni2P/N,PC; g, h surface adsorption free energy diagrams of Ni2P and Ni2P/N,PC, respectively. Reproduced with the permission from ref. [159].
Design of efficient OER electrocatalysts outside the scaling relation between the reaction intermediates
The activity performance indicator or descriptor used in AEM for the design of efficient electrocatalysts relies on the utilization of scaling relation among the OER intermediates, i.e., HO*, O*, and HOO* species, which is determined by a volcano cap on the OER activity scale that indicates that theoretical overpotential cannot go beyond 0.37 eV. However, in the recent time, three mechanistic approaches have now been conceived to break the earlier conventional use of scaling relation for achieving better OER activity. The newly introduced strategies including (1) stabilization of OER intermediate, i.e., HOO* without undermining the surface adsorption of HO*; (2) secondly, the strategy involves the introduction of proton (H+) acceptor to alter the reaction pathway and (3) it includes the lattice oxygen participation of an electrocatalyst in use, which can be activated for direct O–O radical coupling and commonly is referred to as lattice oxygen mediation mechanism (LOM). The LOM strategy bypasses the formation of HOO*, by which the constraint ordinarily imposed by the adsorption-energy scaling relation between HO* and HOO* is avoided completely [101, 162, 163]. Although the LOM thermodynamic has not been studied, but could be an alternative reaction pathway of OER with much less energy consumption than the energy use in AEM.
Stabilization of HOO*
This is an attempt towards breaking the interaction between HOO* and HO* over an electrocatalyst with overall aim of reducing the OER overpotential closer to 0 V h. Several efforts are underway towards finding a way to independently change the adsorption behaviors of HOO* and HO*. Up to the present time, dual-site mechanism and hydrogen bond formation [164] are two the most effective methods for stabilizing HOO*.
-
i.
Dual site-mechanism As previously mentioned, AEM proceeds through the OER intermediates, i.e., HO*, O*, HOO* and HOO* and O2*. Therefore, by modifying the adsorption/biding of HO* and HOO* separately on two different surface active sites would be an advantage in an attempt to end the intermediates scaling relation in AEM. In an example, it has been reported that the synthesis of single-atom transition metals either of Fe, Co, and/or Ni embedded in N-doped graphene (M-NHGFs) coupled with an identical MN4C4 moiety, of which their electrocatalytic properties and behavior are found to be different as a result of the modified ligand effect caused by different metal centers [163]. In MN4C4, both M and N are initiated as potential surface active sites for the OER intermediates. However, regardless of whether the created dual sites participated in the adsorption of OER intermediates, the contribution of the dual-sites largely depends on the number of d electrons of the transition metal in the electrocatalysts. Accordingly, this reaction mechanism describing herein is different from the single atom site of Co and Fe examined in the AEM mechanism. Here, the OER proceeded through a dual-site mechanism on MN4C4 moieties (Fig. 15a–c). Explicitly, the OER intermediates particularly the O* and HO* were preferentially adsorbed on the C site whereas HOO* on the Ni site, which ultimately led to different adsorption energy between HOO* and HO* that was not the same value as the commonly reported 3.2 eV in the conventional AEM. This was how clearly confirmed that Ni-NHGF followed dual-site electrocatalytic than those of Co-NHGF and Fe-NHGF. So, carbon support can be instrumental in dual-site mechanism, as in this case raised the OER activity by acting as an additional active site. Introducing carbon support regulated the electronic structure and facilitated local chemical coordination environment between Ni sites of the synthesized electrocatalyst and thus improved the intrinsic OER activity. Hence, because of the remarkable carbon properties which including good mechanical and thermal stability, high electrical conductivity as well as flexible and tunable electronic structure, heteroatom doped-graphene showed promising potential in this situation. Likewise, other forms of carbon supports might also be useful and worth to be investigated.
-
ii.
Initiation of hydrogen bond formation From the molecular structural point of view, the structure of HOO* is electronically more sensitive than HO* owing to additional pendulous O in HOO* and perhaps responsible for its engagement in extra interaction with a different electrocatalyst structure compared to HO*. As such, the HOO* adsorbate is more readily to be stabilized than HO* and thus, it is feasible to break their scaling relationship. In response to that, though several efforts were made, however, one group of researchers proposed establishing a 3D nanoscopic structure of an electrocatalyst to break the scaling relation between OER intermediates, which as a result create limited surface reaction environment capable of selective interaction with specific OER intermediate [165]. In the proposed molecular scheme, the nanoscopic electrocatalyst structure was mainly designed to allow selective interaction for HOO* intermediate with surface active sites sluggishly while the adsorption of HO* remained unaffected. In the schematic model of the proposal, RuO2 and IrO2 were used as model surfaces and joined with a second surface to create a network (Fig. 15d). In this study, the relationship between HO*/HOO* adsorption with the formed network width were investigated and the observed results disclosed that the adsorption energies of HO*remained constant for a network greater than 6 A. In contrast, the adsorption energy of HOO* was reduced at about 6 A (Fig. 15e), which was an indication that a stabilized interaction between HOO*and O atom in the opposite network surface was established through a specified hydrogen bond formation. Consequently, the overpotential for RuO2 was deceased to ≈ 0.2 eV for the network at about 7 A, which was quite below the optimum theoretical overpotential of 0.4 eV based on the scaling relation prediction. Although this has presented a fundamental significance of the reaction mechanism through the hydrogen bond formation, but the proposed mechanism needs thorough and advanced investigation, so as to strengthen the claim. Because it seems quite challenging to maintain the weak hydrogen bond between the electrocatalyst channel/network layer and HOO* under the OER operational conditions. However, with the presence of carbon support richly enough with the large number of terminal oxygen-containing functional groups, such as –OH, –CO, and –COO could be helpful and therefore, in the rational design of carbon-tunable alloys of transition metal electrocatalysts, functionalization of such surface functional groups in the carbon support framework could be promising towards this direction.

Schematic reaction mechanisms where the reaction intermediates are presented in the optimized geometry of a single-site and b dual-site mechanisms in OER process; c free energy profile diagram for OER over Fe-NHGF, Co-NHGF, and Ni-NHGF electrocatalysts with single-site mechanism and Ni-NHGF with a dual-site mechanism at 1.23 V vs RHE; d atomic side view of the model system used to simulate confinement, where the reaction is assumed to take place as the network/channel width (dmm) decreased; e adsorption free energies of HO* and HOO* in response to change in the network/channel width. Reproduced with the permission from ref. [164] and [165] for Figure a–c and d, e, respectively. Copyright of Nature Publishing Group, 2018 and Wiley-VHC, 2015 for ref. a–c and d, e, respectively
Approaches of introducing of proton acceptors
As earlier observed and mentioned above, the formation of hydrogen bond to exert stabilization effect on HOO* is quite limited. However, introduction of proton acceptor to stabilize HOO* is one of the most efficient methods, through which the HOO* intermediate will be made to produce OO* and H* (OO* + H*). In this method, an introduced proton acceptor will be a fully independent site, which is used to stabilize the OER intermediates (OO* + H*) and thus, resulting in a significant increase in OER electrocatalytic activity [166,167,168]. The electrocatalytic reaction cycle is initiated by a single metal coupled with proton acceptor sites. Accordingly, two ways for incorporating proton acceptor into the OER schematic process have been recommended. In one of the ways, it involves substitution of elements with higher electronegativity that can activate oxygen as a proton acceptor on the surface of an electrocatalyst. In the second one, it involves functionalization of the surface of electrocatalyst with strong nucleophilic groups such as phosphate, amine, for the acceptance of proton. Details of the mechanism of occurrence in the two the methods are discussed below.
-
i.
Metals substitution with higher electronegativity Modification of surface active sites of an electrocatalyst can break the typical scaling relation between the OER intermediates. It has been proposed that by replacing Ru with Ni or Co, the oxygen atoms on the bridge or edge positions of the RuO2 surface can be activated as proton acceptors [169] Upon this substitution of Ru with Ni or Co, the H atoms in HO* and HOO* can be made to relocate to the neighboring activated oxygen to form O* + H*and OO* + H*, respectively. As a result, the OER intermediates are made to behave different from the conventional HO* and HOO* intermediates and in effect, the adsorption free energy difference between O* + H* and OO* + H* was reportedly reduced lower than 3.2 eV [169]. This resulted in the largest free energy steps of Ni- and Co-substituted RuO2 to be 1.49 and 1.33 eV, respectively that are evidently less than 3.2 eV. Similarly, overpotential of 0.26 and 0.10 eV were recorded for Ni- and Co-substituted RuO2, respectively, which were considerably lower than the utmost value of the volcano curve (0.37 eV). With respect to that, X-ray scattering measurements together with DFT calculations were used to substantiate on the claim based on the surface structural influence on the single-crystal RuO2 in acidic electrolytic solution [166]. At the OER potential window, the OO* intermediates were identified on the uncoordinated Ru sites and stabilized by nearby HO* groups on the same site or bridge site on the surface of the electrocatalyst. This was followed by deprotonation of HO* in the reaction pathway, which was regarded as the potential-determining step. Further confirmations of the similar observation were also reported for the single metal and alloyed forms of transition metals such as Co3O4, CaMnO3, and NiFeOx surfaces (Fig. 16a) [157, 169, 170]. In addition, it was revealed that in γ-NiOOH melded with some fraction of nanoclusters of γ-FeOOH, Fe functioned and served as the oxygen-evolving center (OO*) and the nearby terrace O site on the γ-NiOOH acted as a proton acceptor (H*) [171]. This provides an insight and is a useful information in the design and development of carbon-tunable alloys of transition metal electrocatalysts for OER, in which a metal substitution with higher electronegativity influence can be incorporated and target for enhanced OER.
-
ii.
Introduction of nucleophilic group Owing that phosphate ion groups have been shown to exhibit high nucleophilic characteristics coupled with their suitable pKa value of 12.67, are used as electron acceptors in electrocatalytic reactions. With its high pKa value, it means much readily to accept proton and therefore can be appropriately used for functionalization of the surface of electrocatalyst. As presented in Fig. 16b, it was found that surface phosphate functionalization on perovskite enhanced the interfacial proton transfer significantly [51]. This was practically demonstrated with functionalized form of La0.5Sr0.5CO3–δ, which in activity performance was almost two times that of non-functionalized La0.5Sr0.5CO3–δ and one order of magnitude that of LaCO3. In a similar effort, a constructed hybrid electrocatalyst of Sr3B2O6 was designed to be proton acceptor with a functionalized A-site-deficient Sr0.8Co0.8Fe0.2O3-δ [172]. Therefore, on a similar scenario, supporting transition metal alloyed nanoparticles on carbon supports could be undoubtedly an advantageous, particularly on the functionalized carbon supports rich with nucleophiles, such as -NH2 and –CN, etc.

Copyright of American Chemical Society, 2018 and 2017 for a, b, respectively
a Illustrative modelling route of HOO* via activation of surface oxygen as a proton acceptor for breaking the conventional scaling relation on CaMnO3, b surface functionalization of perovskite with phosphate ion as proton acceptors for the enhancement of OER activity. Reproduced with the permission from ref. [170] and [51].
Lattice oxygen mediation mechanism (LOM)
By virtue of in-depth understanding of the fact that different transition metal alloyed electrocatalysts activity activation mechanism is strongly associated with supports, surface conductivity, mixed valence oxidation states, it is important to further gain better fundamental understanding of OER based on LOM codes. Because LOM is directly derived from the redox chemistry of lattice oxygen, it is rationalized based on intrinsic activity and surface reconstruction of electrocatalysts characterized with high covalency of metal and oxygen.
Before driving the discussion deeply into the newly proposed LOM, it is noteworthy to mention that multimetal-based electrocatalysts are typically endow with more superior activity than the single-based electrocatalysts due to the interatomic electronic interactions that efficiently modulating the electronic structure of metal sites, which is unlikely to be achieved with single-metal electrocatalysts. For this reason, combination of two or more metal together can still offer and serve as a useful approach for tuning the redox electrochemistry and designing highly oxidized metal electrocatalysts [132, 173, 174]. However, because of structural complicacy associated with multimetal electrocatalysts, logical design of effective multimetal OER electrocatalysts with abundant valence metal sites along with the fundamental understanding of their structure–activity nexus still remains a daunting challenge. On the other hand, because of their unique intrinsic properties, multi-constituent alloyed materials have recently gained a lot interest [175, 176], which in particular transition metals are of special interest. This is because of the fact that surface redox electrochemistry is directly connected to the chemical affinity between oxygen-containing OER intermediate species and metal sites, which through variable or multiples of electronic oxidation states can be used to modulate it efficiently [177,178,179,180]. This is a part of interesting properties possessed by the transition metals. In addition, flexibility to adjust components of transition metal alloys based variability of oxidation states in the design of their electrocatalysts is another added advantage against many other noble metal-free electrocatalysts. Therefore, with logical modulation and engineering of electronic structure of transition metal alloys, it is practicable to tune highly oxidized metal species for LOM.
In agreement with preceding section, the most promising noble metal-free electrocatalysts have been thought to be 3d transition metals especially in alkaline-based OER process due to their ability to form oxyhydroxide intermediates, controlled by their electronic surface structure, as favorable active sites for OER [105, 177, 180,181,182,183]. Meanwhile, as surface structure through reconstruction into oxyhydroxides plays a vital role in regulating OER activity kinetics, core metal supported-electrocatalysts also offer a promising approach to optimize OER activity through an interfacial interaction of core metals and the supports. This obviously calls for studies to be carried out to discover the possible underlying link between electronic structures of carbon-tunable alloys of transition metal electrocatalysts and preferential mechanism for OER based on the concepts of LOM. A large number of transition metal alloyed clusters decorated on carbon supports exhibited enhanced OER activity performance, which are attributed to improved electronic conductivity, optimized interface, large density of active sites, and many abound features that responsible for their outstanding OER activity. On the other hand, although routes to enhance OER activity performance of carbon-tunable alloys of transition metal electrocatalysts based on the novel LOM are still not discussed in the literature, but understanding how the LOM for OER process works and relate to carbon-tunable alloys of transition metal electrocatalysts is of significant importance to the future rational design of the materials.
As for the novel LOM, the mechanistic pathway in LOM is sharply different from the conventional AEM, all of which meant for OER process. In the OER process through LOM pathway, electrocatalytic surface is not thermodynamically stable, but it allows surface dynamic changes in accompany with OER process. Contrary to the conventional AEM, the LOM proceeds through oxidation of electrocatalyst core metals, follows by exchange and release of lattice oxygen ligands on the surface of electrocatalyst, which are all necessary for conserving and continuing OER cycling process. In the cyclic process, the OH−/H2O are the adsorbing/binding species at the oxygen vacancy sites to act as the newly bred lattice oxygen for the next cycle of OER. This is how the LOM process occurs repeatedly. Accordingly, lattice oxygen activation is the initiation part of the mechanism and is a prerequisite if its reaction pathway is needed to be successfully triggered, which requires the host electrocatalyst to possess distinct surface electronic structure, particularly the lattice oxygen itself. This means to act as a reservoir of oxygen, storing and releasing it for OER process at the electrocatalyst surface. Although these are the fundamental routes towards LOM, however, several dominant OER pathways through LOM with different surface active centers have also been recently proposed (Fig. 17a–c) [25, 28, 41, 47, 55, 98]. These are namely, oxygen-vacancy-site mechanism (OVSM, Fig. 17a), which directly receives OH− and form HOO* through nucleophilic attack [28]. In this pathway, the release of O2 creates Ovac sites and subsequently be refilled by OH− species. In another reported mechanism, it involves the participation of single metal site as the electrocatalytic center that adsorb OH− and then follows the deprotonation step, commonly known as single-metal-site-mechanism (SMSM, Fig. 17b) [41, 55]. The activation of lattice oxygen occurs via surface reconstruction, which allows direct coupling of O* intermediate and activated lattice oxygen and form OO* species through surface reaction that is energetically favorable particularly for the high-valence metal cations. The species of OO*are regarded as a newly generated O2 molecules readily to undergo subsequent oxygen production. Although it has already been mentioned in the subsection of stabilization of HOO*, there is a situation of dual-metal-site mechanism (DMSM) that has been deduced to rationalize LOM (Fig. 17c) [35]. The intermolecular oxygen coupling occurs between two adjacent activated lattice oxygen, which are expected to form a M–OO–M and follows by propagation of *OO* species that typically behaves as peroxy-like species. Due to structural flexibility of metal oxyhydroxides, the dual-metal scenario has been well documented in the literature [47].

Reproduced with permission from ref. [184]. Copyright of Royal Society of Chemistry, 2021
Schematic illustration of different OER mechanisms a oxygen-vacancy-site mechanism (OVSM), b single-metal-site mechanism (SMSM), and c dual-metal-site mechanism (DMSM). The participation of chemically inert lattice oxygen, active lattice oxygen in OER process and oxygen from the exterior electrolyte are marked in black, blue and red colours, respectively.
LOM in carbon-tunable alloys of transition metal electrocatalysts
Generally, with respect to the conventional AEM, the OER proceeds through involvement of four concerted proton–electron transfer steps over electrocatalysts. With this mechanism, the high OER electrolytic activity is always achieved through modulation of the surface adsorption strength of OER intermediates over the surfaces of electrocatalysts and limited by volcano scaling relationships. Contrary to AEM, the LOM is channeled and accelerated through direct coupling of lattice oxygen that bypasses and circumvents the generation of HOO* and eventually limiting energy barrier caused by such reaction intermediate surface adsorption free energy [185]. This offers valuable insights not limited to modulation of surface adsorption strength when it comes to the logical design of high performance OER electrocatalysts. With the insights from LOM, high performance OER transition metal alloyed electrocatalysts such as Co–Zn oxyhydroxide [55], NiFeCu/metal oxide [186] and FeCoCrNi alloy [47] were successfully designed. However, despite the ideal candidature of conductive carbon supports in the design and development of transition metal alloyed electrocatalysts for OER, it seems up to now research has not been opened to elucidate on the mechanism of activity of OER over carbon-tunable alloys of transition metal electrocatalysts via LOM. This may be associated with the avoidance of structural complicacy [187]. Notwithstanding, it is important to investigate carbon-tunable alloys of TMs electrocatalysts activity performance relative to stability issues by following the LOM strategic route. This is because of the positive effects between conductive carbon support and core metal alloys that are directly connected to increase of surface area, improvement of electrical conductivity, and facilitation of good dispersion uniformity that prevents core metal nanoparticles from aggregation. In addition, porous structure of carbon framework provides suitable pathways for mass diffusion within the carbon matrix, and more importantly derives excellent long-lasting stability that prevents leaching during electrochemical process [188, 189]. Also, the oxidation of carbon could be advantage to practical application of carbon-tunable alloys of transition metal electrocatalysts for OER through LOM, as the oxidation may exert synergistic effect between the oxygen-rich sites of carbon and the core metal sites for enhanced activity compare to unsupported transition metal alloyed electrocatalysts. This may offer opportunity to manipulate and boost OER process at low overpotential, leading to economic production of hydrogen. The window still remains open for investigation.
Others
Computational high-throughput screening approach (HTPS)
The use of mathematical approach via data manipulation to develop simple and fast screen process of functional materials is a viable option that can also be adapted in the screening of parameter required to guide designing of potential electrocatalysts rationally for overall water splitting process based on quantitative structure—or composition—property relationship. The approach is becoming more popular in the field of materials information as a useful tool for discovery of materials and thus, it could accelerate rational design of materials. The modern data science allows material scientists to pursue a rational design that is promising to mitigate many of the prevalent inefficiencies and limitations of the conventional approaches. Recently, embracing automation as a tool from data science has led to an acceleration and easy streamlining of the discovery process of materials. In particular, high-throughput screening (HTPS) is now becoming a mainstream technique use to search for materials with properties that are tailored for specific applications [190, 191]. Owing to its efficiency coupled with the increasing availability of operational codes, open-source and large computational resources have made it a powerful and attractive tool in material research.
HTPS is a process involves the use of large numbers of compounds that are characterized and assessed in an automated fashion. For example, the approach was applied in the drug discovery context for activity as inhibitor or activator of a particular biological target. In addition to its experimental approach, the tool can be virtually used in the enumeration of candidate compounds and their characterization and thus, the use of virtual HTPS has witnessed tremendous growth. This is because the key challenge in identifying and designing new materials previously is that their behavior is determined by complex structure–property and structure–activity relationships within the chemical space that is practically infinite [191,192,193], while equally advanced materials required more and more intricate property profiles [194,195,196]. However, recent efforts have clearly demonstrated that the combination of HTPS and modern data-science techniques are able to overcome such challenges that the ill-equipped conventional methods failed to meet. Now, the combined HTPS and data-science technique allows rational design of materials and lessens the limitations of the traditional approaches [197, 198]. In the application of the approach, the general strategy in screening material candidates lies on a divide-and-conquer hierarchy, in which a given candidate library is filtered in sequential steps. The approach employs a series of modelling protocols in evaluation of different properties of desired interest and then sorted out based on computational importance (Fig. 18) [199]. As depicted in the diagram below, the candidate materials are characterized, assessed, and screened at each level based on different target property.

Copyright of World Scientific, 2020
Schematic illustration of screening of a library of molecules to identify promising candidates. Reproduced with the permission from ref. [198].
With respect to catalytic materials, virtual high-throughput screening techniques in the design of catalyst have received great attention in the last decade. Although advancement has gone far, however, the prerequisite to perform HTPS for the design of an efficient catalyst still requires thorough preliminary investigation on the reactions of desired interests [200]. As described in the previous section above, this technique also applies descriptor of catalytic activity and other structure–property relationship, which can be extended over a library of candidate catalysts for their evaluation through a form of high-throughput computational screening. As a result, several research groups have utilized this approach to model catalytic reactions, e.g., OER over metal surfaces [201,202,203,204,205,206,207,208]. In a clear demonstration, one particular research group pioneered the application of HTPS using above-mentioned reaction activity descriptor for electrocatalysts, which involved screening of 736 distinct binary transition metal alloys [209]. In the process, the electrocatalysts were ranked on the basis of free energy of surface adsorption, described as the closer the free energy to zero, the better the electrocatalytic efficiency, as demonstrated in Fig. 19a. In a similar work, a comparison of the activity of 256 binary surface alloys (Fig. 19b) was also made with application of HTPS. The plot shows the numerous binary alloys with high predicted electrocatalytic activity. Besides the activity screening, the group computed the stability of the alloys based on four parametric tests, namely, (1) estimation of the free-energy change associated with segregation events; (2) formation of island of nanoparticles causes by/and surface de-alloying; and (3) the probability of dissolution of the alloy in acidic electrolytic reaction environments. Interestingly, with these tests, the group were able to identify several alloyed metal candidates that are both electrochemically stable and active (Fig. 19c). In short, these studies were able to map out the electrocatalytic activity to a single descriptor, for example, surface adsorption energies of the key reaction intermediates to the surface of the electrocatalyst. The mapping confirmed that the most active electrocatalysts had favorable values of the descriptors. Therefore, to find an active electrocatalyst, useful structure–property relations that will serve as sufficient descriptors of electrocatalytic activity for discovery of new electrocatalysts must be identified. Although since the demonstration of the feasibility of the application of HTPS in electrocatalytic water splitting process has been exemplified [210,211,212,213], the advancements made in the electrocatalytic water splitting process using of HTPS, to date, only medium scale throughput studies have been reported in the literature. The challenge lies in defining clear electrocatalytic OER mechanism and creation of chemical descriptors that are fit to evaluate the electrocatalytic efficiency. Because a good descriptor need to be clear and simple, but also with capacity to provide physical insights, which can help guide and speed up the design and development of new electrocatalysts for water splitting process.

Copyright of Nature Publishing Group, 2006
a Free energy of adsorption versus electrocatalytic activity where maximum activity is observed when the adsorption energy is close to 0 eV, b the calculated hydrogen adsorption free energies on surfaces of alloyed electrocatalysts, c transformed free energy values determined from each alloy plotted against the absolute magnitude of free energy of hydrogen adsorption. Reproduced with the permission of ref. [208].
The conductive carbon-tunable alloys of TMs electrocatalysts are important chemical family of electrocatalysts for OER applications, which are in high demand because of their earth abundance and are low-cost materials, as well as their suitability for renewable and sustainable hydrogen energy production from water splitting process. Thus, the activity performance descriptor(s) can likewise be used to describe the activities of carbon-tunable alloys of transition metal electrocatalysts, as demonstrated by various studies over the last decade. The application of such technique can be extended to its fullest potential, as all it requires is just appropriate and justifiable identification of the OER electrocatalyst for it to be successfully employed.
Identification of descriptors of electrocatalytic activity can be achieved through the following steps, namely, (1) clear identification and classification of elementary steps involved in the OER process; (2) energy evaluation of individual step in the mechanisms of OER, this including the dissociation and adsorption energies and reaction barrier, for example, all the transition metal surfaces and this is followed by identification of active sites and the sites with the lowest energy barrier; and (3) creation of scaling relationships between adsorption energies and corresponding activation energies of the transition states [214, 215]. It is the scaling relationships that serve as descriptor and used to quantify electrocatalytic activity without need to evaluate all the thermodynamic parameters involved in the overall mechanism. Once all of the OER reaction steps are quite understood and the descriptors are well identified, the next line of action involves evaluation of activities for a library of promising candidates. As these are made readily available, activity performance of electrocatalysts based on their electronic structure can be evaluated and therefore, activity performance can be tailored by their chemical structure. The technique is an interpretable machine learning methodology that simultaneously seeks for the best mathematical formulas of functional set parameters in the formula [187, 216]. As such, the technique is capable to delivering easily understandable mathematical formulas and calculations that provide direct material design guidance, which could also be applicable for electrocatalysts. Accordingly, by employment of HTPS approach, it is possible to tailor the electronic structure of an electrocatalyst in a large-scale fashion. Overall, with HTPS strategy, design and development of high-efficient, low-cost electrocatalytic systems for OER process are attainable. Anyway, despite the great potential of the technique that can construct an automated descriptor that allows speeding up of electrocatalysts design process for better activity performance, its application in the research arena of electrocatalysis and overall material science is still scanty. Therefore, there remain numerous avenues that have not been explored in this effort of utilizing automatic system to optimize rational design and development of electrocatalysts for the OER process in overall water splitting technology.
Statistical approaches
Statistical phase approach was introduced into modern material science particularly in electrocatalysis to achieve optimum combination of parameters that guide to best design of electrocatalysts based on structure–activity relationships, which is targeted to control waste resources, time, and tediousness in the process of selection of best candidate electrocatalyst. This informs us that statistical analysis is not, as most often believed, mainly for analysis of research results, but should emerge as a basic tool in the planning phase of experiments. The goal of design is to produce materials that are to do their function effectively well, safely and at reasonable cost [217]. This also applies to material science, particularly in the subject matter under discussion, the electrocatalysis. The rational design of electrocatalysts for OER is most often based on single-parameter descriptor, but sometimes involves multiple parameter descriptors, which are all used to simplify design of electrocatalysts.
Correlative analysis between single-parameter descriptor and electrocatalytic activity performance
It is quite challenging to examine heterogeneous catalysts along with their catalytic mechanisms explicitly. This is simply because of the heterogeneity nature of their active sites, which are constraints to rational design and growing demand for precise electrocatalyst and electrocatalysis for OER process. Fortunately, such constraints can be overcome by the advanced single-parameter descriptor of activity performance of electrocatalysts. Although several theoretical concepts, such as d-band centers (εd), ℮g-filling, the valance band peak position (p-band model) and some other descriptors that focus on the chemical coordination environment, have been proposed for the quantitative evaluation of activity performance of electrocatalysts in water splitting process [218,219,220], however, they suffer from certain limitations. Precisely, for instance, the d-band center (εd) theory that was proposed by Nørskov et al. [221] is widely applied for the evaluation of catalytic activity performance of metal-based electrocatalysts. Although the general acceptability of the theory is a success, but not applicable to transition metal-based alloyed materials and some single forms of the metal family due to d-band energy splitting issue associated with such materials. In the case of ℮g-filling, it is an efficient descriptor for predicting the electrocatalytic activity performance of transition metal materials [218]. Unfortunately, it is not a universal descriptor, as it is mainly limited to materials with perovskite structure. In the same vein, the p-band model is only applied to explaining the electrocatalytic performance of perovskite oxides [219, 220]. In addition, some of such descriptors are much associated with the chemical coordination environment [218]. Therefore, a universal single-parameter descriptor to evaluate the activity performance of graphene-metal based electrocatalyst for HER, OER and ORR was proposed [219], which is in perfect agreement with the local structural and chemical surroundings of the core atoms and their neighbors. In short, despite the fact that the earlier mentioned descriptors were successful in some ways, their multiple-parameter characteristics necessarily make the design of efficient electrocatalysts more complicated. Because the contribution of each parameter is difficult to prove and distinguish from one another. So, to avoid the problem of multiple-parameter descriptors, single-parameter descriptor has also been proposed and recognized as a promising tool for designing highly efficient electrocatalysts for OER process.
In application of single-parameter descriptor using statistical approach, it was demonstrated that adsorption free energy (Ead) in OER process was conveniently utilized in the evaluation of electrocatalyst activity performance for OER process [222]. In this context, correlative analysis between ∆GO*, ∆GHO* and ∆GHOO* was used in the logical quest for highly active electrocatalysts. A correlative linear relationship between OER intermediate species was studied, as shown in Fig. 20a, b. From the illustration of the plots, a fantastic linear correlative relationship between ∆GHO* and ∆GHOO* was observed, which was far better than that of ∆GHO* and ∆GO*. The well-fitted linear relationship between ∆GHO* and ∆GHOO* resulted in due to the nature of adsorption behavior of the single metal–oxygen bond of HO* and HOO* species but different from that of the O* species [223]. This means that the mode of adsorption over the electrocatalyst surface by the two species was almost the same and is consistent with the previously reported DFT results. As a result, compared with ∆GO*, ∆GHOO* presents a better linear relationship with ∆GHO*. For this case, it means that the correlative linear relationship between ∆GHO* and ∆GHOO* is the descriptor here and can be used to fine-tune the structure of the electrocatalyst electronically towards optimum adsorption free energies of HO* and HOO* intermediates for much better performance. Therefore, as it is commonly known that OER activity performance of a given electrocatalyst is governed by the surface adsorption free energy of the intermediates (∆GO*, ∆GHO*, ∆GHOO*), correlative linear relationship is also sufficient enough to guide on designing highly efficient electrocatalyst.

Reproduced with permission from ref. [221]. Copyright of the Royal Society of Chemistry, 2021
a Correlative scaling relationship between ∆GHO* and ∆GHOO*, b between ∆GHO* and ∆GO*, c the correlative scaling relationship between adsorption energy (Ead) and overpotential.
The research was further shifted forward, where the research group drew the relationship between the surface adsorption energy (Ead) and the overpotential of the OER process (Fig. 20c). Interestingly, it is clear that there is a strong linear link between the two variables that is evidently glaring. It serves as an excellent way to bridge relationship between Ead and overpotential, because Ead can be a good descriptor of OER activity since either too strong adsorption or too weak influences overpotential in term of phase transition of adsorption and desorption of reaction intermediates coming onto and leaving out the surface of electrocatalyst, respectively. This can be evidently established as the point of the correlative association can be easily deduced from the plotted graph (Fig. 20c) that overpotential approaches to be less than 0 eV when Ead is greater than 7.42 eV, which in comparison to reported values in the literature is relatively arbitrary. In fact, from the previous studies it has been demonstrated that for Ead to be greater than 7 eV is quite very rare [224, 225]. However, this issue cannot affect the utilization of Ead as descriptor. Because, in general, some inferences can still be drawn from the correlation. From the theoretical background, it is reasonable to suggest that all electrocatalyst samples with Ead values ranging between 5 and 6 eV represent excellent OER electrocatalytic activity performance. Accordingly, Ead as a descriptor can be helpful to rapidly screen the very promising electrocatalysts for OER process. Thus, this serves as an insight and can provide convenient route to fast rational design of carbon-tunable alloys of transition metal electrocatalysts.
Multiple descriptors of activity performance of electrocatalysts in water splitting process
The central issue associated to rational design of an electrocatalyst with optimum activity performance is the development of quantitative analysis and establishment of structure–activity relationship. This is primarily directed to link the electrocatalysts’ structural and/or elemental descriptors to intended electrocatalytic behavior. The major objective for such correlation is to make use of the nexus to inform decision and guide material design appropriately. The general idea in statistical approach in modern material science is basically to identify materials with desired properties tailored to specific applications.
Recently, the operational rationality of single-parameter descriptor has been challenged, as single descriptors are usually sufficient primarily for particular catalytic systems and reactions. The limitation associated with single-parameter descriptors is its inability to effectively correlate different catalytic systems, particularly for in situ cyclic catalytic systems. This has remained as a formidable challenge for a single-parameter descriptors, as complicated electrocatalysts with multiple components or heterostructures operating in an electrolytic systems cannot be described by single-parameter activity descriptor [226]. Therefore, multiple-parameters descriptors have been proposed, particularly to describe electrocatalytic OER activity performance. In the field, several researchers have reported OER activity performance descriptors differently, for example, Shao-Horn and his co-workers studied the electrocatalytic behavior of 14 descriptors on the strength of the M–O bond based on 101 inherent OER activity of multiple tenths of perovskites. The group reported that the 14 descriptors were used, which were further grouped into five families, out of which established that the electron occupancy and M–O covalency were the principal factors influenced the OER process. However, other descriptors, such as the number of d-electrons, charge-transfer energy, and ℮g-filling, all were also played a vital role, as well as geometric factors such as M–O–M bond angle and tolerance factor, were also found partly relevant [218]. In another different research group, they reported that local structure and chemical surroundings of the active centers of a graphene-based single metal electrocatalyst. This including the coordination number, electronegativity and electronegativity of the nearest neighbor atoms, which all can be used as a universal descriptor to predict the OER electrocatalytic activity [227]. To them, it was implied that the single atom of transition metal together with the closest neighbor as the active center and pronounced them as a more universal descriptor ϕ than even the commonly known ∆GHO*, which is quite elusive. As complicated as the issues of activity descriptors are in electrocatalytic OER process, in general, in systems with multiple constituents or heterostructures, activity descriptors are required be connected with each other and combined together to develop more robust predictive descriptors fit to accommodate several electrocatalytic systems. This has imperatively called for the development of broader and more inclusive descriptors for the prediction of optimum electrocatalysts for OER process. Hence, it is expected that computer-aided statistical approach through the application of machine learning jointly with artificial intelligence will quicken the technological incubation of high-tech descriptors to eradicate ambiguity in the route of rational design of optimum electrocatalysts for OER process.
Based on the advancement reached so far, behavior and performance of 14 multiple descriptors of the M–O bond strength using number of statistical approaches, including factor analysis and linear regression model are being tested and reported [218]. These are quite promising as the approach outperformed both single and dual descriptors and their universality is superb. In this approach, relative OER activity was plotted against the multiple parameters that are directly associated the OER electrocatalytic performance (Fig. 21a), which including number of d-electrons, number of ℮g electrons, tolerance factor, and charge-transfer energy. As shown the plots, despite the scattered nature of the data, the general trend in the plots are in good agreement with the previous results, of which a linear trend with number of d-electrons [228, 229] and optimum range of values for the number of ℮g electrons, were all in agreement with the reported volcano trend of results in the literature. In short, there was trend between OER activity and associated descriptors’ parameters, as well as trends in the subset of the data points. In addition, with this approach, strong correlation among the OER activity descriptors was also established, which suggested that none of the multiple parameters was independent of the other. This indicates and confirms the feasibility of common characteristics of one family descriptors to influence OER activity.

Reproduced with permission from ref. [227]. Copyright of the American Chemical Society, 2016
a Plots of trends in the relative OER activity by four different performance descriptors, namely number of d-electrons, number of ℮g electrons, tolerance factor, and charge-transfer energy determined by confidence band, b plots of trends in relative OER activity for four descriptor expatiated by loading magnitudes for the 14 sub-descriptors obtained by factor analysis, using Kaiser criterion to determine the optimum number of factors (5). The larger the radial component, the higher the contribution of a descriptor to the factor. The factors are regarded as descriptor families related to the covalency, electrostatic, structure, exchange interaction and electron occupancy are represented by green, gray, yellow, red, and dark gray colors, respectively.
In a similar effort, factor analysis was also used to classify the relationship among the descriptors and identified the most effective in describing any observed chemistry variation among the used electrocatalysts. With this approach, because each parameter represented an underlying physical phenomenon that relates the descriptors, descriptors’ family were identified and grouped each family as an entity, e.g., covalency, electrostatics, structure, exchange interaction, and electron occupancy, were all categorized as families [218]. In a very typical example, the interpretation of the factor analysis was demonstrated as such, although M–O–M bond formed in the cause of OER process followed through LOM as a descriptor with a structural definition. However, it is more closely related to covalency descriptors. Accordingly, the factor analysis explains vividly the physical consequence of fine-tuning a descriptor and identifies the most essential descriptors for each factor, as well as the relative importance to each other (Fig. 21b). Hence, this study clearly demonstrates that statistical approach is a powerful tool that can be employed for descriptor-based design strategies for OER electrocatalysts with a better estimate of error magnitude. Thus, it can be useful in rational design of carbon-tunable alloys of electrocatalysts, as particularly that such kind of material comprises of multiple components or heterostructure made up of multiple elemental compositions.
Summary and outlook
The review summarized and discussed the progress of alloys of transition metals tuned with carbon materials for electrocatalytic OER in water splitting process towards sustainable hydrogen production. The reaction mechanisms of OER was thoroughly discussed with full inclusion of all the general OER mechanistic pathways. On the basis of majority of research findings, it seems reasonable to infer that the integration of one transition metal with another anchored on conductive carbon materials offer great potential to, (1) enhance the activity through offering routes to modification of d-band centers by insertion of the 3-d of one metal into another in the alloyed system that facilitates modulation of the surface adsorption free energies of the reaction intermediates; (2) modify the interface that exposes and augments surface area and active sites by hybridization between metal alloyed atomic orbitals and carbon nanostructure that promotes adsorption by downshifting the activation barrier, mass transport of reactants and intermediates, as well as transfer of electrons; (3) improve the electronic state of metals d-orbital close to Fermi level, and (4) enhance dispersion of core metal nanoparticles and long-lasting stability through supporting and coating effect of the carbon supports. Some compositions, FeCo and FeNi clusters supported carbon for instance, exhibited promising electrocatalytic activity for OER process (Table 1). However, to further enhance their performance to comparable levels of the benchmark Pt/C, RuO2 and IrO2 electrocatalysts, more efforts are needed towards (1) exploration of vital insights on the basis of activity descriptors need to be complemented with reaction kinetics data to thoroughly investigate the OER mechanistic route under realistic conditions with aim to lead to the rational design of the highly efficient earlier mentioned electrocatalysts; (2) in order not to recede the electrocatalytic performances of the mentioned electrocatalysts, increase mechanical robustness, long-lasting durability and reliability of carbon materials by glazing with antioxidant could make improvement of oxidation resistance of carbon supports under high anodic overpotentials; (3) tailoring the shape to expose optimum facet of the metal alloy nanoparticles of the electrocatalysts that can regulate activity by providing different atomic arrangements and electronic structure to affect adsorption and activation energies of reactants and intermediates at the their surfaces. Similarly, the ternary alloyed systems of Fe, Co, and Ni anchored on N-doped-carbon materials also showed promise for OER process, particularly the ternary alloyed system of the three metals encapsulated in N-doped carbon nanofiber (NiCoFe@N-CNFs) (Table 1). However, such electrocatalysts are not kinetically favored because of their high Tafel slope values suggesting that charge transfer gap or barrier was high, which led to their observed overpotentials. Hence, there is further need to identify relevant descriptors for OER activity performance to serve as route to reveal their best characteristics for maximum electrocatalytic activity. For example, the needed attention can be centered towards (1) modulation of charge transfer gap or barrier by particle size manipulation and configuration to achieve optimum interatomic spacing; (2) modulation of magnetic properties of such electrocatalysts could facilitate fast formation of O2 molecules, as O2 molecule is magnetically ordered in triplet state and is much readily to be formed when the spin orientation of its precursors (O and/or OH) are appropriately brought together in the right orientation by an electrocatalyst; (3) modulation of mole ratio of the three components of ternary alloyed system could increase the degrees of freedom of the alloyed nanoparticles that can lead to enhanced OER activity. All these insights can be complemented with kinetics studies, so as to lead to the rational design of highly efficient ternary alloyed electrocatalysts of the three metals.
Overall, in pursuance of further optimization of the carbon-tunable alloys of transition metal electrocatalysts with desired catalytic behavior for OER process, greater efforts are required.
-
i.
Although significant progress in gainful insight into the detail mechanisms of electrocatalytic process of OER has been made in the past decade. However, direct in situ observation on the routes of the identifiable mechanisms through which OER follows during the electrolysis of water based on the structural design and transformation of carbon-tunable alloys of TMs electrocatalysts is still lacking. Therefore, integration of in situ characterization couple with theoretical modelling in an advanced approach to gain insight into what would significantly help in rational design of electronic structure of electrocatalysts with specificity to a particular mechanistic route towards achieving a desired reaction product with low overpotential is required.
-
ii.
To achieve an overall water splitting process with low overpotential, understanding the working mechanism is essential. Therefore, ex situ characterization techniques to probe the active sites and substantiate with distinction between the three mechanistic routes of LOM, which are the oxygen-vacancy-site mechanism (OVSM), single-metal-site mechanism (SMSM), and dual-metal-site mechanism (DMSM) is highly needed and will be of great importance if it could be made realizable in the future. This will provide vital information on the reaction intermediates adsorbed on the surface of electrocatalysts during the OER process. This is very essential in the logical development of experimental strategies required to target the most favorable mechanism among all the possibilities in OER process.
-
iii.
At the moment, mainly DFT calculations are used for the rational design of electrocatalysts based on the interpretations of the classical theories derived from the activity of the noble-metal electrocatalysts. This may not necessarily and adequately be enough to reflect the real electrocatalytic operational conditions and thus, investigation under realistic conditions need to be accompanied.
-
iv.
It has been commonly shown that the surface adsorption free energies of the reaction intermediate of OER, i.e., ∆GHO* is connected and correlated with the electrocatalytic activity of the OER process, of which obeys the volcano relationship. However, the correlation between the inherent characteristics of the surface active sites of electrocatalysts that govern the adsorption strength of the intermediates still remains indescribable. In addition, currently to modulate the adsorption energy of reaction intermediates towards desirable electrocatalytic activity is certainly unrealistic experimentally. Because direct measurements of the adsorption energies of the reaction intermediates is impracticable under the operational conditions of water electrocatalysis. Hence, there is need in the future to make it practical to easily engineer the surface active sites of carbon-tunable alloys of transition metal electrocatalysts to optimize the ∆HO* based on the relationships between the intrinsic activity and adsorption energy value of the adsorbate. This is primarily target to achieve low overpotential.
-
v.
In procedural stabilization of HOO* in OER mechanistic pathway, conductive carbon-material can be functionalized with organic peroxides to achieve multiple O sites to act as proton acceptors (H*), as exemplified in γ-NiOOH melded with nanoclusters of γ-FeOOH. This could provide multiple replication effects of stabilization of HOO* alongside with long durability of repetitive and accelerated reproduction of oxygen-evolving (OO*) species with much low cost than the metallic peroxide for enhanced OER process. This could make a difference, as most of the current and widespread understanding of the OER is based on the metal ions as the mainly active sites.
-
vi.
As the bulk of the challenges of water splitting process is concentrated more on OER process that even brought up the surge of interests more recently in lattice oxygen engineering with respect to LOM over electrocatalysts. It is therefore expected that such could stimulate interests to drive surface scientists and catalysis research communities to new advancements in the design of highly electrochemically low-cost, active, and stable carbon-tunable alloys of transition metal electrocatalysts. The oxygen rich environment in the carbon framework could offer complementary support on the supply of oxygen. This conceivably leads to a route for enhanced LOM through oxidation of and spill-over from oxygen-containing functional groups in the carbon structure.
-
vii.
It is true that water splitting process is a promising strategy to produce hydrogen, but the OER is the major bottleneck that limiting the efficiency of the process due the sluggish kinetics and therefore, design of well-to-do OER electrocatalyst is crucial to solving this efficiency bottleneck in water splitting process. Hence, it is expected that in the future to know which of the AEM and the three types of LOM (single-metal-site mechanism (SMSM), oxygen-vacancy-site mechanism (OVSM), and dual-metal-site mechanism (DMSM)) is/are kinetically and thermodynamically favorable. Consequently, the future rational design of carbon-tunable electrocatalysts for OER can be constructed based on the known favorable kinetics and thermodynamics identities.
-
viii.
To further quicken the advancement of LOM, in the future direction should there be emphasis to investigate its underlying thermodynamics and relate with the reaction kinetics of all the three (3) types of LOM, by which that would create bridge between model situation and practical electrocatalysts behaviors in reality rather just to be guided by theory. Currently, the thermodynamics data on LOM is quite lacking. Therefore, generation of such data is very necessary considering the potentiality of LOM in OER.
-
ix.
Because that the adsorption energies of the reaction intermediates of OER process cannot be determined experimentally, the use of ∆GO* and ∆GHO* as well as ∆GHO* and ∆GHOO* are inconvenient for the fast high-throughput screening of electrocatalysts. Because they lack predictive guidance for the design of new carbon-tunable alloys of transition metal electrocatalysts rationally. This has now become a key issue in the electrocatalytic water splitting process and therefore, it is essential in the future to identify and determine the underlying electronic structure of active sites on the surface of the electrocatalysts. Thus can be easily fine-tuned and manipulated experimentally to directly adjust the adsorption energies of the reaction intermediates and electrocatalytic activity.
-
x.
As there has been ongoing debates on which of the single-parameter descriptor and multiple-parameter descriptors is rationally suitable and effective in real application when it comes to automated preparation for design of efficient electrocatalysts for OER. This is because of their versatility, machine learning and high-throughput computational approach will greatly help and accelerate the process of breaking the scaling relationships and ease routes for identifying universal descriptors, which can be used to rationally design efficient carbon-tunable alloys of transition metal electrocatalysts. In short, in the future, machine learning technology through high-throughput computational approach will play an important role in developing new descriptors and unveiling possible electrocatalytic routes. As a result, the approach could help in rational design of more suitable carbon-tunable alloys of transition metal electrocatalysts for overall water splitting process.
References
Laajimi, M., Go, Y.I.: Energy storage system design for large-scale solar PV in Malaysia: technical and environmental assessments. J. Energy Storage 26, 100984 (2019)
Kumar, A., Chaudhary, D.K., Parvin, S., Bhattacharyya, S.: High performance duckweed-derived carbon support to anchor NiFe electrocatalysts for efficient solar energy driven water splitting. J. Mater. Chem. A 6, 18948–18959 (2018)
Eisenberg, R., Gray, H.B., Crabtree, G.W.: Addressing the challenge of carbon-free energy. Proc. Natl. Acad. Sci. 117, 12543–12549 (2020)
Le Quéré, C., Andrew, R.M., Friedlingstein, P., Sitch, S., Pongratz, J., Manning, A.C., Korsbakken, J.I., Peters, G.P., Canadell, J.G., Jackson, R.B., Boden, T.A.: Global carbon budget 2017. Earth Syst. Sci. Data 10, 405–448 (2017)
Wang, S., Lu, A., Zhong, C.J.: Hydrogen production from water electrolysis: role of catalysts. Nano Converg. 8, 1–23 (2021)
Bernt, M., Gasteiger, H.A.: Influence of ionomer content in IrO2/TiO2 electrodes on PEM water electrolyzer performance. J. Electrochem. Soc. 163, F3179 (2016)
Lu, F., Zhou, M., Zhou, Y., Zeng, X.: First-row transition metal based catalysts for the oxygen evolution reaction under alkaline conditions: basic principles and recent advances. Small 13, 1701931 (2017)
Hu, Q., Li, G., Li, G., Liu, X., Zhu, B., Chai, X., Zhang, Q., Liu, J., He, C.: Trifunctional electrocatalysis on dual-doped graphene nanorings–integrated boxes for efficient water splitting and Zn–air batteries. Adv. Energy Mater. 9, 1803867 (2019)
Hu, C., Zhang, L., Gong, J.: Recent progress made in the mechanism comprehension and design of electrocatalysts for alkaline water splitting. Energy Environ. Sci. 12, 2620–2645 (2019)
Sardar, K., Petrucco, E., Hiley, C.I., Sharman, J.D., Wells, P.P., Russell, A.E., Kashtiban, R.J., Sloan, J., Walton, R.I.: Water-splitting electrocatalysis in acid conditions using ruthenate-iridate pyrochlores. Angew. Chem. 126, 11140–11144 (2014)
He, L., Liu, J., Liu, Y., Cui, B., Hu, B., Wang, M., Tian, K., Song, Y., Wu, S., Zhang, Z.: Titanium dioxide encapsulated carbon nitride nanosheets derived from MXene and melamine-cyanuric acid composite as a multifunctional electrocatalyst for hydrogen and oxygen evolution reaction and oxygen reduction reaction. Appl. Catal. B Environ. 248, 366–379 (2019)
Gopi, S., Giribabu, K., Kathiresan, M.: Porous organic polymerderived carbon composite as a bimodal catalyst for oxygen evolution reaction and nitrophenol reduction. ACS Omega 3, 6251–6258 (2018)
Gopi, S., Giribabu, K., Kathiresan, M., Yun, K.: Cobalt (ii) ions and cobalt nanoparticle embedded porous organic polymers: an efficient electrocatalyst for water-splitting reactions. Sustain. Energy Fuels 4, 3797–3805 (2020)
Zuo, Z., Li, Y.: Emerging electrochemical energy applications of graphdiyne. Joule 3, 899–903 (2019)
Fang, Y., Xue, Y., Li, Y., Yu, H., Hui, L., Liu, Y., Xing, C., Zhang, C., Zhang, D., Wang, Z.: Graphdiyne interface engineering: highly active and selective ammonia synthesis. Angew. Chem.-Int. Ed. 59, 13021–13027 (2020)
Sankar, S.S., Karthick, K., Sangeetha, K., Karmakar, A., Kundu, S.: Transition-metal-based zeolite imidazolate framework nanofibers via an electrospinning approach: a review. ACS Omega 5, 57–67 (2019)
Zou, X., Zhang, Y.: Noble metal-free hydrogen evolution catalysts for water splitting. Chem. Soc. Rev. 44, 5148–5180 (2015)
Huynh, M., Ozel, T., Liu, C., Lau, E.C., Nocera, D.G.: Design of template-stabilized active and earth-abundant oxygen evolution catalysts in acid. Chem. Sci. 8, 4779–4794 (2017)
Ursua, A., Gandıa, L.M., Sanchis, P.: Hydrogen production from water electrolysis: current status and future trends. Proc. IEEE 100, 410–426 (2012)
Chen, Z., Qing, H., Zhou, K., Sun, D., Wu, R.: Metal-organic framework-derived nanocomposites for electrocatalytic hydrogen evolution reaction. Prog. Mater. Sci. 108, 100618 (2020)
Wu, Z.P., Lu, X.F., Zang, S.Q., Lou, X.W.: Non-noble-metal-based electrocatalysts toward the oxygen evolution reaction. Adv. Funct. Mater. 30, 1910274 (2020)
Cui, C., Hu, X., Wen, L.: Recent progress on nanostructured bimetallic electrocatalysts for water splitting and electroreduction of carbon dioxide. J. Semicond. 41, 091705 (2020)
Tang, C., Wang, H.F., Zhu, X.L., Li, B.Q., Zhang, Q.: Advances in hybrid electrocatalysts for oxygen evolution reactions: rational integration of NiFe layered double hydroxides and nanocarbon. Part Part Syst Charact. 33, 473–486 (2016)
Suen, N.T., Hung, S.F., Quan, Q., Zhang, N., Xu, Y.J., Chen, H.M.: Electrocatalysis for the oxygen evolution reaction: recent development and future perspectives. Chem. Soc. Rev. 46, 337–365 (2017)
Zhu, Y.P., Guo, C., Zheng, Y., Qiao, S.Z.: Surface and interface engineering of noble-metal-free electrocatalysts for efficient energy conversion processes. Acc. Chem. Res. 50, 915–923 (2017)
Nørskov, J.K., Rossmeisl, J., Logadottir, A., Lindqvist, L.R., Kitchin, J.R., Bligaard, T., Jonsson, H.: Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004)
Man, I.C., Su, H.Y., Calle-Vallejo, F., Hansen, H.A., Martínez, J.I., Inoglu, N.G., Kitchin, J., Jaramillo, T.F., Nørskov, J.K., Rossmeisl, J.: Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3, 1159–1165 (2011)
Grimaud, A., Diaz-Morales, O., Han, B., Hong, W.T., Lee, Y.L., Giordano, L., Stoerzinger, K.A., Koper, M.T., Shao-Horn, Y.: Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457–465 (2017)
Wang, X., Vasileff, A., Jiao, Y., Zheng, Y., Qiao, S.Z.: Electronic and structural engineering of carbon-based metal-free electrocatalysts for water splitting. Adv. Mater. 31, 1803625 (2019)
Nørskov, J.K., Abild-Pedersen, F., Studt, F., Bligaard, T.: Density functional theory in surface chemistry and catalysis. Proc. Natl Acad. Sci. 108, 937–943 (2011)
Kim, J.S., Kim, B., Kim, H., Kang, K.: Recent progress on multimetal oxide catalysts for the oxygen evolution reaction. Adv. Energy Mater. 8, 1702774 (2018)
Pedersen, J.K., Batchelor, T.A., Yan, D., Skjegstad, L.E., Rossmeisl, J.: Surface electrocatalysis on high-entropy alloys. Curr. Opin. Electrochem. 26, 100651 (2021)
Song, J., Wei, C., Huang, Z.F., Liu, C., Zeng, L., Wang, X., Xu, Z.J.: A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 49, 2196–2214 (2020)
Hwang, J., Rao, R.R., Giordano, L., Katayama, Y., Yu, Y., Shao-Horn, Y.: Perovskites in catalysis and electrocatalysis. Science 358, 751–756 (2017)
Grimaud, A., Hong, W.T., Shao-Horn, Y., Tarascon, J.M.: Anionic redox processes for enhanced battery and water splitting devices. Nat. Mater. 15, 121–126 (2016)
Matsumoto, Y., Manabe, H., Sato, E.: Oxygen evolution on La1− x Srx CoO3 electrodes in alkaline solutions. J. Electrochem. Soc. 127, 811 (1980)
Giordano, L., Han, B., Risch, M., Hong, W.T., Rao, R.R., Stoerzinger, K.A., Shao-Horn, Y.: pH dependence of OER activity of oxides: current and future perspectives. Catal. Today 262, 2–10 (2016)
Shi, Y., Xie, R., Liu, X., Zhang, N., Aruta, C., Yang, N.: Tunable pH-dependent oxygen evolution activity of strontium cobaltite thin films for electrochemical water splitting. Phys. Chem. Chem. Phys. 21, 16230–16239 (2019)
Rossmeisl, J., Qu, Z.W., Zhu, H., Kroes, G.J., Nørskov, J.K.: Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607, 83–89 (2007)
Rong, X., Parolin, J., Kolpak, A.M.: A fundamental relationship between reaction mechanism and stability in metal oxide catalysts for oxygen evolution. ACS Catal. 6, 1153–1158 (2016)
Mefford, J.T., Rong, X., Abakumov, A.M., Hardin, W.G., Dai, S., Kolpak, A.M., Johnston, K.P., Stevenson, K.J.: Water electrolysis on La1− x Srx CoO3− δ perovskite electrocatalysts. Nat. Commun. 7, 11053 (2016)
Zhu, Y., Zhou, W., Shao, Z.: Perovskite/carbon composites: applications in oxygen electrocatalysis. Small 13, 1603793 (2017)
Cheng, X., Fabbri, E., Nachtegaal, M., Castelli, I.E., Kazzi, M., Haumont, R., Marzari, N., Schmidt, T.J.: Oxygen evolution reaction on La1–xSrxCoO3 perovskites: a combined experimental and theoretical study of their structural, electronic and electrochemical properties. Chem. Mater. 27, 7662–7672 (2015)
Grimaud, A., May, K.J., Carlton, C.E., Lee, Y.L., Risch, M., Hong, W.T., Zhou, J., Shao-Horn, Y.: Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat. Commun. 4, 2439 (2013)
Lee, Y.L., Kleis, J., Rossmeisl, J., Shao-Horn, Y., Morgan, D.: Prediction of solid oxide fuel cell cathode activity with first-principles descriptors. Energy Environ. Sci. 4, 3966–3970 (2011)
Goodenough, J.B.: Perspective on engineering transition-metal oxides. Chem. Mater. 26, 820–829 (2014)
Zhang, N., Feng, X., Rao, D., Deng, X., Cai, L., Qiu, B., Long, R., Xiong, Y., Lu, Y., Chai, Y.: Lattice oxygen activation enabled by high-valence metal sites for enhanced water oxidation. Nat. Commun. 11, 4066 (2020)
Zhu, Y., Tahini, H.A., Hu, Z., Yin, Y., Lin, Q., Sun, H., Zhong, Y., Chen, Y., Zhang, F., Lin, H.J., Chen, C.T.: Boosting oxygen evolution reaction by activation of lattice-oxygen sites in layered Ruddlesden-Popper oxide. EcoMat. 2, e12021 (2020)
Diaz-Morales, O., Ferrus-Suspedra, D., Koper, M.T.: The importance of nickel oxyhydroxide deprotonation on its activity towards electrochemical water oxidation. Chem. Sci. 7, 2639–2645 (2016)
Gao, J., Xu, C.Q., Hung, S.F., Liu, W., Cai, W., Zeng, Z., Jia, C., Chen, H.M., Xiao, H., Li, J., Huang, Y.: Breaking long-range order in iridium oxide by alkali ion for efficient water oxidation. J. Am. Chem. Soc. 141, 3014–3023 (2019)
Yang, C., Laberty-Robert, C., Batuk, D., Cibin, G., Chadwick, A.V., Pimenta, V., Yin, W., Zhang, L., Tarascon, J.M., Grimaud, A.: Phosphate ion functionalization of perovskite surfaces for enhanced oxygen evolution reaction. J. Phys. Chem. Lett. 8, 3466–3472 (2017)
Yoo, J.S., Rong, X., Liu, Y., Kolpak, A.M.: Role of lattice oxygen participation in understanding trends in the oxygen evolution reaction on perovskites. ACS Catal. 8, 4628–4636 (2018)
Pan, Y., Xu, X., Zhong, Y., Ge, L., Chen, Y., Veder, J.P., Guan, D., O’Hayre, R., Li, M., Wang, G., Wang, H.: Direct evidence of boosted oxygen evolution over perovskite by enhanced lattice oxygen participation. Nat. Commun. 11, 2002 (2020)
She, Z.W., Kibsgaard, J., Dickens, C.F., Chorkendorff, I.B., Nørskov, J.K., Jaramillo, T.F.: Combining theory and experiment in electrocatalysis: insights into materials design. Science 355, eaad4998 (2017)
Huang, Z.F., Song, J., Du, Y., Xi, S., Dou, S., Nsanzimana, J.M., Wang, C., Xu, Z.J., Wang, X.: Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329–338 (2019)
Kim, B.J., Fabbri, E., Abbott, D.F., Cheng, X., Clark, A.H., Nachtegaal, M., Borlaf, M., Castelli, I.E., Graule, T., Schmidt, T.J.: Functional role of Fe-doping in Co-based perovskite oxide catalysts for oxygen evolution reaction. J. Am. Chem. Soc. 141, 5231–5240 (2019)
Zhu, Y., Tahini, H.A., Hu, Z., Chen, Z.G., Zhou, W., Komarek, A.C., Lin, Q., Lin, H.J., Chen, C.T., Zhong, Y., Fernandez-Diaz, M.T.: Boosting oxygen evolution reaction by creating both metal ion and lattice-oxygen active sites in a complex oxide. Adv. Mater. 32, 1905025 (2020)
Hua, B., Li, M., Pang, W., Tang, W., Zhao, S., Jin, Z., Zeng, Y., Amirkhiz, B.S., Luo, J.L.: Activating p-blocking centers in perovskite for efficient water splitting. Chem. 4, 2902–2916 (2018)
Jakšić, M.M.: Electrocatalysis of hydrogen evolution in the light of the Brewer–Engel theory for bonding in metals and intermetallic phases. Electrochim. Acta 29, 1539–1550 (1984)
Zhang, R., Sun, Z., Feng, R., Lin, Z., Liu, H., Li, M., Yang, Y., Shi, R., Zhang, W., Chen, Q.: Rapid adsorption enables interface engineering of PdMnCo alloy/nitrogen-doped carbon as highly efficient electrocatalysts for hydrogen evolution reaction. ACS Appl. Mater. Interfaces 9, 38419–38427 (2017)
Oh, A., Sa, Y.J., Hwang, H., Baik, H., Kim, J., Kim, B., Joo, S.H., Lee, K.: Rational design of Pt–Ni–Co ternary alloy nanoframe crystals as highly efficient catalysts toward the alkaline hydrogen evolution reaction. Nanoscale 8, 16379–16386 (2016)
Wang, Y.J., Zhao, N., Fang, B., Li, H., Bi, X.T., Wang, H.: Carbon-supported Pt-based alloy electrocatalysts for the oxygen reduction reaction in polymer electrolyte membrane fuel cells: particle size, shape, and composition manipulation and their impact to activity. Chem. Rev. 115, 3433–3467 (2015)
Hou, Y., Cui, S., Wen, Z., Guo, X., Feng, X., Chen, J.: Strongly coupled 3D hybrids of N-doped porous carbon nanosheet/CoNi alloy-encapsulated carbon nanotubes for enhanced electrocatalysis. Small 11, 5940–5980 (2015)
Shen, Y., Zhou, Y., Wang, D., Wu, X., Li, J., Xi, J.: Nickel–copper alloy encapsulated in graphitic carbon shells as electrocatalysts for hydrogen evolution reaction. Adv. Energy Mater. 8, 1701759 (2018)
Jiang, P., Chen, J., Wang, C., Yang, K., Gong, S., Liu, S., Lin, Z., Li, M., Xia, G., Yang, Y., Su, J.: Tuning the activity of carbon for electrocatalytic hydrogen evolution via an iridium-cobalt alloy core encapsulated in nitrogen-doped carbon cages. Adv. Mater. 30, 1705324 (2018)
Zhu, Y., Chen, G., Xu, X., Yang, G., Liu, M., Shao, Z.: Enhancing electrocatalytic activity for hydrogen evolution by strongly coupled molybdenum nitride@ nitrogen-doped carbon porous nano-octahedrons. ACS Catal. 7, 3540–3547 (2017)
Anantharaj, S., Ede, S.R., Sakthikumar, K., Karthick, K., Mishra, S., Kundu, S.: Recent trends and perspectives in electrochemical water splitting with an emphasis on sulfide, selenide, and phosphide catalysts of Fe Co, and Ni: a review. ACS Catal. 6, 8069–8097 (2016)
Gupta, S., Qiao, L., Zhao, S., Xu, H., Lin, Y., Devaguptapu, S.V., Wang, X., Swihart, M.T., Wu, G.: Highly active and stable graphene tubes decorated with FeCoNi alloy nanoparticles via a template-free graphitization for bifunctional oxygen reduction and evolution. Adv. Energy Mater. 6, 1601198 (2016)
Fu, Y., Yu, H.Y., Jiang, C., Zhang, T.H., Zhan, R., Li, X., Li, J.F., Tian, J.H., Yang, R.: NiCo alloy nanoparticles decorated on N-doped carbon nanofibers as highly active and durable oxygen electrocatalyst. Adv. Funct. Mater. 28, 1705094 (2018)
Yu, J., Zhong, Y., Zhou, W., Shao, Z.: Facile synthesis of nitrogen-doped carbon nanotubes encapsulating nickel cobalt alloys 3D networks for oxygen evolution reaction in an alkaline solution. J. Power Sources 338, 26–33 (2017)
Tang, C., Zhong, L., Zhang, B., Wang, H.F., Zhang, Q.: 3D mesoporous van der Waals heterostructures for trifunctional energy electrocatalysis. Adv. Mater. 30, 1705110 (2018)
Hu, Q., Liu, X., Tang, C., Fan, L., Chai, X., Zhang, Q., Liu, J., He, C.: Facile fabrication of a 3D network composed of N-doped carbon-coated core–shell metal oxides/phosphides for highly efficient water splitting. Sustain. Energy Fuels 2, 1085–1092 (2018)
Li, G., Sun, Y., Rao, J., Wu, J., Kumar, A., Xu, Q.N., Fu, C., Liu, E., Blake, G.R., Werner, P., Shao, B.: Carbon-tailored semimetal MoP as an efficient hydrogen evolution electrocatalyst in both alkaline and acid media. Adv. Energy Mater. 8, 1801258 (2018)
Li, Y., Duerloo, K.A., Wauson, K., Reed, E.J.: Structural semiconductor-to-semimetal phase transition in two-dimensional materials induced by electrostatic gating. Nat. Commun. 7, 10671 (2016)
Hu, Q., Li, G., Han, Z., Wang, Z., Huang, X., Yang, H., Zhang, Q., Liu, J., He, C.: Recent progress in the hybrids of transition metals/carbon for electrochemical water splitting. J. Mater. Chem. A 7, 14380–14390 (2019)
Hu, Q., Li, G., Liu, X., Zhu, B., Li, G., Fan, L., Chai, X., Zhang, Q., Liu, J., He, C.: Coupling pentlandite nanoparticles and dual-doped carbon networks to yield efficient and stable electrocatalysts for acid water oxidation. J. Mater. Chem. A 7, 461–468 (2019)
Sun, H., Lian, Y., Yang, C., Xiong, L., Qi, P., Mu, Q., Zhao, X., Guo, J., Deng, Z., Peng, Y.: A hierarchical nickel–carbon structure templated by metal–organic frameworks for efficient overall water splitting. Energy Environ. Sci. 11, 2363–2371 (2018)
Hu, C., Li, M., Qiu, J., Sun, Y.P.: Design and fabrication of carbon dots for energy conversion and storage. Chem. Soc. Rev. 48, 2315–2337 (2019)
Deng, J., Ren, P., Deng, D., Bao, X.: Enhanced electron penetration through an ultrathin graphene layer for highly efficient catalysis of the hydrogen evolution reaction. Angew. Chem. Int. Ed. 54, 2100–2104 (2015)
Hinnemann, B., Moses, P.G., Bonde, J., Jørgensen, K.P., Nielsen, J.H., Horch, S., Chorkendorff, I., Nørskov, J.K.: Biomimetic hydrogen evolution: MoS2 nanoparticles as catalyst for hydrogen evolution. J. Am. Chem. Soc. 127, 5308–5309 (2005)
Corona, B., Howard, M., Zhang, L., Henkelman, G.: Computational screening of core@ shell nanoparticles for the hydrogen evolution and oxygen reduction reactions. J. Chem. Phys. 145, 244708 (2016)
Deng, J., Yu, L., Deng, D., Chen, X., Yang, F., Bao, X.: Highly active reduction of oxygen on a FeCo alloy catalyst encapsulated in pod-like carbon nanotubes with fewer walls. J. Mater. Chem. A 1, 14868–14873 (2013)
Hu, Y., Jensen, J.O., Zhang, W., Cleemann, L.N., Xing, W., Bjerrum, N.J., Li, Q.: Hollow spheres of iron carbide nanoparticles encased in graphitic layers as oxygen reduction catalysts. Angew. Chem. 126, 3749–3753 (2014)
Yang, Y., Lin, Z., Gao, S., Su, J., Lun, Z., Xia, G., Chen, J., Zhang, R., Chen, Q.: Tuning electronic structures of nonprecious ternary alloys encapsulated in graphene layers for optimizing overall water splitting activity. ACS Catal. 7, 469–479 (2017)
Liu, Y., Jiang, H., Zhu, Y., Yang, X., Li, C.: Transition metals (Fe Co, and Ni) encapsulated in nitrogen-doped carbon nanotubes as bi-functional catalysts for oxygen electrode reactions. J. Mater. Chem. A 4, 1694–1701 (2016)
Wang, J., Wu, H., Gao, D., Miao, S., Wang, G., Bao, X.: High-density iron nanoparticles encapsulated within nitrogen-doped carbon nanoshell as efficient oxygen electrocatalyst for zinc–air battery. Nano Energy 13, 387–396 (2015)
Jia, G., Zhang, W., Fan, G., Li, Z., Fu, D., Hao, W., Yuan, C., Zou, Z.: Three-dimensional hierarchical architectures derived from surface-mounted metal-organic framework membranes for enhanced electrocatalysis. Angew. Chem. 129, 13969–13973 (2017)
Sivanantham, A., Ganesan, P., Estevez, L., McGrail, B.P., Motkuri, R.K., Shanmugam, S.: Water electrolysis: a stable graphitic, nanocarbon-encapsulated, cobalt-rich core-shell electrocatalyst as an oxygen electrode in a water electrolyzer. Adv. Energy Mater. 8, 1870065 (2018)
Su, Y., Zhu, Y., Jiang, H., Shen, J., Yang, X., Zou, W., Chen, J., Li, C.: Cobalt nanoparticles embedded in N-doped carbon as an efficient bifunctional electrocatalyst for oxygen reduction and evolution reactions. Nanoscale 6, 15080–15089 (2014)
Fu, G., Chen, Y., Cui, Z., Li, Y., Zhou, W., Xin, S., Tang, Y., Goodenough, J.B.: Novel hydrogel-derived bifunctional oxygen electrocatalyst for rechargeable air cathode. Nano Lett. 16, 6516–6522 (2016)
Zhang, X., Xu, H., Li, X., Li, Y., Yang, T., Liang, Y.: Facile synthesis of nickel–iron/nanocarbon hybrids as advanced electrocatalysts for efficient water splitting. ACS Catal. 6, 580–588 (2016)
Zhang, J.W., Zhang, H., Ren, T.Z., Yuan, Z.Y., Bandosz, T.J.: FeNi doped porous carbon as an efficient catalyst for oxygen evolution reaction. Front. Chem. Sci. Eng. 15, 279–287 (2021)
Hou, C.C., Zou, L., Xu, Q.: A hydrangea-like superstructure of open carbon cages with hierarchical porosity and highly active metal sites. Adv. Mater. 31, 1904689 (2019)
Deng, J., Ren, P., Deng, D., Yu, L., Yang, F., Bao, X.: Highly active and durable non-precious-metal catalysts encapsulated in carbon nanotubes for hydrogen evolution reaction. Energy Environ. Sci. 7, 1919–1923 (2014)
Wang, C., Yang, H., Zhang, Y., Wang, Q.: NiFe alloy nanoparticles with hcp crystal structure stimulate superior oxygen evolution reaction electrocatalytic activity. Angew. Chem. Int. Ed. 58, 6099–6103 (2019)
Nam, G., Son, Y., Park, S.O., Jeon, W.C., Jang, H., Park, J., Chae, S., Yoo, Y., Ryu, J., Kim, M.G., Kwak, S.K.: A ternary Ni46Co40Fe14 nanoalloy-based oxygen electrocatalyst for highly efficient rechargeable zinc-air batteries. Adv. Mater. 30, 1803372 (2018)
Cao, F., Yang, X., Shen, C., Li, X., Wang, J., Qin, G., Li, S., Pang, X., Li, G.: Electrospinning synthesis of transition metal alloy nanoparticles encapsulated in nitrogen-doped carbon layers as an advanced bifunctional oxygen electrode. J. Mater. Chem. A 8, 7245–7252 (2020)
Cui, X., Ren, P., Deng, D., Deng, J., Bao, X.: Single layer graphene encapsulating non-precious metals as high-performance electrocatalysts for water oxidation. Energy Environ. Sci. 9, 123–129 (2016)
Bu, F., Chen, W., Gu, J., Agboola, P.O., Al-Khalli, N.F., Shakir, I., Xu, Y.: Microwave-assisted CVD-like synthesis of dispersed monolayer/few-layer N-doped graphene encapsulated metal nanocrystals for efficient electrocatalytic oxygen evolution. Chem. Sci. 9, 7009–7016 (2018)
Liu, H., Xi, C., Xin, J., Zhang, G., Zhang, S., Zhang, Z., Huang, Q., Li, J., Liu, H., Kang, J.: Free-standing nanoporous NiMnFeMo alloy: An efficient non-precious metal electrocatalyst for water splitting. Chem. Eng. J. 404, 126530 (2021)
Koper, M.T.: Theory of multiple proton–electron transfer reactions and its implications for electrocatalysis. Chem. Sci. 4, 2710–2723 (2013)
Wu, A., Xie, Y., Ma, H., Tian, C., Gu, Y., Yan, H., Zhang, X., Yang, G., Fu, H.: Integrating the active OER and HER components as the heterostructures for the efficient overall water splitting. Nano Energy 44, 353–363 (2018)
Trześniewski, B.J., Diaz-Morales, O., Vermaas, D.A., Longo, A., Bras, W., Koper, M.T., Smith, W.A.: In situ observation of active oxygen species in Fe-containing Ni-based oxygen evolution catalysts: the effect of pH on electrochemical activity. J. Am. Chem. Soc. 137, 15112–15121 (2015)
Trasatti, S.: Work function, electronegativity, and electrochemical behaviour of metals: III. Electrolytic hydrogen evolution in acid solutions. J. Electroanal. Chem. Interfac. Electrochem. 39, 163–184 (1972)
Suntivich, J., May, K.J., Gasteiger, H.A., Goodenough, J.B., Shao-Horn, Y.: A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 334, 1383–1385 (2011)
Suntivich, J., Hong, W.T., Lee, Y.L., Rondinelli, J.M., Yang, W., Goodenough, J.B., Dabrowski, B., Freeland, J.W., Shao-Horn, Y.: Estimating hybridization of transition metal and oxygen states in perovskites from Ok-edge x-ray absorption spectroscopy. J. Phys. Chem. C 118, 1856–1863 (2014)
Calle-Vallejo, F., Díaz-Morales, O.A., Kolb, M.J., Koper, M.T.: Why is bulk thermochemistry a good descriptor for the electrocatalytic activity of transition metal oxides? ACS Catal. 5, 869–873 (2015)
Hibbert, D.B., Churchill, C.R.: Kinetics of the electrochemical evolution of isotopically enriched gases. Part 2.—18O 16O evolution on NiCo2O4 and Lix Co3–xO4 in alkaline solution. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 80, 1965–1975 (1984)
Fierro, S., Nagel, T., Baltruschat, H., Comninellis, C.: Investigation of the oxygen evolution reaction on Ti/IrO2 electrodes using isotope labelling and on-line mass spectrometry. Electrochem. Commun. 9, 1969–1974 (2007)
Macounova, K., Makarova, M., Krtil, P.: Oxygen evolution on nanocrystalline RuO2 and Ru0.9Ni0.1O2−δ electrodes–DEMS approach to reaction mechanism determination. Electrochem. Commun. 11, 1865–1868 (2009).
Wohlfahrt-Mehrens, M., Heitbaum, J.: Oxygen evolution on Ru and RuO2 electrodes studied using isotope labelling and on-line mass spectrometry. J. Electroanal. Chem. Interfac. Electrochem. 237, 251–2560 (1987)
Surendranath, Y., Kanan, M.W., Nocera, D.G.: Mechanistic studies of the oxygen evolution reaction by a cobalt-phosphate catalyst at neutral pH. J. Am. Chem. Soc. 132, 16501–16509 (2010)
Mavros, M.G., Tsuchimochi, T., Kowalczyk, T., McIsaac, A., Wang, L.P., Voorhis, T.V.: What can density functional theory tell us about artificial catalytic water splitting? Inorg. Chem. 53, 6386–6397 (2014)
Wang, L.P., Van Voorhis, T.: Direct-coupling O2 bond forming a pathway in cobalt oxide water oxidation catalysts. J. Phys. Chem. Lett. 2, 2200–2204 (2011)
Betley, T.A., Wu, Q., Van Voorhis, T., Nocera, D.G.: Electronic design criteria for O−O bond formation via metal−oxo complexes. Inorg. Chem. 47, 1849–1861 (2008)
Montoya, J.H., Seitz, L.C., Chakthranont, P., Vojvodic, A., Jaramillo, T.F., Nørskov, J.K.: Materials for solar fuels and chemicals. Nat. Mater. 16, 70–81 (2017)
Bader, R.F., Carroll, M.T., Cheeseman, J.R., Chang, C.: Properties of atoms in molecules: atomic volumes. J. Am. Chem. Soc. 109, 7968–7979 (1987)
Hong, W.T., Risch, M., Stoerzinger, K.A., Grimaud, A., Suntivich, J., Shao-Horn, Y.: Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 8, 1404–1427 (2015)
McCrory, C.C., Jung, S., Ferrer, I.M., Chatman, S.M., Peters, J.C., Jaramillo, T.F.: Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices. J. Am. Chem. Soc. 137, 4347–4357 (2015)
Pan, Y., Sun, K., Liu, S., Cao, X., Wu, K., Cheong, W.C., Chen, Z., Wang, Y., Li, Y., Liu, Y., Wang, D.: Core–shell ZIF-8@ ZIF-67-derived CoP nanoparticle-embedded N-doped carbon nanotube hollow polyhedron for efficient overall water splitting. J. Am. Chem. Soc. 140, 2610–2618 (2018)
Zhang, C., Yang, H., Zhong, D., Xu, Y., Wang, Y., Yuan, Q., Liang, Z., Wang, B., Zhang, W., Zheng, H., Cheng, T.: A yolk–shell structured metal–organic framework with encapsulated iron-porphyrin and its derived bimetallic nitrogen-doped porous carbon for an efficient oxygen reduction reaction. J. Mater. Chem. A 8, 9536–9544 (2020)
Xiong, Y., Yang, Y., DiSalvo, F.J., Abruña, H.D.: Metal–organic-framework-derived Co–Fe bimetallic oxygen reduction electrocatalysts for alkaline fuel cells. J. Am. Chem. Soc. 141, 10744–10750 (2019)
Ning, H., Li, G., Chen, Y., Zhang, K., Gong, Z., Nie, R., Hu, W., Xia, Q.: Porous N-doped carbon-encapsulated CoNi alloy nanoparticles derived from MOFs as efficient bifunctional oxygen electrocatalysts. ACS Appl. Mater. Interfaces 11, 1957–1968 (2018)
Li, Z., He, H., Cao, H., Sun, S., Diao, W., Gao, D., Lu, P., Zhang, S., Guo, Z., Li, M., Liu, R.: Atomic Co/Ni dual sites and Co/Ni alloy nanoparticles in N-doped porous Janus-like carbon frameworks for bifunctional oxygen electrocatalysis. Appl. Catal. B Environ. 240, 112–121 (2019)
Huang, L., Gao, G., Zhang, H., Chen, J., Fang, Y., Dong, S.: Self-dissociation-assembly of ultrathin metal-organic framework nanosheet arrays for efficient oxygen evolution. Nano Energy 68, 104296 (2020)
Zhang, B., Zheng, Y., Ma, T., Yang, C., Peng, Y., Zhou, Z., Zhou, M., Li, S., Wang, Y., Cheng, C.: Designing MOF nanoarchitectures for electrochemical water splitting. Adv. Mater. 33, 2006042 (2021)
Li, S., Li, E., An, X., Hao, X., Jiang, Z., Guan, G.: Transition-metal-based catalysts for electrochemical water splitting at high current density: current status and perspectives. Nanoscale 13, 12788–12817 (2021)
Zou, X., Su, J., Silva, R., Goswami, A., Sathe, B.R., Asefa, T.: Efficient oxygen evolution reaction catalyzed by low-density Ni-doped Co3O4 nanomaterials derived from metal-embedded graphitic C3 N4. Chem. Commun. 49, 7522–7524 (2013)
Qian, G., Chen, J., Yu, T., Luo, L., Yin, S.: N-doped graphene-decorated NiCo alloy coupled with mesoporous NiCoMoO nano-sheet heterojunction for enhanced water electrolysis activity at high current density. Nano-micro Lett. 13, 77 (2021)
Fan, K., Ji, Y., Zou, H., Zhang, J., Zhu, B., Chen, H., Daniel, Q., Luo, Y., Yu, J., Sun, L.: Hollow iron–vanadium composite spheres: a highly efficient iron-based water oxidation electrocatalyst without the need for nickel or cobalt. Angew. Chem. Int. Ed. 56, 3289–3293 (2017)
Pi, Y., Shao, Q., Wang, P., Lv, F., Guo, S., Guo, J., Huang, X.: Trimetallic oxyhydroxide coralloids for efficient oxygen evolution electrocatalysis. Angew. Chem. Int. Ed. 56, 4502–4506 (2017)
Zhang, B., Zheng, X., Voznyy, O., Comin, R., Bajdich, M., García-Melchor, M., Han, L., Xu, J., Liu, M., Zheng, L., de Arquer, F.P.: Homogeneously dispersed multimetal oxygen-evolving catalysts. Science 352, 333–337 (2016)
Xiang, R., Peng, L., Wei, Z.: Tuning interfacial structures for better catalysis of water electrolysis. Chem. A Euro. J. 25, 9799–9815 (2019)
Ma, X., Shi, Y., Wang, K., Yu, Y., Zhang, B.: Solid-state conversion synthesis of advanced electrocatalysts for water splitting. Chem. A Euro. J. 26, 3961–3972 (2020)
Feng, J., Lv, F., Zhang, W., Li, P., Wang, K., Yang, C., Wang, B., Yang, Y., Zhou, J., Lin, F., Wang, G.C.: Iridium-based multimetallic porous hollow nanocrystals for efficient overall-water-splitting catalysis. Adv. Mater. 29, 1703798 (2017)
Jiang, J., Sun, F., Zhou, S., Hu, W., Zhang, H., Dong, J., Jiang, Z., Zhao, J., Li, J., Yan, W., Wang, M.: Atomic-level insight into super-efficient electrocatalytic oxygen evolution on iron and vanadium co-doped nickel (oxy) hydroxide. Nat. Commun. 9, 2885 (2018)
Liao, P., Keith, J.A., Carter, E.A.: Water oxidation on pure and doped hematite (0001) surfaces: prediction of Co and Ni as effective dopants for electrocatalysis. J. Am. Chem. Soc. 134, 13296–13309 (2012)
Liu, J., Ji, Y., Nai, J., Niu, X., Luo, Y., Guo, L., Yang, S.: Ultrathin amorphous cobalt–vanadium hydr(oxy) oxide catalysts for the oxygen evolution reaction. Energy Environ. Sci. 11, 1736–1741 (2018)
Ye, S.H., Shi, Z.X., Feng, J.X., Tong, Y.X., Li, G.R.: Activating CoOOH porous nanosheet arrays by partial iron substitution for efficient oxygen evolution reaction. Angew. Chem. Int. Ed. 57, 2672–2676 (2018)
Gerken, J.B., Shaner, S.E., Massé, R.C., Porubsky, N.J., Stahl, S.S.: A survey of diverse earth abundant oxygen evolution electrocatalysts showing enhanced activity from Ni–Fe oxides containing a third metal. Energy Environ. Sci. 7, 2376–2382 (2014)
Schwanke, C., Stein, H.S., Xi, L., Sliozberg, K., Schuhmann, W., Ludwig, A., Lange, K.M.: Correlating oxygen evolution catalysts activity and electronic structure by a high-throughput investigation of Ni1-yzFeyCrzOx. Sci. Rep. 7, 4710 (2017)
Speck, F.D., Kim, J.H., Bae, G., Joo, S.H., Mayrhofer, K.J., Choi, C.H., Cherevko, S.: Single-atom catalysts: a perspective toward application in electrochemical energy conversion. J. Am. Chem. Soc. 1, 1086–1100 (2021)
Perez-Rodriguez, S., Sebastian, D., Lazaro, M.J.: Electrochemical oxidation of ordered mesoporous carbons and the influence of graphitization. Electrochim. Acta 303, 167–175 (2019)
Filimonenkov, I.S., Bouillet, C., Kerangueven, G., Simonov, P.A., Tsirlina, G.A., Savinova, E.R.: Carbon materials as additives to the OER catalysts: RRDE study of carbon corrosion at high anodic potentials. Electrochim. Acta 321, 134657 (2019)
Alegre, C., Sebastián, D., Lázaro, M.J.: Carbon xerogels electrochemical oxidation and correlation with their physico-chemical properties. Carbon 144, 382–394 (2019)
Jorge, A.B., Jervis, R., Periasamy, A.P., Qiao, M., Feng, J., Tran, L.N., Titirici, M.M.: 3D carbon materials for efficient oxygen and hydrogen electrocatalysis. Adv. Energy Mater. 10, 1902494 (2020)
Li, G., Li, K., Yang, L., Chang, J., Ma, R., Wu, Z., Ge, J., Liu, C., Xing, W.: Boosted performance of Ir species by employing TiN as the support toward oxygen evolution reaction. ACS Appl. Mater. Interfaces 10, 38117–38124 (2018)
Qu, H.Y., He, X., Wang, Y., Hou, S.: Electrocatalysis for the oxygen evolution reaction in acidic media: progress and challenges. Appl. Sci. 11, 4320 (2021)
Wang, M., Wang, Y., Mao, S.S., Shen, S.: Transition-metal alloy electrocatalysts with active sites modulated by metal-carbide heterophases for efficient oxygen evolution. Nano Energy 88, 106216 (2021)
Stelmachowski, P., Duch, J., Sebastián, D., Lázaro, M.J., Kotarba, A.: Carbon-based composites as electrocatalysts for oxygen evolution reaction in alkaline media. Materials 14, 4984 (2021)
Tang, C., Wang, H.S., Wang, H.F., Zhang, Q., Tian, G.L., Nie, J.Q., Wei, F.: Spatially confined hybridization of nanometer-sized NiFe hydroxides into nitrogen-doped graphene frameworks leading to superior oxygen evolution reactivity. Adv. Mater. 27, 4516–4522 (2015)
Chen, S., Duan, J., Bian, P., Tang, Y., Zheng, R., Qiao, S.Z.: Three-dimensional smart catalyst electrode for oxygen evolution reaction. Adv. Energy Mater. 5, 1500936 (2015)
Wu, J., Xue, Y., Yan, X., Yan, W., Cheng, Q., Xie, Y.: Co3O4 nanocrystals on single-walled carbon nanotubes as a highly efficient oxygen-evolving catalyst. Nano Res. 5, 521–530 (2012)
Jiang, J., Zhang, C., Ai, L.: Hierarchical iron nickel oxide architectures derived from metal-organic frameworks as efficient electrocatalysts for oxygen evolution reaction. Electrochim. Acta 208, 17–24 (2016)
Long, X., Li, J., Xiao, S., Yan, K., Wang, Z., Chen, H., Yang, S.: A strongly coupled graphene and FeNi double hydroxide hybrid as an excellent electrocatalyst for the oxygen evolution reaction. Angew. Chem. 126, 7714–7718 (2014)
Lv, J.J., Zhao, J., Fang, H., Jiang, L.P., Li, L.L., Ma, J., Zhu, J.J.: Incorporating nitrogen-doped graphene quantum dots and Ni3S2 nanosheets: a synergistic electrocatalyst with highly enhanced activity for overall water splitting. Small 13, 1700264 (2017)
Zoller, F., Häringer, S., Böhm, D., Luxa, J., Sofer, Z., Fattakhova-Rohlfing, D.: Carbonaceous oxygen evolution reaction catalysts: from defect and doping-induced activity over hybrid compounds to ordered framework structures. Small 4, 2007484 (2021)
Wang, H.F., Tang, C., Zhang, Q.: A review of precious-metal-free bifunctional oxygen electrocatalysts: rational design and applications in Zn−air batteries. Adv. Funct. Mater. 28, 1803329 (2018)
Yuan, M., Sun, Y., Yang, Y., Zhang, J., Dipazir, S., Zhao, T., Li, S., Xie, Y., Zhao, H., Liu, Z., Zhang, G.: Boosting oxygen evolution reactivity by modulating electronic structure and honeycomb-like architecture in Ni2P/N, P-codoped carbon hybrids. Green Energy Environ. 6, 866–874 (2021)
Shao, Y., Xiao, X., Zhu, Y.P., Ma, T.Y.: Single-crystal cobalt phosphate nanosheets for biomimetic oxygen evolution in neutral electrolytes. Angew. Chem. Int. Ed. 58, 14599–14604 (2019)
Shinde, S.S., Lee, C.H., Yu, J.Y., Kim, D.H., Lee, S.U., Lee, J.H.: Hierarchically designed 3D holey C2N aerogels as bifunctional oxygen electrodes for flexible and rechargeable Zn-air batteries. ACS Nano 12, 596–608 (2018)
Shinde, S.S., Lee, C.H., Sami, A., Kim, D.H., Lee, S.U., Lee, J.H.: Scalable 3-D carbon nitride sponge as an efficient metal-free bifunctional oxygen electrocatalyst for rechargeable Zn–air batteries. ACS Nano 11, 347–357 (2017)
Kulkarni, A., Siahrostami, S., Patel, A., Nørskov, J.K.: Understanding catalytic activity trends in the oxygen reduction reaction. Chem. Rev. 118, 2302–2312 (2018)
Fei, H., Dong, J., Feng, Y., Allen, C.S., Wan, C., Volosskiy, B., Li, M., Zhao, Z., Wang, Y., Sun, H., An, P.: General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities. Nat. Catal. 1, 63–72 (2018)
Doyle, A.D., Montoya, J.H., Vojvodic, A.: Improving oxygen electrochemistry through nanoscopic confinement. ChemCatChem 7, 738–742 (2015)
Rao, R.R., Kolb, M.J., Halck, N.B., Pedersen, A.F., Mehta, A., You, H., Stoerzinger, K.A., Feng, Z., Hansen, H.A., Zhou, H., Giordano, L.: Towards identifying the active sites on RuO2 (110) in catalyzing oxygen evolution. Energy Environ. Sci. 10, 2626–2637 (2017)
Frydendal, R., Busch, M., Halck, N.B., Paoli, E.A., Krtil, P., Chorkendorff, I., Rossmeisl, J.: Enhancing activity for the oxygen evolution reaction: the beneficial interaction of gold with manganese and cobalt oxides. ChemCatChem 7, 149–154 (2015)
Pham, H.H., Cheng, M.J., Frei, H., Wang, L.W.: Surface proton hopping and fast-kinetics pathway of water oxidation on Co3O4 (001) surface. ACS Catal. 6, 5610–5617 (2016)
Halck, N.B., Petrykin, V., Krtil, P., Rossmeisl, J.: Beyond the volcano limitations in electrocatalysis–oxygen evolution reaction. Phys. Chem. Chem. Phys. 16, 13682–13688 (2014)
Qiu, T., Tu, B., Saldana-Greco, D., Rappe, A.M.: Ab initio simulation explains the enhancement of catalytic oxygen evolution on CaMnO3. ACS Catal. 8, 2218–2224 (2018)
Song, F., Busch, M.M., Lassalle-Kaiser, B., Hsu, C.S., Petkucheva, E., Bensimon, M., Chen, H.M., Corminboeuf, C., Hu, X.: An unconventional iron nickel catalyst for the oxygen evolution reaction. ACS Central Sci. 5, 558–568 (2019)
She, S., Zhu, Y., Chen, Y., Lu, Q., Zhou, W., Shao, Z.: Realizing ultrafast oxygen evolution by introducing proton acceptor into perovskites. Adv. Energy Mater. 9, 1900429 (2019)
Detsi, E., Cook, J.B., Lesel, B.K., Turner, C.L., Liang, Y.L., Robbennolt, S., Tolbert, S.H.: Mesoporous Ni60 Fe30 Mn10-alloy based metal/metal oxide composite thick films as highly active and robust oxygen evolution catalysts. Energy Environ. Sci. 9, 540–549 (2016)
Zuo, Y., Rao, D., Ma, S., Li, T., Tsang, Y.H., Kment, S., Chai, Y.: Valence engineering via dual-cation and boron doping in pyrite selenide for highly efficient oxygen evolution. ACS Nano 13, 11469–11476 (2019)
Yao, Y., Huang, Z., Xie, P., Lacey, S.D., Jacob, R.J., Xie, H., Chen, F., Nie, A., Pu, T., Rehwoldt, M., Yu, D.: Carbothermal shock synthesis of high-entropy-alloy nanoparticles. Science 359, 1489–1494 (2018)
George, E.P., Raabe, D., Ritchie, R.O.: High-entropy alloys. Nat. Rev. Mater. 4, 515–534 (2019)
Wu, T., Sun, S., Song, J., Xi, S., Du, Y., Chen, B., Sasangka, W.A., Liao, H., Gan, C.L., Scherer, G.G., Zeng, L.: Iron-facilitated dynamic active-site generation on spinel CoAl2O4 with self-termination of surface reconstruction for water oxidation. Nat. Catal. 2, 763–772 (2019)
Bergmann, A., Jones, T.E., Moreno, E.M., Teschner, D., Chernev, P., Gliech, M., Reier, T., Dau, H., Strasser, P.: Unified structural motifs of the catalytically active state of Co (oxyhydr)oxides during the electrochemical oxygen evolution reaction. Nat. Catal. 1, 711–719 (2018)
Chen, J.Y., Dang, L., Liang, H., Bi, W., Gerken, J.B., Jin, S., Alp, E.E., Stahl, S.S.: Operando analysis of NiFe and Fe oxyhydroxide electrocatalysts for water oxidation: detection of Fe4+ by Mossbauer spectroscopy. J. Am. Chem. Soc. 137, 15090–15093 (2015)
Trotochaud, L., Young, S.L., Ranney, J.K., Boettcher, S.W.: Nickel–iron oxyhydroxide oxygen-evolution electrocatalysts: the role of intentional and incidental iron incorporation. J. Am. Chem. Soc. 136, 6744–6753 (2014)
Hunter, B.M., Gray, H.B., Muller, A.M.: Earth-abundant heterogeneous water oxidation catalysts. Chem. Rev. 116, 14120–14136 (2016)
Cheng, W., Zhao, X., Su, H., Tang, F., Che, W., Zhang, H., Liu, Q.: Lattice-strained metal–organic-framework arrays for bifunctional oxygen electrocatalysis. Nat. Energy 4, 115–122 (2019)
Qiu, B., Wang, C., Zhang, N., Cai, L., Xiong, Y., Chai, Y.: CeO2-induced interfacial Co2+ octahedral sites and oxygen vacancies for water oxidation. ACS Catal. 9, 6484–6490 (2019)
Zhang, N., Chai, Y.: Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647–4671 (2021)
Zhang, P., Li, L., Nordlund, D., Chen, H., Fan, L., Zhang, B., Sheng, X., Daniel, Q., Sun, L.: Dendritic core-shell nickel-iron-copper metal/metal oxide electrode for efficient electrocatalytic water oxidation. Nat. Commun. 9, 381 (2018)
Liu, P.F., Yin, H., Fu, H.Q., Zu, M.Y., Yang, H.G., Zhao, H.: Activation strategies of water-splitting electrocatalysts. J. Mater. Chem. A 8, 10096–10129 (2020)
Lu, S., Zhou, Q., Ouyang, Y., Guo, Y., Li, Q., Wang, J.: Accelerated discovery of stable lead-free hybrid organic-inorganic perovskites via machine learning. Nat. Commun. 9, 3405 (2018)
Xue, D., Balachandran, P.V., Hogden, J., Theiler, J., Xue, D., Lookman, T.: Accelerated search for materials with targeted properties by adaptive design. Nat. Commun. 7, 11241 (2016)
Xue, D., Balachandran, P.V., Yuan, R., Hu, T., Qian, X., Dougherty, E.R., Lookman, T.: Accelerated search for BaTiO3-based piezoelectrics with vertical morphotropic phase boundary using Bayesian learning. Proc. Natl. Acad. Sci. 113, 13301–13306 (2016)
Schmidt, J., Marques, M.R., Botti, S., Marques, M.A.: Recent advances and applications of machine learning in solid-state materials science. NPJ Computl. Mater. 5, 1–36 (2019)
Lipinski, C., Hopkins, A.: Navigating chemical space for biology and medicine. Nature 432, 855–861 (2004)
Kirkpatrick, P., Ellis, C.: Chemical space. Nature 432, 823–834 (2004)
Dobson, C.M.: Chemical space and biology. Nature 432, 824–828 (2004)
Zvinavashe, E., Murk, A.J., Rietjens, I.M.: Promises and pitfalls of quantitative structure–activity relationship approaches for predicting metabolism and toxicity. Chem. Res. Toxicol. 21, 2229–2236 (2008)
Scior, T., Medina-Franco, J.L., Do, Q.T., Martínez-Mayorga, K., Yunes, R.J.A., Bernard, P.: How to recognize and workaround pitfalls in QSAR studies: a critical review. Curr. Med. Chem. 16, 4297–4313 (2009)
Schneider, G.: Virtual screening: an endless staircase? Nat. Rev. Drug Discov. 9, 273–276 (2010)
Green, M.L., Choi, C.L., Hattrick-Simpers, J.R., Joshi, A.M., Takeuchi, I., Barron, S.C., Campo, E., Chiang, T., Empedocles, S., Gregoire, J.M., Kusne, A.G.: Fulfilling the promise of the materials genome initiative with high-throughput experimental methodologies. Appl. Phys. Rev. 4, 011105 (2017)
Afzal, M.A., Hachmann, J.: High-throughput computational studies in catalysis and materials research, and their impact on rational design. In: Handbook on Big Data and Machine Learning in the Physical Sciences: Big Data Methods in Experimental Materials Discovery vol. 1, pp. 1–44 (2020)
Kang, J., Yu, J.S., Han, B.: First-principles design of graphene-based active catalysts for oxygen reduction and evolution reactions in the aprotic Li–O2 battery. J. Phys. Chem. Lett. 7, 2803–2808 (2016)
Nørskov, J.K., Bligaard, T., Rossmeisl, J., Christensen, C.H.: Towards the computational design of solid catalysts. Nat. Chem. 1, 37–46 (2009)
Valdes, A., Brillet, J., Grätzel, M., Gudmundsdottir, H., Hansen, H.A., Jonsson, H., Klüpfel, P., Kroes, G.J., Le, F., Man, I.C., Martins, R.S.: Solar hydrogen production with semiconductor metal oxides: new directions in experiment and theory. Phys. Chem. Chem. Phys. 14, 49–70 (2012)
Skúlason, E., Tripkovic, V., Björketun, M.E., Gudmundsdottir, S., Karlberg, G., Rossmeisl, J., Bligaard, T., Jónsson, H., Nørskov, J.K.: Modeling the electrochemical hydrogen oxidation and evolution reactions on the basis of density functional theory calculations. J. Phys. Chem. C 114, 18182–18197 (2010)
Nørskov, J.K., Bligaard, T., Logadottir, A., Kitchin, J.R., Chen, J.G., Pandelov, S., Stimming, U.: Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 152, J23 (2005)
Greeley, J., Nørskov, J.K., Kibler, L.A., El-Aziz, A.M., Kolb, D.M.: Hydrogen evolution over bimetallic systems: understanding the trends. ChemPhysChem 7, 1032–1035 (2006)
Greeley, J., Nørskov, J.K.: Combinatorial density functional theory-based screening of surface alloys for the oxygen reduction reaction. J. Phys. Chem. C 113, 4932–4939 (2009)
Hou, Y., Abrams, B.L., Vesborg, P.C., Björketun, M.E., Herbst, K., Bech, L., Setti, A.M., Damsgaard, C.D., Pedersen, T., Hansen, O., Rossmeisl, J.: Bioinspired molecular co-catalysts bonded to a silicon photocathode for solar hydrogen evolution. Nat. Mater. 10, 434–438 (2011)
Hou, Y., Vesborg, P.C., Bech, L., Seger, B., Dahl, S., Chorkendorff, I., Abrams, B.L., Herbst, K., Bjørketun, M., Rossmeisl, J., Nørskov, J.K.: Photoelectrocatalysis and electrocatalysis on silicon electrodes decorated with cubane-like clusters. J. Photon. Energy 2, 026001 (2012)
Greeley, J., Jaramillo, T.F., Bonde, J., Chorkendorff, I.B., Nørskov, J.K.: Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 5, 909–913 (2006)
Singh, A.K., Mathew, K., Zhuang, H.L., Hennig, R.G.: Computational screening of 2D materials for photocatalysis. J. Phys. Chem. Lett. 6, 1087–1098 (2015)
Ling, C., Shi, L., Ouyang, Y., Wang, J.: Searching for highly active catalysts for hydrogen evolution reaction based on O-terminated MXenes through a simple descriptor. Chem. Mater. 28, 9026–9032 (2016)
Luo, G.G., Li, X.C., Wang, J.H.: Visible light-driven hydrogen evolution from aqueous solution in a noble-metal-free system catalyzed by a cobalt phthalocyanine. Chem. Select. 1, 425–429 (2016)
Abghoui, Y., Skúlason, E.: Hydrogen evolution reaction catalyzed by transition-metal nitrides. J. Phys. Chem. C 121, 24036–24045 (2017)
Nørskov, J.K., Bligaard, T., Logadottir, A., Bahn, S., Hansen, L.B., Bollinger, M., Bengaard, H., Hammer, B., Sljivancanin, Z., Mavrikakis, M., Xu, Y.: Universality in heterogeneous catalysis. J. Catal. 209, 275–278 (2002)
Bligaard, T., Nørskov, J.K., Dahl, S., Matthiesen, J., Christensen, C.H., Sehested, J.: The Brønsted–Evans–Polanyi relation and the volcano curve in heterogeneous catalysis. J. Catal. 224, 206–217 (2004)
Ouyang, R., Curtarolo, S., Ahmetcik, E., Scheffler, M., Ghiringhelli, L.M.: SISSO: a compressed-sensing method for identifying the best low-dimensional descriptor in an immensity of offered candidates. Phys. Rev. Mater. 2, 083802 (2018)
Tarcolea, C., Paris, A., Demetrescu-Tarcolea, A.: Statistical methods applied for materials selection. APPS. Appl. Sc. 11, 145–150 (2009)
Wang, X., Gao, X.J., Qin, L., Wang, C., Song, L., Zhou, Y.N., Zhu, G., Cao, W., Lin, S., Zhou, L., Wang, K.: Eg occupancy as an effective descriptor for the catalytic activity of perovskite oxide-based peroxidase mimics. Nat. Commun. 10, 704 (2019)
Xu, H., Cheng, D., Cao, D., Zeng, X.C.: A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 1, 339–348 (2018)
Luc, W., Jiang, Z., Chen, J.G., Jiao, F.: Role of surface oxophilicity in copper-catalyzed water dissociation. ACS Catal. 8, 9327–9333 (2018)
Hammer, B.J., Nørskov, J.K.: Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 343, 211–220 (1995)
Huang, H.C., Li, J., Zhao, Y., Chen, J., Bu, Y.X., Cheng, S.B.: Adsorption energy as a promising single-parameter descriptor for single atom catalysis in the oxygen evolution reaction. J. Mater. Chem. A 9, 6442–6450 (2021)
Gao, G., Waclawik, E.R., Du, A.: Computational screening of two-dimensional coordination polymers as efficient catalysts for oxygen evolution and reduction reaction. J. Catal. 352, 579–585 (2017)
Li, L., Li, B., Guo, Q., Li, B.: Theoretical screening of single-atom-embedded MoSSe nanosheets for electrocatalytic N2 Fixation. J. Phys. Chem. C 123, 14501–14507 (2019)
Cai, L., Zhang, N., Qiu, B., Chai, Y.: Computational design of transition metal single-atom electrocatalysts on PtS2 for efficient nitrogen reduction. ACS Appl. Mater. Interfaces 12, 20448–20455 (2020)
Cao, Y., Wang, T., Li, X., Zhang, L., Luo, Y., Zhang, F., Asiri, A.M., Hu, J., Liu, Q., Sun, X.: A hierarchical CuO@ NiCo layered double hydroxide core–shell nanoarray as an efficient electrocatalyst for the oxygen evolution reaction. Inorg. Chem. Front. 8, 3049–3054 (2021)
Gong, M., Zhou, W., Tsai, M.C., Zhou, J., Guan, M., Lin, M.C., Zhang, B., Hu, Y., Wang, D.Y., Yang, J., Pennycook, S.J.: Nanoscale nickel oxide/nickel heterostructures for active hydrogen evolution electrocatalysis. Nat. Commun. 5, 4695 (2014)
Hong, W.T., Welsch, R.E., Shao-Horn, Y.: Descriptors of oxygen-evolution activity for oxides: a statistical evaluation. J. Phys. Chem. C 120, 78–86 (2016)
Calle-Vallejo, F., Inoglu, N.G., Su, H.Y., Martinez, J.I., Man, I.C., Koper, M.T., Kitchin, J.R., Rossmeisl, J.: Number of outer electrons as descriptor for adsorption processes on transition metals and their oxides. Chem. Sci. 4, 1245–1249 (2013)
Bockris, J.O., Otagawa, T.: The electrocatalysis of oxygen evolution on perovskites. J. Electrochem. Soc. 131, 290 (1984)
Acknowledgements
M. Qamar extends his appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work (No. DRI 598). He also acknowledges the funding support provided by the King Abdullah City for Atomic and Renewable Energy (K.A.CARE). Similarly, H. Adamu gratefully acknowledges Abubakar Tafawa Balewa University, Bauchi, Nigeria for grating him postdoctoral research fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared that no conflicts of interest exist in any form(s).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Adamu, H., Yamani, Z.H. & Qamar, M. Modulation to favorable surface adsorption energy for oxygen evolution reaction intermediates over carbon-tunable alloys towards sustainable hydrogen production. Mater Renew Sustain Energy 11, 169–213 (2022). https://doi.org/10.1007/s40243-022-00214-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40243-022-00214-3