
Abstract
This study validates three carbon allotropes, such as carbon nanotubes, graphene and carbon nanohorns that are covalently bound via a Kevlar-type linker (amide-bound aryl) with polypyrrole. In the proposed synthesis, the linker moiety is formed onto the carbon surface via combined electrochemical and chemical reactions and the polymer is attached during the final phase. Overall, the highest specific capacitance (~350 F/g) and thermal and electrochemical stability were observed for the MWCNT-based system, followed by Nanohorn-g-PPy (-g- stands for grafted) and the least stable with the smallest capacitance for the Graphene-based composite. The MWCNT-grafted material demonstrated a non-continuous and very thin polypyrrole coating onto the carbon surface. These structures are rigid, stable and reveal good accessibility of ions towards both the carbon and polymer. This system showed also the highest diffusion coefficient for the ion doping owing to the polymer effective diffusion length. The fast degradation of Graphene-g-PPy is associated with the carbon morphology and its high activity towards electrochemical oxygen reduction due to the presence of catalytic sp2 carbon atoms at the edges of the graphene. The presented work highlights an effect of carbon morphology in designing chemically grafted components for ultra-capacitor electrodes.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The important requirement that needs to be fulfilled for an effective capacitor is fast energy uptake and release [1, 2]. Other important features are a long-lasting cycle life, stability and high specific capacitance. They all can be improved by proper chemical, structural and morphological modification of the electrode materials [3]. Currently, in field of energy storage/conversion electronics, one of the most important task is to replace/decrease the usage of toxic heavy metals by carbonaceous materials that operate with relatively mild electrolytes [4]. Carbon allotropes such as carbon nanotubes and graphene are the most promising, and thus became essential components that could reach characteristics similar or greater than metallic electrodes [5]. This is however very challenging since carbons have lower energy density, and consequently, smaller specific capacitance [6]. On the other hand, carbon-based supercapacitors can store energy and be charged very fast during the reversible adsorption/desorption process of ions in double-layer capacitors, or by the counter-ion doping in pseudo-capacitors [7, 8]. Both pseudo-capacitors and electrical double-layer capacitors (EDLCs) have possibilities to improve their specific capacitance by expanding the specific surface and increasing the number of redox active centers (for pseudo-capacitors) [9, 10]. Many studies have shown that a combination of both types results in a synergistic improvement of the total capacitance, especially when pseudo-capacitors from the group of conjugated polymers (e.g., polypyrrole or polyaniline) are used. These carbon-polymer hybrid materials or composites outperform their individual components [11–13]. The synergistic improvement is generated by two effects. First, is the increase in conductivity for the pseudo-capacitors resulting in faster ion diffusion that restricts the capacitance [14]. This process is particularly effective when a strong covalent bond (e.g., amide-type linker) between the polymer and carbon is created. The covalently grafted polymer benefits from improved mechanical stability provided by the carbon support. This combination also demonstrates more dynamic (flexible) structure that can accommodate volumetric changes taking place upon the ion uptake/release in the polymer pseudo-capacitor. The high surface area carbon facilitates uniform distribution of the polymer particles, which is critical during the extensive charge–discharge (shrinking–stretching). Such volumetric changes for a thick and uneven polymer electrode accelerate its degradation due to the local inhomogeneity of the charge distribution (surface with less conductive fractions). All these phenomena shorten the electrode cycle life [15]. Another important factor is the electronic interaction at the carbon-linker-polymer junction. In the reverse donor–acceptor system, the covalently bound conductive polymer acts as an electron acceptor and the carbon allotrope as the electron donor, resulting in a more electrochemically stable system [16].
Regarding the carbon type, combinations of conjugated polymers with the carbon nanotubes and graphene have been broadly studied in respect to the synergistic improvement of capacitance [17–19]. Also, the type of bonding between molecular components (electronic interaction via orbital stacking or hydrogen-bridge bonding [20] or via additional covalent bonding [21]) has been investigated in details. In respect to the electrode stability, the recommendation is to apply the chemical grafting of the polymer and carbon, i.e., with a molecular linker such as 3-aminobenzoic acid [22, 23].
This work demonstrates studies on a new covalent anchor, aryl amide grafted onto the commercial EDLC carbon materials such as the multi-walled carbon nanotubes (MWCNT), graphene and a new form of carbon, which are carbon nanohorns (CNH). The grafted carbons are further functionalized with the polypyrrole pseudo-capacitor material (the choice of polymer is based on its electrochemical stability [24, 25]). The carefully tailored morphology with a new organic linker is expected to generate synergistic improvement of the capacitance. Also, the strong carbon-polymer bonding will improve the rigidness of the structure for the grafted materials, while sustaining a good electrochemical stability. The new carbon nanohorns are particularly interesting in this study as they show branched porous structure that should significantly influence the morphology of combined systems.
Experimental
Materials and methods
Unless otherwise stated, all chemicals have been used as received without further purification. 3-aminobenzoic acid (98%, Sigma-Aldrich Canada, 127671 ALDRICH) was recrystallized twice from 95% ethanol (Home source- UNB Fredericton science store). 2-propanol (for molecular biology, BioReagent, ≥99.5%, I9516 SIGMA), ammonium persulfate (ACS reagent, ≥98%, 248614 Sigma-Aldrich), carbon nanohorns (as-grown, 804118 ALDRICH, BET 400 m2/g), DCM (ACS reagent, ≥99.5%, contains 50 ppm amylene as stabilizer, 443484 Sigma-Aldrich), dimethylformamide (anhydrous, 99.8%, 227056 Sigma-Aldrich), Multi-walled Carbon Nanotubes (O. D. × L 6–9 nm × 5 µm, >95% (carbon), 724769 ALDRICH, BET ~220 m2/g), poly(vinylidene difluoride) average M w ~180.000 by GPC, 427152 ALDRICH), potassium bromide (For IR spectroscopy, 34919 FLUKA), potassium chloride (analytical grade, p.a., ≥99.5% (AT) 60130 Sigma-Aldrich), p-phenylenediamine (P6001 Sigma), sodium nitrite (ACS reagent, ≥97.8%, 237213 Sigma-Aldrich), pyrrole (reagent grade, 98%, 131709 ALDRICH), sodium hydroxide (reagent grade, ≥98%, pellets (anhydrous), S5881 Sigma-Aldrich), THF (anhydrous, anhydrous, ≥99.9%, inhibitor-free, 401757 Sigma-Aldrich) and thionyl chloride (reagent grade, 97%, 320536 Sigma-Aldrich) were purchased from Sigma-Aldrich Canada. Single-layer graphene flakes were obtained from Graphene Supermarket (grade AO-3, 12 nm flakes, specific surface area ~80 m2/g, purity 99.2%, Calverton, NY, USA). Pyrrole was distilled over calcium chloride under reduced pressure and stored under dry nitrogen. Hydrochloric acid (37%) was supplied by VWR International, Canada. Carbon black (Super P Conductive, 99%, metal basis) was purchased from Alfa Aesar, UK. TEM imaging was carried out using Jeol 2100 sTEM operating at 200 kV. XPS studies were performed at Dalhousie University on VG Microtech MultiLab ESCA 2000 System with 100 µm analyzer spatial resolution, 10 meV energy resolution and Mg Kα linewidth. FTIR spectra on a KBr pellet were recorded with the Nicolet™ iS™ 50 FTIR, 4 cm−1 resolution, 100 scan per sample (KBr was dried in oven at 80 °C prior the use). Raman spectra were recorded with a Renishaw inVia Raman spectrometer, 785 nm excitation source (Renishaw Inc, UK). The spectral range was from 1000 to 3000 cm−1, and 0.1 mW laser power using 50 times magnification on the microscope. Thermal analysis was carried out on the TGA Q500 model TA Instruments from 25 to 650 °C at the heating rate of 5 °C/min in N2 in 100 mL alumina crucible. All electrochemical experiments were carried out using a CH Instruments electrochemical workstation model C760 and with an electrochemical cell containing a platinum wire counter electrode (CE), Ag/AgCl reference electrode (RE), and the ink-deposited glassy carbon as a working electrode (WE), which was mounted to the Pine AFMSRCE rotating disk electrode station. All potentials are quoted vs. Ag/AgCl (222 mV vs. standard hydrogen electrode). All electrochemical measurements were carried out in 1 M KCl purged with N2 for 30 min prior to experiments (N2 gas blanket was kept during the measurement). The glassy carbon working electrode disk of 0.5 cm diameter (PINE Instrument Company, USA) was mechanically polished with 0.05 µm Al2O3 slurry (Cypress Systems Inc., USA), rinsed in double-distilled water, sonicated for 5 min, finally rinsed in ultrapure isopropanol and acetone and dried under a stream of air. The ink was prepared by dissolving PVDF (2 mg) in isopropanol (2 mL), sonicated for 0.5 h together with a carbon black (2 mg) and 17 mg of an active material. The resulting ink was dropped (17, 8.5 µL each cast) onto the polished glassy carbon working electrode (1 cm2 area) and dried for 2 min under a 60 W lamp. The electrochemical reduction of the 3-diazonium aryl salts was performed onto the similar glassy carbon disk (working electrode). The CV scan was conducted in the potential range from 0.65 to −0.65 V (vs. Ag/AgCl 0.222 V based on the reduction potentials for a 4-diazobenzcarboxylic acid [26]). The charge–discharge (CD) test was carried out at the current load of 0.25 mA/g with time intervals of 30 s, and the data storage interval of 0.1 s. Fifty scans were applied for each electrode and the CV was recorded afterwards and compared with the CV curve before CD test (stability test). A chronocoulometry was examined at 0.3 V at the onset potential of the polypyrrole oxidation wave.
Synthesis
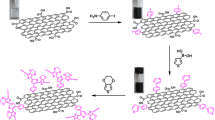
Figure 1a demonstrates a multi-step synthesis procedure of the carbon allotrope-grafted polypyrrole taking MWCNTs as an example. This synthesis route falls into four phases:

A multi-step synthesis pathway for the carbon-grafted polypyrrole composite (a). Step II is presented in the CV scan (b)
The electrochemical derivation of in situ generated 3-diazo aryl chlorides
In this step, 4 mmol of 3-substituted aniline was dissolved in 12.5 mL 1 M HCl and 7.5 mL distilled water and the solution was brought to 0 °C in the 3-neck electrochemical cell (Gamry, Fig S1). 6 mmol (1.5 eq.) of sodium nitrite in 5 mL cold distilled water was added dropwise through the dropping funnel to the solution and stirred in an ice bath for 5 min (Fig. 1a, step I). In next step the funnel was removed and the cell was equipped with the carbon-coated RDE (rotating disk electrode) glassy carbon or the glassy carbon (GC) plate, Ag/AgCl reference electrode and Pt wire (counter electrode). Afterwards the cyclic voltammetry scan was carried out in the potential range from +0.978 to −1.022 V at the scan rate of 100 mV/s. In order to observe irreversibility of reduction process (position and intensity of cathodic peaks) two voltammograms were recorded (Fig. 1a, step II). The reactant concentration was evaluated in the range of 5–45 mmol and the optimum of derivatization was assessed experimentally based on the CV peak area and correlated with the XPS surface analysis. The reverse CV scan of the cathodic wave was validated for each concentration as demonstrated in the supporting information (Fig. S2).
Synthesis of the carbon-benzoyl chloride
In this reaction, 100 mg of the functionalized carbon allotrope (MWCNTs, graphene or carbon nanohorns) and 25 ml of a thionyl chloride (in excess) were placed in the 50 ml round-bottom flask equipped with a Liebig reflux condenser, a tube filled with a calcium chloride connected to the gas absorption trap and heated on a steam bath for 6 h. Afterwards, the excess of thionyl chloride was removed during the distillation (Fig. 1a, step III). The reaction product was collected by ultrafiltration (Millipore Ultrafiltration system) and washed with a dry THF (three times 10 mL), dried in vacuum and used as the reactant in the Schotten–Baumann reaction.
Schotten–Baumann reaction with a p-amino aniline
100 mg of a p-aminoaniline was mixed with the carbon allotrope-benzoyl chloride in 15 mL CH2Cl2 and an equimolar amount of NaOH (dissolved in a very small volume of water). Thereafter the mixture was sonicated for 15 min and then refluxed for 24 h. The final product was collected by ultra-filtration, washed with dichloromethane and dried in oven at 40 °C. This reaction is shown as the step IV in Fig. 1a.
Polymerization of pyrrole onto the functionalized carbon
After the purification of functionalized carbon (MWCNTs, graphene, carbon nanohorns), the oxidative/free radical polymerization of the pyrrole was initiated using ammonium persulfate (APS) in 1 M HCl [16]. In preliminary phase of the synthesis, ammonium persulfate reacts with hydrochloric acid resulting in the ammonium chloride precipitate [27, 28]. Furthermore, the persulfate dianion is a source of reactive oxygen species, which undergo polymerization via an ionic bonding, as a result of protonation. Overall, ammonium persulfate generates two sulfate radicals that are combined with a hydrogen from the basic polymer chains, resulting in polymer radicals [27]. The polymerization was conducted at 5 °C in order to quench the formation of oligomeric poly(pyrrole) units that were not attached to the linker (organic functionality onto the carbon surface, generated in steps 1–3). The step V in Fig. 1a represents the polymerization of the pyrrole onto the functionalized carbon.
Results and discussion
Material characterization
The electrochemical grafting of carbon with the respective anchoring mote (in this case benzoic acid) starts with an in situ diazotiazation of a 3-aminobenzoic acid in aqueous/acidic media and the sub-sequential electrochemical reduction followed by a sp2-sp2 carbon-carbon bond formation between the carbon allotrope and the reduced 3-carboxybenzenediazonium chloride. This process can be controlled by a cyclic voltammetry that demonstrates a characteristic cathodic peak corresponding to the reduction (first irreversible peak at 0.28 V shown in Fig. 1b). The covalent grafting results in the formation of C sp2 –C sp2 bond (second irreversible peak at 0.068 V). An irreversibility of the process indicates on a successful derivatization [2]. The reduction peak was analyzed in order to estimate the amount of the deposited linker, by calculating the area under the peak (Fig. S2). The obtained coulombic charge was further used to calculate the mass of deposit according to Faraday’s first law of electrolysis (further details are provided in the supporting information). Furthermore, the concentration of an organic moiety synthesized in the step II was correlated with the XPS analysis, taking into consideration the C 1s and O 1s signals. The detailed kinetic studies of the step II derivatization including the optimization of the concentration precursor and the time of deposition process for all carbon allotropes is discussed in our recent work [29]. This study includes also quantitative analysis of the electro-generated product of the step II phase. The surface concentration of the organic linker varies with a type of carbon allotrope. For example, surface functionalization of MWCNTs is ~15% according to the electrochemical estimation using Faraday’s law, and 14.99% based on the XPS analysis, while graphene showed 30.01% of the surface concentration quantified by both electrolysis and XPS (concentration of the product generated in phase II onto the carbon nanohorns is higher than for MWCNTs, but less than observed for the graphene). This difference is apparently correlated with the accessibility of the carbon surface towards the reactant, and is affected by the carbon particle shape (preferential deposition onto the graphene flakes as compared to the less accessible nanotube, or with some preference onto the nanohorn porous structure). The carboxylic acid was transformed to acid chloride using thionyl chloride to ensure the formation of the amide bond in proposed Schotten–Baumann reaction. In next phase, the carbon allotrope/acid chloride reacts with a p-phenylenediamine in Schotten-Baumann reaction as demonstrated in Fig. 1a (steps III–IV). The product of this reaction is further used for an oxidative/radical polymerization of pyrrole (step V). The carbon-polymer grafting with the amide linker demonstrates geometry similar to the Kevlar structure that is known from its chemical stability [27].
X-ray photoelectron spectroscopy was employed in order to study the chemical composition of the final product. Figure 2 shows the XPS narrow scans for C1s, O1s, N1s and the survey analysis of MWCNTs-g-PPy as an example. C1s scan of the MWCNT-g-PPy (Fig. 2a) consists of two major components, one at 286.74 eV corresponding to the carbonyl bond [30] and at 284.58 eV that represents both the graphitic C–C bonding and C=N bonds of the polymer backbone (signals are very close thus difficult to separate). N1s peak (Fig. 2b) at 397.463 eV corresponds to the C–N and C=N pyrrole units [31]. The peak at 395.615 eV represents the amine bond between pyrrole and linker anchor [32] and the amide bond of the linker moiety is seen at 400.385 eV [33]. There are traces of nitrite [34] that might be leftover reactant of the step I reaction. The O1s peak at 532.428 eV in Fig. 2c represents the carbonyl bond in the polymeric structure [35] and at 530.704 eV corresponds to amide in the linker. The XPS spectra of the Graphene-g-PPy and CNH-g-PPy did not show significant difference in the peak position and the area of the fitted signals (concentration of respective chemical bonds), leading to the conclusion that the chemical composition of the final product and the corresponding carbon–polymer–organic linker interactions are comparable for all allotropes.

XPS C1s (a), N1s (b), O1s (c) signals and a survey analysis (d) for MWCNT-g-PPy
Figure 3 demonstrates FTIR (left) and Raman (right) analysis of the grafted carbon allotropes revealing some spectral differences. Regarding the chemical composition analyzed by FTIR, for the CNH-based composite (A), a weak C–H out of plane vibrational deformation corresponding to the cyclic aromatic rings is observed at 670.5 cm−1 [36]. Another vibrational out of plane deformation at 805.1 cm−1 arises from three neighboring H atoms in the aromatic ring attached to the carbon nanohorns [37]. N–H and C–H deformation vibrations associated with the poly(pyrrole) ring are observed at 1019.2 cm−1 [38]. Furthermore, a strong stretching vibration corresponding to the phenolic carbon in ketone (or amide) structure appears at 1098.7 cm−1 and refers to the attached linker [39]. The weak stretching vibration at 1159.8 cm−1 is associated with the C–N in aryl amide and poly(pyrrole) [40]. A strong vibrational stretching signal at 1270.1 cm−1 corresponds to the urethane-like amide bond of the linker [41]. Also, a weak N–H deformation vibration of the aryl amide is observed at 1398.5 cm−1 [41]. The two bands at 1453.2 and 1520.8 cm−1 represent the C–H vibrational deformation of the amide [41] together with a weak C–N deformation at 1569.7 cm−1 (also amide signal) [42]. A strong carbonyl stretching signal—typical for the chemically modified carbonaceous materials appears at 1649.25 cm−1 [43]. The C–H stretching vibration of the carbon nanohorn starts at 2841.3 and ends at 2669.1 cm−1 [44]. Similar functionalities are identified for Graphene-g-PPy (B) and MWCNT-g-PPy (C). A residual CO2 from the background subtraction is still present at 2358.1 cm−1. A significant difference in the FTIR spectrum is observed for the Graphene-g-PPy (B). This includes the presence of a medium C–H wagging vibration at 591.8 cm−1 [45] and a medium vibration deformation from the amide structure that appear at 616.2 cm−1 [46]. At 658.3 cm−1 another medium vibration deformation is related to the –N–C=O bending [47]. In case of CNH-g-PPy, a similar vibration from –C–N–H is observed at 798.9 cm−1 [48]. Also, an aliphatic stretching vibration appears at 927.4 cm−1 and the medium C–N stretching vibration at 1043.6 cm−1. The neighboring H atoms in the poly(pyrrole) ring can be seen at 1104.8 cm−1 [48]. The coupled C–N and C–O vibrations are clearly visible at 1263.8 cm−1 [47]. The amide structure vibration at 1366.7 and 1386.2 cm−1 [47], and the C–N stretching vibration from the poly(pyrrole) appear at 1459.6 and 1469.3 cm−1 [48]. The conjugated C=C vibration in the extended π-system of graphene can be seen at 1618.7 cm−1 [49], and at 1716.54 cm−1 a strong C=O stretch is identified [50]. A broad protonated nitrogen amide bond appears at 2364.2 cm−1 (last step of the synthesis was conducted in 1 M hydrochloric acid) [51]. C–H deformations from a large conjugated system of the graphene can be seen in the range from 2853.5 to 2962.9 cm−1 [49]. As the morphology (nano-flakes versus branched porous particles) and the structure (3- versus 2-dimensional carbon) differ significantly for the CNH and Graphene, the corresponding spectral differences can be expected. The general observation is that for Graphene some signals are stronger as compared to the CNH (A) and MWCNT (B), which corresponds to the higher concentration of the organic linker (and thus higher percentage of functionalization) onto the Graphene, due to its large accessible surface (as confirmed by the electrochemical and XPS quantifications, Table S1).

FTIR (left) and Raman spectra (right) of the CNH-g-PPy (a, d), Graphene-g-PPy (b, e) and MWCNT-g-PPy (c, f)
The FTIR spectrum of MWCNT-g-PPy (Fig. 3c) consists of all functionalities identified for the graphene-based composite. There are minor shifts such as the signal at 610.1 cm−1 assigned to the aliphatic amide structure of the linker, at 939.6 cm−1 aliphatic stretching vibration, and at 1086.5 cm−1 there is a medium C–N stretching vibration. Also, C–N and C–O vibrations are identified at 1282.2 cm−1, and in the range from 1404.6 to 1453.5 cm−1 the C–N stretching vibrations correspond to the pyrrole. The shifted (as compared to graphene-based sample) are (1) the peak at 1581.9 cm−1 associated with the conjugated C=C vibration from the extended π-system in MWCNT, and (2) the strong C=O signal at 1637.1 cm−1. Due to the structure of MWCNT there are no clear C–H deformations, which were visible for the graphene and CNH composites.
The Raman spectroscopy was further applied to analyze the possible changes in the carbon topology and the presence of Stone–Wales defects upon its chemical modification [52]. A good indication for such changes is the correlation of the intensities between D and G bands (Fig. 3d, f). For the pure carbons, the D and G band signals occur at ~1300 and ~1600 cm−1, respectively. Almost 1:1 intensity ratio is observed for the Graphene-g-PPy (D:G = 1.15, Fig. 3e) and it significantly varies for the MWCNT-g-PPy (D:G = 1.88, Fig. 3f) and for the CNH-g-PPy (D:G = 2.12, Fig. 3d), indicating on the presence of crystalline defects. The G band signal is associated with the crystalline structure and a high frequency of the optical phonon (E2g), while the D band represents the A1g breathing mode in a Brillouin zone [53]. The general observation is that Graphene-based composite showed very weak changes of D:G intensity ratio indicating on the highly crystalline interface. This shows that the carbon surface is significantly modified by the overlaying amorphous phase.
TEM observations are further carried out in order to investigate the morphology of the grafted materials (Fig. 4). The general observation is that for chosen carbons the polymer coating looks completely different. This is also correlated with the distribution the organic linker on the carbon surface that affects the monomer nucleation and a particle growth in the final step of the synthesis. For the graphene-based system (Fig. 4a), spherical particles of the polyryrrole are evenly distributed onto the carbon surface and the size of polymer is in the range 20–80 nm (this range of the polymer particle size favors an ion doping and thus the charge storage process). The distribution of polypyrrole around multi-walled carbon nanotube is completely different. The polymer coils the carbon tube and this spiral-like polymer structure is expected to benefit in the capacitance improvement since (1) the polypyrrole thickness is less than 80 nm and thus in the range of the effective diffusion length, and (2) the carbon surface is not fully covered by the macromolecular chain, thus freely accessible for the ion (the important double-layer capacitance of the carbon is not obstructed). Such morphology favours the synergy of pseudo- and double-layer capacitance. Another important observation for the MWCNT-grafted polypyrrole is the separation of tubes. This considerably improves an access of the electrolyte to the capacitive interface. Opposite, a significant agglomeration is observed for the graphene-based material (Fig. 4a), and is confirmed by an in situ selected area electron diffraction (SAED, Fig. 4d). The characteristic hexagonal diffraction patterns of the aromatic carbon rings are present in the single graphene layers and are well resolved before functionalization. The additional diffraction spots are detected for the final product as a result of agglomeration (also observed in Fig. 4a). Most probably the organic functionalities generate the cross-linking between the adjacent graphene layers. These stacked, chemically bond sheets are not accessible for the electrolyte and for the pyrrole, and have significant impact on the electrochemical features of the final product. The results of SAED test are shown in the Table 1 and demonstrate variations of the Miller’s hkl indices for unmodified and functionalized graphene. The changes in hkl are generated by both the π–π* interactions in graphene sheets due to the layer stacking, and by the chemical modification of the carbon surface and the formation of new bonds between carbon and the organic moiety. The carbon nanohorn-grafted-PPy showed on the other hand, very uniform distribution of the polymer onto the carbon. The carbon nanohorns are porous (Fig. 4c insert) and relatively easy to access for both the organic linker and the monomer. However, since the individual “horns” are very small (thickness of 2 nm and the length about 10 nm), they are too fragile to host larger polymer beads. This results in the total coverage of the carbon structures (nanohorns are not separated, they exist in colonies in bare carbon, as shown in the Fig. 4c insert). For such morphology of the grafted material, the carbon double-layer capacitance is obstructed, which will considerably affect its electrochemical response.

Transmission electron microscope (TEM) image of the Graphene-g-PPy (a), MWCNT-g-PPy (b), carbon nanohorn-g-PPy (c), and the selected area electron diffraction (SAED) patterns of a pure graphene (right, top) and the final product
Only pure graphene and the modified graphene were compared by SAED, as CNH lacks a well-defined crystallographic structure and did not show any significant crystallographic difference upon grafting (only diffused rings indicating amorphous structure). Nevertheless, modification of the graphene by attaching the poly(pyrrole) demonstrates stacking of the graphene layers (more rings with multiple spots in Fig. 4, insert d).
Figure 5 represents the TGA curves for all composites in the range from 25 to 650 °C. According to literature, grafted carbons decompose in this temperature regime (excluding water loss) starting from the detachment of the carbon-amide linker, followed by the degradation of polymer-linker, and the degradation of polypyrrole at higher temperatures [56, 57]. The corresponding transitions are projected in Fig. 5 (gray vertical lines). Overall, the MWCNT-g-PPy sample degrades at 290.52 °C (A), Graphene-g-PPy decomposes at 188.44 °C (B) and CNH-g-PPy at 200 °C (C). As the concentration of organic functionalities is the highest for CNH and Graphene, these degradation steps are more clearly observed. The MWCNT-based sample demonstrated the highest thermal stability. This effect is generated by the rigidness of the structure and the fact that roughly a half of the nanotubes are evenly covered by the polymer (Fig. 4b). This morphology stabilizes the structure of an individual coated-tube, resulting in the protection of soft organic linker from the thermal attack. The graphene-based composite demonstrates a completely different morphology, with the polymer nano-spheres scattered evenly and the large uncovered carbon surface. This exposure generates faster degradation for both carbon and the organic fractions (polypyrrole particle size is also smaller as compared to PPy coating onto the MWCNTs). As the nanohorn-grafted composites showed the smallest particle size and also due to the fact that whole carbon is completely covered by the organic components (Fig. 4c), its degradation is the fastest. These structures are at least thermally stable, probably due to the very small size of the carbon particles and a significantly lower degree of the carbon graphitization (amorphous carbons are less stable under the thermal, chemical or electrochemical oxidative treatment [58]).

TGA/1st derivative of MWCNT-g-PPy (a), Graphene-g-PPy (b) and CNH-g-PPy (c) at the heating rate of 10 °C/min
Electrochemical analysis
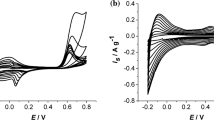
Figure 6 represents the cyclic voltammogram of the grafted material with the MWCNTs as the carbon core. This spectrum shows a typical feature of the double-layer capacitor of MWCNTs with a square-like CV and the oxidation wave with the maximum at −0.48 V associated with the redox activity of polypyrrole. In this process, the positive charge generated during the oxidation of the polymer chain is compensated by chloride ions from the electrolyte. Consequentially, reverse cathodic scan introduces electrons into the poly(pyrrole) and conveys it to the neutral state. The oxidation peak at −0.15 V is related to the redox activity of the amide linker [59]. Furthermore, the influence of carbon type or/and the morphology of the grafted material is studied using a differential voltammetry that allows to better resolve the corresponding peaks. DPV curves of the cathodic scan (the activity of polymer, carbon and the amide linker is reversible) in Fig. 7 show three major redox processes. At −0.48 V, reduction of polymer occurs on MWCNT-g-PPy (Fig. 7c), and is shifted towards positive potential (−0.23 V) for the Graphene-g-PPy (Fig. 7c) and for the CNH-g-PPy (−0.22 V, Fig. 7a). Possibly, the positive shift of the onset potential of this redox is a catalytic effect of the sp2 edges in the graphene and CNHs. Another important observation is the catalytic activity towards oxygen reduction at 0.21 V, taking place onto the oxygen-containing functionalities and the sp2 carbon active centers [59, 60, 61] for the graphene (Fig. 7b), and occurs as a very weak signal at 0.27 V for the CNH-based capacitor (Fig. 7a). This peak is stronger and shifted towards higher potential for MWCNTs-g-PPy (Fig. 7c). Again, the shift in onset potential can be associated with the different surface concentration and the type of these catalytic functionalities onto various carbons. The intensity of corresponding signals is also affected by the morphology of a final product. For instance, in the MWCNTs-g-PPy and Graphene-g-PPy, the carbon itself is more exposed to the electrolyte and thus the response towards ORR is stronger (less polymer coating or uneven polymer coating allow the carbon surface to be in contact with the electrolyte). An opposite, for the CNH-based sample this redox peak is very weak as almost the whole carbon is covered by the polymer. The cathodic peaks at −0.84 V for the Graphene-g-PPy and at −1.01 for the MWCNT-g-PPy correspond to the reduction of amino bond. This peak is nevertheless not visible for the CNH-g-PPy as the carbon surface is fully covered by the PPy and thus the ion mobility is hindered [62].

Cyclic voltammogram of the MWCNT-g-polypyrrole in KCl; insert represent the redox activity of polypyrrole

Cathodic DPV scans for CNH-g-PPy (a), Graphene-g-PPy (b) and MWCNT-g-PPy (c)
Specific capacitance of the grafted capacitors was estimated by integrating the area under CV curve (Fig. 9, as prepared electrodes) and standardizing it to the mass of the active components (m in g) at the applied scan rate (\(\nu\) in V/s) and in the potential window (E 2–E 1 in V) according to Eq. 1 [63]:
and after subtraction of the background double-layer current (glassy carbon, carbon black and PVdF binder). The mass of deposited polymer (as the polymer and linker content varies significantly for these carbons) was further calculated from the integration of voltammograms allowing to obtain the charge cumulated into the polymer fraction, and by applying Faraday–Matteucci’s first law of electrolysis (details and corresponding equations are provided in supporting information, similar approach was used for the quantification of amide aryl linker). The calculated quantities are presented in Table 2. For the specific capacitance the highest value of 350.49 F/g was obtained for MWCNT-g-PPy that is significant improvement as compared to the pure MWCNT (90 F/g) [3] and indicates on the synergistic effect of combined pseudo- and double-layer capacitances. This is also observed for the Graphene-g-PPy (33.20 versus 11.60 F/g for pure Graphene in this work) and for the CNH-g-PPy (43.61 versus 15 F/g for pure CNHs in this work). In case of graphene and CNH-based samples the calculated capacitance is close to that of pure PPy or carbons [20]. In case of graphene and CNH-based samples the calculated capacitance is close to that of pure PPy or carbons [20]. The unmodified CNH carbon is very porous, and its large BET surface is manly generated by needle-like structures (“horns”) of ~15 nm length and 2–5 nm thickness, as shown on the insert to Fig. 4c. The grafting of these structures with the organic linker and further polypyrrole leads to the blocking of an active surface and the total coverage of the carbon with the pseudo-capacitive polymer (this is why the capacitance of grafted CNH is close to that of pure polypyrrole). Once the carbon is fully covered with organic components is no longer accessible for the KCl electrolyte and thus the double-layer capacitance is hindered. This is rather expected for the CNH-g-PPy, as the whole carbon is covered by the polymer, indicating that the carbon morphology and the carbon particle size (too small to host the macromolecule and organic linkers) is discriminating factor in fabrication of the well-performing capacitive composite. For the future work, this material will be investigated at much lower polymer and the organic linker concentrations. This needs to be adjusted in order to sustain the benefit from the high surface area and graphitized carbon nano-fingers (horns).
As the BET surface area for all carbons is high (~220 m2/g MWCNT, 400 m2/gCNH and the ~80 m2/gGraphene; as provided by the carbon supplier), the conclusion is that the carbon morphology plays a critical rule in electrochemical performance of the combined electrode materials. For Graphene-g-PPy, the prognosis is similar to CNHs-based sample as graphene flakes are too thin and fragile (3–12 nm) to support the macromolecules attached via organic linker, their concentration have to be adjusted. The specific capacitance of MWCNT-g-PPy is very high as compared to the state-of-the-art capacitor materials. This significant capacitance enhancement is generated by the synergy of the double-layer (MWCNT) and pseudo-(PPy) capacitances and the presence of very effective charging interface at the carbon-polymer junctions [64]. In our study, this is further improved by expanding this interface (thin non-continuous polymer layer). The organic linker has two functions in this system, one is the stabilization and the chemical bonding between the capacitive components (carbon and the polymer) and the second is to improve the charge separation/distribution at the carbon and polymer interface, similar to the function of dielectric in the conventional electrolytic capacitor [65]. This can be particularly important for the development of the molecular-scale capacitor devices that are applied in the field of memory storage or the nano-electronic circuits and the low-charge store-pump applications.
Furthermore, the diffusion coefficient of chloride storage in composites was analyzed according to the Cottrell equation (Eq. 2) applied to the linear function of the charge (Q) and the square root of time (t 1/2). This correlation is presented in Fig. 8 (Anson plots) allowing to calculate D (where n is a number of electrons exchanged in the polymer unit, 1.58; F corresponds to the Faraday constant 96,485 C/mol K, and A is the geometric area of the electrode, 0.1963 cm2):

Anson plots for MWCNT-g-PPy (a), Graphene-g-PPy (b) and CNH-g-PPy (c)
The MWCNTs-g-PPy demonstrated significantly higher diffusion coefficient as compared to the competing samples, indicating on an effective mass transport in MWCNT-based system. This is directly related to its superior capacitance and the improved rate of the ion exchange due to several reasons. First is the higher specific surface of MWCNTs and thus the higher polymer content—this automatically gives larger electrochemical interface. Second is the thickness of the polymer film (it has been already proven that PPy layer thickness or its particle size that is less than 40 nm is optimal and generates so called an effective diffusion length for the doping ions). Another important factor is the PPy distribution over the carbon surface—the non-continuous coating also improves the ion exchange at the carbon and facilitates a fast anion doping due to the redox activity of the PPy. This further reflects on the charge–discharge characteristics. As shown in Fig. S3, the chronopotentiograms recorded at 0.25 mA/g for all composites demonstrate that MWCNT-g-PPy can reach the electrode potential plateau faster and the potential is significantly higher as compared to the competing composites (1.05 V for MWCNT-g-PPy, 0.6 V for Graphene-g-PPy and 0.58 V for CNH-g-PPy). This indicated on the fast mass transfer of the doping ions into the polymer facilitated by the morphology and the polymer distribution onto the MWCNT’s surface.
Figure 9 demonstrates the CVs scans for the fresh electrode (black curves) and after charge–discharge test (red curves). Generally, MWCNT-g-PPy and CNH-g-PPy both showed good stability with less than 3% of the capacitance lost. In contrary, Graphene-based electrode lost more than 50% of the initial capacitance. As the potential window applied in this work is in the range of electrochemical stability of the carbon, polypyrrole and the amide linker. The degradation of the Graphene-g-PPy could be thus associated with its morphology and the chemical composition of the graphene surface. Firstly, the thickness of the single graphene flake is very small as compared with the size of the polymer particles—this may cause detachment and phase segregation. This can be also accelerated during the activity towards the electrochemical oxygen reduction reaction onto the sp2 catalytic centers around graphene edges (the sp2 C is identified at 284.8–283.0 eV in XPS spectra of Graphene-g-PPy, weak and broad signals are due to the presence of PPy and amid aryl, Fig. S4). Since the ration for edges and corners to the flat surface in graphene flake is high, possibly the simultaneous sideways processes accelerate the material degradation.

CV scans of MWCNT-g-PPy (a), CNH-g-PPy (b) and Graphene-g-PPy (c) as prepared (black curve) and after charge–discharge at 0.25 mA/g (red curve)
Conclusions
Carbon allotropes combined with various electronically conductive polymers or metal oxides are important components of lightweight capacitor devices. These type of hybrid or composites demonstrate an improved total capacitance as compared to their individual constituents due to the synergy between the double-layer capacitance (carbon) and pseudo-capacitance from redox active fractions (polymer or oxide). The critical aspect in achieving the capacitance improvement is to design a combined system on the molecular level, allowing to control precisely the morphology, and thus tailoring their electrochemical properties. In this work we validate three allotropes, carbon nanotubes, graphene and carbon nanohorns that are covalently bound via a Kevlar-type linker (amide-bound aryl) with the polypyrrole. In the proposed synthesis procedure, the linker moiety is created onto the carbon surface via combined electrochemical and chemical steps and the polymer is attached at the final phase. The following differences were observed for composites synthesized with various carbon allotropes:
-
1.
the highest specific capacitance and thermal and electrochemical stability among carbon-grafted materials were observed for the MWCNT-based samples, followed by Nanohorn-g-PPy and the least stable with the smallest capacitance was observed for the Graphene-based composite;
-
2.
the fast degradation of Graphene-g-PPy is associated with the carbon morphology and its high activity towards electrochemical oxygen reduction due to the presence of catalytic sp2 at the edges of the carbon; also the small thickness of the graphene flakes as compared to the size of polymer particles and the simultaneous side process are responsible for the detachment/deactivation of the capacitive electrode;
-
3.
the highest diffusion coefficient of ion doping into the polymer and thus the best capacitance was observed for the MWCNT-g-PPy; this is related to the composite morphology formed by a non-continuous and thin (less than 40 nm) thickness PPy film coated onto the MWCNTs, such structure facilitates good access of electrolyte to the carbon and polymer (double layer- and pseudo capacitance synergy)
-
4.
the particle size of carbon nanohorns is too small to host the polymer; the synthesis procedure proposed in this study should be modified to generate PPy of size less than 1 nm in order to manufacture useful electrode material.
References
González, A., Goikolea, E., Barrena, J.A., Mysyk, R.: Review on supercapacitors: technologies and materials. Renew. Sustain. Energy Rev. 58, 1189–1206 (2016)
Ke, Q., Wang, J.: Graphene-based materials for supercapacitor electrodes—a review. J. Materiom. 2, 1189–1206 (2016)
Pan, H., Li, J., Ping, Feng Y.: Carbon nanotubes for supercapacitor. Nanoscale Res. Lett. 5, 654–668 (2010)
Maheswari, N., Muralidharan, G.: Supercapacitor behavior of cerium oxide nanoparticles in neutral aqueous electrolytes. Energy Fuels 29, 8246–8253 (2015)
Balogun, M., Luo, Y., Qiu, W., Liu, P., Tong, Y.: A review of carbon materials and their composites with alloy metals for sodium ion battery anodes. Carbon 98, 162–178 (2016)
Mirri, F., Orloff, N.D., Forster, A.M., Ashkar, R., Headrick, R.J., Bengio, E.A., Long, C.J., Choi, A., Luo, Y., Hight Walker, A.R., Butler, P., Migler, K.B., Pasquali, M.: Lightweight, flexible, high-performance carbon nanotube cables made by scalable flow coating. ACS Appl. Mater. Interfaces 8, 4903–4910 (2016)
Ullah, H., Shah, A.A., Bilal, S., Ayub, K.: Doping and dedoping processes of polypyrrole: DFT study with hybrid functionals. J. Phys. Chem. C 118, 17819–17830 (2014)
Augustyn, V., Simon, P., Dunn, B.: Pseudocapacitive oxide materials for high-rate electrochemical energy storage. Energy Environ. Sci. 7, 1597–1614 (2014)
Liu, T., Finn, L., Yu, M., Wang, H., Zhai, T., Lu, X., Tong, Y., Li, Y.: Polyaniline and polypyrrole pseudocapacitor electrodes with excellent cycling stability. Nano Lett. 14, 2522–2527 (2014)
Chen, S., Pei, W., Gui, Q., Tang, R., Chen, Y., Zhao, S., Wang, H., Chen, H.: PEDOT/MWCNT composite film coated microelectrode arrays for neural interface improvement. Sens. Actuators A 193, 141–148 (2013)
Guldi, D.M., Martin, N.: Carbon Nanotubes and Related Structures: Synthesis, Characterization, Functionalization, and Applications. Wiley-VCH, Weinheim (2010)
Dyke, C.A., Stewart, M.P., Tour, J.M.: Separation of single-walled carbon nanotubes on silica gel. materials morphology and raman excitation wavelength affect data interpretation. J. Am. Chem. Soc. 127, 4497–4509 (2005)
Tiwari, A., Uzun, L.: Advanced Functional Materials. Wiley-Scrivener, Salem (2015)
Velraj, S., Zhu, J.H.: Cycle life limit of carbon-based electrodes for rechargeable metal–air battery application. J. Electroanal. Chem. 736, 76–82 (2015)
Simon, P., Gogotsi, Y.: Materials for electrochemical capacitors. Nat. Mater. 7, 845–854 (2008)
Radtke, M., McMillan, D.G.G., Schröter, B., Höppener, S., Dietzek, B., Schubert, U.S.S., Ignaszak, A.: The effect of 3-amino benzoic acid linker and the reversal of donor–acceptor pairs on the electrochemical performance and stability of covalently bonded poly(pyrrole) nanotubes. Polymer 77, 289–296 (2015)
Bai, Y., Xu, Y., Wang, J., Gao, M., Wang, J.: Interface effect on the electropolymerized polypyrrole films with hollow micro/nanohorn arrays. ACS Appl. Mater. Interfaces. 6(7), 4693–4704 (2014)
Zhou, M., Heinze, J.: Electropolymerization of pyrrole and electrochemical study of polypyrrole: 1. evidence for structural diversity of polypyrrole. Electrochim. Acta 44, 1733–1748 (1999)
Peng, P., Zhang, S., Jewell, D., Chen, G.Z.: Carbon nanotube and conducting polymer composites for supercapacitors. Prog. Nat. Sci. 18, 777–788 (2008)
Mosch, H., Höppener, S., Paulus, R., Schröter, B., Schubert, U.S., Ignaszak, A.: The correlation of binding mechanism of polypyrrole-carbon capacitive interphase with electrochemical stability of composite electrode. Phys. Chem. Chem. Phys. 17, 13323–13332 (2015)
Bottari, G., de la Torre, G., Guldi, D.M., Torres, T.: Covalent and noncovalent phthalocyanine-carbon nanostructure systems: synthesis, photoinduced electron transfer, and application to molecular photovoltaics. Chem. Rev. 110, 6768–6816 (2010)
Tan, Y., Zhang, H., Liu, H.H., Hou, L.C., Jin, Y.M., Zhang, X.X.: 4-Aminobenzoic acid functionalized PAN-base carbon fibers in mild polyphosphoric acid/phosphorous pentoxide. Adv. Mat. Res. 332–334, 219–222 (2011)
Di Crescenzo, A., Ettorre, V., Fontana, A.: Non-covalent and reversible functionalization of carbon nanotubes. Beilstein J. Nanotechnol. 5, 1675–1690 (2014)
Ruhi, G., Modi, O.P., Dhawan, S.K.: Chitosan-polypyrrole-SiO2 composite coatings with advanced anticorrosive properties. Synt. Me. 200, 7–15 (2015)
Merisalu, M., Kahro, T., Kozlova, J., Niilisk, A., Nikolajev, A., Marandi, M., Floren, A., Alles, H., Sammelselg, V.: Graphene–polypyrrole thin hybrid corrosion resistant coatings for copper. Synt. Met. 200, 6–23 (2015)
Elofson, R.M., Gadallah, F.F.: Substituent effects in the polarography of aromatic diazonium salts. J. Org. Chem. 34, 854–857 (1969)
Wu, A.S., Chou, T.-W.: Carbon nanotube fibers for advanced composites. Mater. Today 15, 302–310 (2012)
Fritz, U.: Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH, Weinheim (2002)
Radtke, M., Ignaszak, A.: Surface grafting of carbon allotropes with in situ generated 3-aryl diazonium chlorides: electrochemical kinetic studies. Electroanalysis 28, 1–11 (2016)
Campbell, D.S., Leary, H.J., Slattery, J.S., Sargent, R.J.: ESCA Surface Analysis of Plasma Exposed Silicon Nitride and Photoresist Polymer, IBM General Technology Division (ed.), Essex Junction, VT 05452, pp. 328–329 (2014)
Pan, F.M., Stair, P.C.: Chemisorption of pyridine and pyrrole on iron oxide surfaces studied by XPS. Surf. Sci. 177, 1–13 (1986)
Jouan, P.Y., Peignon, M.C., Cardinaud, C., Lempérire, G.: Characterisation of TiN coatings and of the TiN/Si interface by X-ray photoelectron spectroscopy and Auger electron spectroscopy. Appl. Surf. Sci. 68, 595–603 (1993)
Everhart, D.S., Reilley, C.N.: Chemical derivatization in electron spectroscopy for chemical analysis of surface functional groups introduced on low-density polyethylene film. Anal. Chem. 53, 665–676 (1981)
Seah, M.P., Briggs, D.: Practical Surface Analysis. Wiley, Chichester (1990)
Goldberg, M.J., Clabes, J.G., Kovac, C.A.: Metal–polymer chemistry. II. Chromium–polyimide interface reactions and related organometallic chemistry. J. Vac. Sci. Technol. A 6, 991–998 (1988)
Zhou, T., Zhoua, T., Zhanga, A.: Separation of the molecular motion from different components or phases using projection moving-window 2D correlation FTIR spectroscopy for multiphase and multicomponent polymers. RSC Adv. 5, 14832–14842 (2015)
Pridham, J.B.: Methods in polyphenol chemistry: proceedings of the Plant Phenolics Group symposium, Oxford, April, 1963. Macmillan, New York (1964)
Xu, C., Sun, J., Gao, L.: Synthesis of novel hierarchical graphene/polypyrrole nanosheet composites and their superior electrochemical performance. J. Mater. Chem. 21, 11253–11258 (2011)
Hara, Y., Yoshida, R.: Self-oscillating polymer fueled by organic acid. J. Phys. Chem. B 112, 8427–8429 (2008)
Huang, Y., Wang, Y., Bi, Y., Jin, J., Ehsan, M.F., Fu, M., He, T.: Preparation of 2D hydroxyl-rich carbon nitride nanosheets for photocatalytic reduction of CO2. RSC Adv. 5, 33254–33261 (2015)
Maisonneuve, L., Lebarbé, T., Nguyen, T.H.G., Cloutet, E., Gadenne, B., Alfos, C., Cramail, H.: Hydroxyl telechelic building blocks from fatty acid methyl esters for the synthesis of poly(ester/amide urethane)s with versatile properties. Polym. Chem. 3, 2583–2595 (2012)
Asefnejad, A., Behnamghader, A., Khorasani, M.T., Farsadzadeh, B.: Polyurethane/fluor-hydroxyapatite nanocomposite scaffolds for bone tissue engineering. Part I: morphological, physical, and mechanical characterization. Int. J. Nanomed. 6, 93–100 (2011)
Zeng, S., Cao, Y., Sang, W., Li, T., Gan, N., Zheng, L.: Enrichment of polychlorinated biphenyls from aqueous solutions using Fe3O4 grafted multiwalled carbon nanotubes with poly dimethyl diallyl ammonium chloride. Int. J. Mol. Sci. 13, 6382–6398 (2012)
Jadhav, A.D., Ogale, S.B., Prasad, B.L.V.: Carbon nanohorn and bovine serum albumin hierarchical composite: towards bio-friendly superhydrophobic protein film surfaces. Mater. Chem. 18, 3422–3425 (2008)
Spinato, C., Giust, D., Vacchi, I.A., Ménard-Moyon, C., Kostarelos, K., Bianco, A.: Different chemical strategies to aminate oxidised multi-walled carbon nanotubes for siRNA complexation and delivery. J. Mater. Chem. B 4, 431–441 (2016)
Murugan, E., Arumugam, S.: New dendrimer functionalized multi-walled carbon nanotube hybrids for bone tissue engineering. RSC Adv. 4, 35428–35441 (2014)
Meera, K.M.S., Sankar, R.M., Paul, J., Jaisankar, S.N., Mandal, A.B.: The influence of applied silica nanoparticles on a bio-renewable castor oil based polyurethane nanocomposite and its physicochemical properties. Phys. Chem. Chem. Phys. 16, 9276–9288 (2014)
Selvaraj, V., Alagar, M., Kumar, K.S.: Synthesis and characterization of metal nanoparticles-decorated PPY–CNT composite and their electrocatalytic oxidation of formic acid and formaldehyde for fuel cell applications. Appl. Catal. B 75, 129–138 (2007)
Tang, Z., Zhang, L., Zeng, C., Lin, T., Guo, B.: General route to graphene with liquid-like behavior by non-covalent modification. Soft Matter 8, 9214–9220 (2012)
Imani, R., Emami, S.H., Faghihi, S.: Synthesis and characterization of an octaarginine functionalized graphene oxide nano-carrier for gene delivery applications. Phys. Chem. Chem. Phys. 17, 6328–6339 (2015)
Llusar, M., Monrós, G., Roux, C., Pozzo, J.L., Sanchez, C.: One-pot synthesis of phenyl- and amine-functionalized silica fibers through the use of anthracenic and phenazinic organogelators. J. Mater. Chem. 13, 2505–2514 (2003)
Jiang, J., Pachter, R., Mehmood, F., Islam, A.E., Maruyama, B., Boeckl, J.J.: A Raman spectroscopy signature for characterizing defective single-layer graphene: defect-induced I(D)/I(D′) intensity ratio by theoretical analysis. Carbon 90, 53–62 (2015)
Cançado, L.G., Jorio, A., Martins Ferreira, E.H., Stavale, F., Achete, C.A., Capaz, R.B., Moutinho, M.V.O., Lombardo, A., Kulmala, T.S., Ferrari, A.C.: Quantifying defects in graphene via raman spectroscopy at different excitation energies. Nano Lett. 11, 3190–3196 (2011)
Meyer, J.C., Geim, A.K., Katsnelson, M.I., Novoselov, K.S., Booth, T.J., Roth, S.: The structure of suspended graphene sheets. Nature 446, 60–63 (2007)
Gray, D., McCaughan, A., Mookerji, B.: Crystal structure of graphite, graphene and silicon. Physics for solid state applications. WVU, Boston (2009)
Liao, Y., Farrell, T.P., Guillen, G.R., Li, M., Temple, J.A.T., Li, X.G., Hoek, E.M.V., Kaner, R.B.: Highly dispersible polypyrrole nanospheres for advanced nanocomposite ultrafiltration membranes. Mater. Horiz. 1, 58–64 (2014)
Yu, Q., Alvarez, N.T., Miller, P., Malik, R., Haase, M.R., Schulz, M., Shanov, V., Zhu, X.: Mechanical strength improvements of carbon nanotube threads through epoxy cross-linking. Materials 9(2), 68–80 (2016)
Cong, K., Radtke, M., Stumpf, S., Schröter, B., McMillan, D.G.G., Rettenmayr, M., Ignaszak, A.: Electrochemical stability of the polymer-derived nitrogen-doped carbon: an elusive goal? Mat. Renew. Sustain. Energy 4(5), 1–11 (2015)
Sánchez, A., Martínez-Mora, O., Martínez-Benavidez, E., Hernández, J., Domínguez, Z., Salas-Reyes, M.: Electrochemical behaviour of new dimeric esters and amides derived from caffeic acid in dimethylsulfoxide. Org. Biomol. Chem. 12, 5981–5989 (2014)
Jiao, Y., Zheng, Y., Jaroniec, M., Qiao, S.Z.: Origin of the electrocatalytic oxygen reduction activity of graphene-based catalysts: a roadmap to achieve the best performance. J. Am. Chem. Soc. 136(11), 4394–4403 (2014)
Sun, L.C., Lee, H.H., Yang, J.M., Wu, C.C.: The simultaneous electrochemical detection of ascorbic acid, dopamine, and uric acid using graphene/size-selected Pt nanocomposites. Biosens. Bioelectron. 33, 3450–3455 (2011)
Zhang, C., Ueno, K., Yamazaki, A., Yoshida, K., Moon, H., Mandai, T., Umebayashi, Y., Dokko, K., Watanabe, M.: Chelate effects in glyme/lithium bis(trifluoromethanesulfonyl)amide solvate ionic liquids. I. Stability of solvate cations and correlation with electrolyte properties. J Phys Chem B 118, 1353–1362 (2014)
Wu, Z., Huang, X.L., Wang, Z.L., Xu, J.J., Wang, H.G., Zhang, X.B.: Electrostatic induced stretch growth of homogeneous β-Ni(OH)2 on graphene with enhanced high-rate cycling for supercapacitors. Sci. Rep. 4, 1–8 (2014)
Zhang, B., Xu, Y., Zheng, Y., Dai, L., Zhang, M., Yang, J., Chen, Y., Chen, X., Zhou, Y.: Facile synthesis of polypyrrole/carbon nanotube composites with ultrathin, uniform and thickness-tunable polypyrrole shells. Nanoscale Res. Lett. 6, 1–9 (2011)
Zhong, C., Deng, Y., Hu, W., Qiao, J., Zhang, L., Zhang, J.: A review of electrolyte materials and compositions for electrochemical supercapacitors. Chem. Soc. Rev. 44, 7484–7539 (2015)
Bard, A.J., Faulkner, L.R.: Electrochemical Methods: Fundamentals and Applications. Wiley, New York (1980)
Acknowledgements
We thank Dr. Louise Weaver (UNB, Fredericton) for TEM imaging and Dr. James Tait for the access to FTIR spectrometer. This work was carried out with the financial support of New Brunswick Foundation for Innovation, Frank J and Norah Toole Graduate Scholarship and UNB President.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Radtke, M., Ignaszak, A. Grafting of the carbon allotropes and polypyrrole via a Kevlar-type organic linker: the correlation of carbon structure/morphology with electrochemistry of the composite electrode. Mater Renew Sustain Energy 6, 1 (2017). https://doi.org/10.1007/s40243-016-0085-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-016-0085-x