
Abstract
Duchenne muscular dystrophy is an inherited, progressive muscle-wasting disorder caused by mutations in the dystrophin gene. An increasing variety of approaches are moving towards clinical testing that all aim to restore dystrophin production and to enhance or preserve muscle mass. Gene therapy methods are being developed to replace the defective dystrophin gene or induce dystrophin production from mutant genes. Stem cell approaches are being developed to replace lost muscle cells while also bringing in new dystrophin genes. This review summarizes recent progress in the field with an emphasis on clinical applications.
Similar content being viewed by others


Introduction
Duchenne muscular dystrophy (DMD) is the most common and severe form of childhood muscular dystrophy, affecting approximately 1 in 5,000 boys [1]. The disease is characterized by progressive muscle weakness with delayed motor and speech milestones and elevated serum creatine kinase levels. Patients demonstrate reduced motor skills by age 3–5 and typically lose ambulation by 12 years of age. Glucocorticoids (prednisone or deflazacort) are prescribed to slow disease progression, but to date there is no cure for this disease. Despite intensive clinical attention to respiratory support and management of cardiac complications [2], most patients do not live beyond age 30.
DMD is caused by loss of function mutations in the X-linked DMD gene (commonly called ‘dystrophin’) resulting in near complete deficiency of dystrophin in cardiac, skeletal and smooth muscles. The types of mutations that lead to DMD include: point mutations that can encode premature termination codons (nonsense mutations, or “stop codons”); deletions or exon–intron splice site mutations that disrupt the normal protein or open reading frame (ORF) of the dystrophin messenger RNA (mRNA); partial gene duplications that disrupt the normal ORF; and, rarely, point mutations or deletions that disrupt critical functional domains of dystrophin. Mutations that enable expression of partially functional dystrophin lead to the milder Becker muscular dystrophy (BMD). Clinical observations of a BMD patient who remained ambulatory past age 70 despite absence of 46 % of the dystrophin gene [3] lent proof of principle that restoring some functional dystrophin expression can ameliorate the disease severity of DMD. Expression of 20–30 % of normal dystrophin levels is sufficient to avoid muscular dystrophy in mice and humans, while lower levels can lead to various severities of BMD [4, 5].
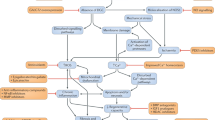
A major challenge for therapy of DMD and other muscular dystrophies is the need to devise a treatment strategy that targets all the major muscles in the body. These include limb muscles, respiratory muscles (intercostal and diaphragm muscles), cardiac muscle and smooth muscles of the gastroesophageal tract. Often emerging therapies are tested by direct intramuscular injection into a single site or into several sites of one muscle. While such an approach is useful for measuring local effects, it is difficult to envision ways to extend intramuscular injection strategies to achieve global muscle targeting. Instead, therapeutic interventions require whole-body targeting of the musculature, an approach generally referred to as systemic delivery. Multiple methods can be used for systemic delivery, depending on the drug or compound being tested, but most current approaches rely on oral or intravascular delivery. This review summarizes the current therapeutic strategies to restore dystrophin expression in DMD animal models and patients, with particular emphasis on the outcomes and discussions arising from the most recent clinical trials, as well as promising new therapies from current pre-clinical studies (Fig. 1).

Functional dystrophin can be restored through rAAV-mediated gene replacement, exon skipping, mutation suppression (not shown) or cell therapies (not shown). For gene replacement, minimally essential regions of dystrophin are removed to fit the limited carrying capacity of rAAV vectors and generate short dystrophin constructs (microdystrophins). Despite the truncation, microdystrophins restored dystrophin protein production and ~90 % of strength in dystrophic mice. Larger dystrophin constructs (minidystrophins) that incorporate additional regions necessary for recruiting important dystrophin binding partners (e.g., nNOS) result in even greater physiological improvements. Minidystrophins are delivered in pieces using multiple rAAV vectors and reassembled in muscle cells by various methods such as homologous recombination. In exon-skipping, synthetic antisense oligonucleotides (AON) are specifically designed to anneal to precursor mRNA (pre-mRNA) and alter RNA splicing to restore normal open-reading frames, resulting in the production of a smaller, but functional dystrophin. These techniques can be used in combination with cell therapies to correct genetic mutations prior to transplantation back into the patient in ex vivo therapies
Small Molecule Therapies
Approximately 10 % of DMD mutations are single-base “point” mutations that introduce premature stop codons into the dystrophin mRNA. One strategy to restore dystrophin expression in patients with such nonsense mutations is mutation suppression, specifically, of premature termination codon (PTC) mutations. Nonsense mutations signal an end to protein translation and result in production of a shortened, non-functional protein that is usually unstable and degraded in the cell. These ‘PTC’ mutations account for ~10 % of DMD cases [6] (see also http://www.umd.be/DMD/W_DMD/index.html). Certain antibiotics such as gentamicin can induce translational read-through of PTC mutations. At high concentrations, the binding of gentamycin to the protein production machinery in cells induces read-through of PTCs in mRNA, inserting a new amino acid to continue translation of the complete protein [7]. However, serious dose-limiting toxicities preclude the use of gentamicin in patients for this purpose. Drug discovery programs led to the identification of PTC124 (also known as ataluren), a compound shown to be more potent in PTC read-through than gentamicin [8], although recent in vitro studies have raised questions regarding the genuine read-through ability of ataluren [9–11]. Regardless of the mechanism, ataluren was somewhat effective in restoring functional dystrophin expression in mdx mice, resulting in improved strength and decreased injury in response to exercise-induced damage [8]. A phase I study in healthy volunteers established safety and tolerability at doses exceeding pre-clinical efficacy [12]. Phase 2b studies over 48 weeks involving DMD and BMD patients aged 5–20 years showed that patients receiving a low-dose treatment (20 mg/kg) experienced a significantly slower disease progression, demonstrating a 29-m improvement in a 6-min walk test (6MWT) compared to patients who received placebo (http://clinicaltrials.gov/show/NCT01182324; http://www.muscular-dystrophy.org/assets/0003/5427/2012-07_ataluren_update.pdf). Intriguingly, patients who received a higher dose (40 mg/kg) showed a similar decline as the placebo group, suggesting that ataluren may have an inverted bell-shaped dose–response curve. As of March 2013, a phase 3 study of ataluren was initiated, with recruitment restricted to DMD patients with PTC (stop codon) mutations. This will be a randomized, double-blind, placebo-controlled study with higher power to determine the potential efficacy and safety of low-dose ataluren (http://clinicaltrials.gov/show/NCT01826487).
The majority of DMD cases arise from partial dystrophin gene deletions or duplications or from mutations that affect the normal splicing of the dystrophin RNA transcript into mRNA. Each of these types of gene mutations is a problem when it disrupts the normal dystrophin mRNA open reading frame such that a functional dystrophin protein can not be produced in muscle cells. An approach to restore the normal mRNA reading frame involves exon skipping. This can be induced by short, synthetic fragments of nucleic acids known as “antisense oligonucleotides” (AONs), which are designed to bind (anneal) with RNA sequences that regulate how a gene’s initial RNA transcript is spliced into a functional mRNA. By altering RNA splicing near the mutation on the precursor mRNA (pre-mRNA), the cell can be tricked into making a slightly shorter than normal mRNA that lacks a mutation and that carries an otherwise normal open reading frame (Fig. 1). This strategy is theoretically applicable to ~80 % of all DMD patients [13]. Recent drug development for exon skipping has targeted exon 51 of the DMD gene, as this could be applied to ~13 % of boys with DMD. Two types of AONs have been taken into clinical trials, each made using different nucleic acid chemistries. Drisapersen (previously known as PRO051, GSK-2402968) was developed by Prosensa and GSK. The other was eteplirsen (AVI-4658) developed by Sarepta Therapeutics. Therapeutic efficacy of these AONs has been demonstrated in several mouse models for DMD (mdx) and the dystrophic cxmd dog [14–17]. Both compounds demonstrated safety and efficacy by direct intramuscular injections in patients, with restoration of dystrophin expression in the majority of muscle fibers near the injection site without severe treatment-related adverse effects [18, 19].
Initial phase 1/2A studies for both drisapersen and eteplirsen appeared promising, but disappointingly, neither compound succeeded in demonstrating significant improvements in the primary outcome measure, the 6MWT, in larger cohorts. In early phase 1/2A drisapersen trials involving 12 patients, a subset of patients who received the higher doses (4.0 and 6.0 mg/kg/week) showed some variable de novo dystrophin expression and demonstrated a mean improvement of 35 m in the 6MWT after 12 weeks of treatment [20••]. However, phase 3 trials where 125 patients (aged 5–16 years) were administered 6 mg/kg/week over 48 weeks did not result in statistically significant improvements in the 6MWD or in secondary measures of motor function when all patients were analyzed. However, Prosensa recently announced that when the analysis was confined to patients younger than 7, a 49-m difference was observed in the 6MWT in patients on a 96-week extension trial. Some of these studies also revealed kidney toxicity and low platelet counts in a few patients at high drug doses. To date, these latest results have only been discussed at conferences and in press releases (http://www.gsk.com/media/press-releases/2013/gsk-and-prosensa-announce-primary-endpoint-not-met-in-phase-iii-.html; http://www.thestreet.com/story/11854354/1/glaxo-dmd-drug-tied-to-serious-side-effects-hospitalizations.html; http://cureduchenne.com/blog/prosensa-reports-initial-findings-from-the-further-clinical-data-analyses-of-drisapersen-for-the-treatment-of-duchenne-muscular-dystrophy/). GSK recently announced that it would not continue studies of drisapersen and is returning rights to the drug to Prosensa, which will continue its development (http://us.gsk.com/html/media-news/pressreleases/2014/prosensa-regains-rights-to-drisapersen-from-gsk-and-retains-righ.html).
Similarly, early phase 2 studies for eteplirsen showed low and variable increases in dystrophin expression in 7 out of 19 patients (all but one who received the highest dose of 10 or 20 mg/kg over 12 weeks). There was a post-treatment increase in dystrophin protein expression from 9 to 16 % of normal controls; three patients showed between 15 and 55 % dystrophin-positive myofibers. Notably, restored dystrophin was shown to be functional and restored β-dystroglycan, α-sarcoglycan and nNOS expression in patients with exon 49–50 deletions, in keeping with the nNOS binding domain being encoded in exons 42–45. Inflammatory cell infiltrate was also reduced in the 10–20 mg/kg cohorts, suggesting that restored dystrophin is tolerated by the immune system [21, 22••]. In a second, more recent phase 2 study, increased dosage of eteplirsen was further assessed in a double-blind placebo-controlled test in 12 DMD boys aged 7–13 years [23]. Patients were administered with placebo, 30 mg/kg/week or 50 mg/kg/week of eteplirsen for 24 weeks, followed by an open-label extension study at the two doses through week 48. Results showed that at least 12 weeks of treatment was required to produce significant increases in dystrophin. At 48 weeks, the mean increase in dystrophin-positive fibers was 47 %, and the four patients in the placebo/delayed group (after 12 weeks of treatment) showed a significant increase of 38 %. Similarly, functional assessment by the 6MWT shows a divergence between the placebo and the eteplirsen groups at week 12, after which the treated group no longer showed a significant decline and stabilized. The placebo/delayed cohort likewise appeared to stabilize by 36 weeks (12 weeks after treatment). Comparison of the 48-week-treated group to the delayed group showed a significant difference of 67 m by week 48 in the 6MWT. Recent natural history data have shown that patients who could walk >350 m at baseline tend to stabilize over 1 year [24], and mean baseline 6MWT for all patients in the eteplirsen study was >350 m [23]. In a very recent press release, Sarepta announced continued stabilization of the 6MWT test in the phase 2b study where patients were continued on high-dose drug (30–60 mg/kg) for 120 weeks (http://investorrelations.sareptatherapeutics.com/phoenix.zhtml?c=64231&p=irol-newsArticle&ID=1891149&highlight=).
Despite these modest gains, the FDA declined to grant accelerated approval for eteplirsen, citing concerns about the study design (http://investorrelations.sareptatherapeutics.com/phoenix.zhtml?c=64231&p=RssLanding&cat=news&id=1875187). Moreover, there was growing concern over the disconnect between increased expression of dystrophin and clinical efficacy, at least in these relatively short trials, raising doubt about the quantification methods used to reliably assay dystrophin expression and the use of dystrophin as a biomarker to reasonably predict clinical benefit over 1 year. Another concern is the use of the 6MWT as the primary clinical endpoint. Although the 6MWT is to date the most reliable and sensitive global measure of lower extremity strength, biochemical inefficiencies, endurance and cardiorespiratory function [24, 25], the 6MWT excludes younger boys who cannot perform such a demanding test and older boys who are no longer ambulatory [26]. Therefore, it was suggested that future studies be designed to mathematically combine findings from a series of tests that measure a broader array of function in a larger range of patients. Redesign of this confirmatory study is currently underway, with dosing expected to initiate mid-2014.
Gene Replacement
Replacement of the defective DMD gene with a synthetic substitute using delivery vehicles such as recombinant adeno-associated virus (rAAV)-based vectors is another promising approach for safe, long-term therapy (Fig. 1). A significant advantage of gene replacement technologies is that they should be applicable to all patients, regardless of their underlying genetic mutation. Wild-type AAV is a small, non-pathogenic parvovirus that contains a 4.7-kb single-stranded DNA genome. Generation of rAAV delivery vectors involves removing all viral genes and replacing them with a therapeutic gene. Advantages of using rAAV-based vectors are that they can be produced at high titers on an industrial scale [27] and can stably persist for years in muscle cells. Of the many known serotypes, AAV6, 8 and 9 exhibit a high tropism for striated muscles, which is ideal for lessening off-target events in the treatment of muscular dystrophies [28, 29].
A significant limitation of rAAV vectors is that they can carry only a limited amount of DNA, i.e., they cannot deliver large genes. To accommodate this minimal carrying capacity of rAAV vectors, a common strategy is to insert miniaturized but highly functional copies of the dystrophin gene into the vector. These microdystrophin genes are deleted for sequences that code for several minimally essential regions in the dystrophin protein [30]. While there are numerous ways to generate a microdystrophin protein, and improved versions are still being developed, all share the feature of retaining only the most crucial functional domains, while taking care to retain structural flexibility and stability of the final protein [31]. Effective treatment of severely dystrophic mice was achieved by intravascular administration of rAAV6 vectors carrying microdystrophin, which restored dystrophin production in the respiratory, cardiac and limb muscles, improving muscle function and extending the lifespan [32–36].
Another strategy to overcome the limited carrying capacity of rAAV vectors is to co-deliver two or more rAAVs that have the gene of interest split between them, with reconstitution of the larger gene occurring inside the muscle cell. Reconstitution can be induced in various ways by specific design. One method is trans-splicing, where reconstruction of the larger gene occurs following a process where the various AAV vectors bind together and form large concatemers. A modification of this method involves splicing together of smaller protein pieces to produce a complete protein [37]. Still another modification involves co-infection of AAV vectors carrying overlapping portions of the desired gene, which reconstitutes the larger gene inside muscle cells by an exchange of DNA pieces known as “homologous recombination” [38••, 39, 40]. These ‘two vector’ methods have been used to deliver larger and more functional “minidystrophins” to dystrophic mice, resulting in somewhat greater physiological improvements compared to microdystrophin delivery [31, 38••, 40]. Recently, delivery of the entire dystrophin gene has been achieved using a triple-AAV vector system, although the efficiency is quite low [41].
Despite efficient gene transfer in mouse models of muscular dystrophy, a major obstacle in the field of AAV-mediated gene transfer is the host immune response. Unlike rodents, large animal models such as dogs and non-human primates, as well as humans, are born with a fully developed immune system, and immune responses against both the viral vector and the delivered therapeutic protein have been observed [42–46, 47••]. This highlights the need for pre-clinical testing in large animals before trialling in humans. In the only human trial to date of rAAV-mediated dystrophin gene transfer, intramuscular injections of a rAAV-minidystrophin into the biceps of six patients induced a minidystrophin-specific T cell immune response from as early as day 15 post-vector treatment in at least one patient, with none of the six patients displaying significant levels of vector-delivered dystrophin [45, 46]. Some of the patients also displayed elevated T-cell immune responses against the AAV vector [42]. Priming of the dystrophin T-cell activity was thought to have occurred through low levels of dystrophin that are produced naturally in rare, so-called ‘revertant fibers’ found in most DMD patients [45]. Recently, it was shown in a study of 70 DMD patients that ~28 % have anti-dystrophin T-cell immunity, with increasing age correlating with increased risk of anti-dystrophin immunity [47••]. Transient immunosuppression has been shown to be effective in preventing activation of T-cell-mediated immune responses and maintains production of vector-delivered microdystrophin in dog DMD models [48, 49], suggesting a need to incorporate an immunosuppressive regimen in future clinical trials.
In addition to cell-mediated responses to the AAV and therapeutic proteins, there have also been reports of pre-existing antibody (humoral) immunity against AAV (including AAV6) in some large animal models, especially in non-human primates and dogs [50, 51•, 52]. However, other groups have found no [53] or low titers of anti-AAV6 pre-existing neutralizing antibodies (NAbs) in dogs [54], suggesting that the source of the animals can skew results. NAbs against AAV2 have also been reported in many patients [46], and cross-reacting antibodies can cause sero-positivity against other serotypes [46, 53–55]. Proper patient screening and careful NAb titering are thus critical prior to future trials, which can also aid in selecting the AAV serotype with the best chance of success for particular patients.
A number of novel methods are being developed to overcome NAb inhibition and enhance gene transfer. Recently, it was found that empty AAV vehicles (i.e., carrying no genes) could act as “decoys” for NAbs when co-delivered with AAV vectors carrying a therapeutic gene [56]. Removal of NAbs by plasmapheresis can also enhance gene transfer [57•]. Directed evolution of AAV vectors is another strategy to enhance muscle tropism, overcome pre-existing neutralizing antibodies and allow for readministration with variants of the original vectors as required [46]. Other methods to reduce immune rejection include the use of muscle-specific gene regulatory switches to prevent protein production in non-muscle cells [58–60], sequence-optimization of the therapeutic gene to be delivered to reduce vector dosage [61], and thorough purification from contaminating viral gene products during vector preparation [27].
An increasing focus is being devoted to developing methods to deliver genes to dispersed muscle groups by using vector perfusion into the vasculature of isolated limbs. This approach includes the use of vascular ligatures, compression bandages or recirculation in the limb to increase gene transfer [48, 62, 63], simultaneous perfusion with agents that promote vector exit from capillaries to underlying muscles, such as histamine [64] and vascular endothelial growth factor [65], or delivering vector using high pressure injections [66, 67]. Importantly, efficient cardiac gene transfer, which is critical for DMD therapies, was achieved by direct “transendocardial injections” of rAAV vectors, with rAAV6 demonstrating the greatest cardiac targeting compared to rAAV8 and rAAV9 vectors in canine and non-human primate models [54, 68•]. In this method, vector is injected directly into cardiac muscle using catheters inserted into the cardiac vasculature. Direct infusion of vector via the jugular vein using rAAV6 and rAAV8 also resulted in good transgene delivery to canine heart and diaphragm [48, 69], with homogeneous gene expression being obtained in the heart 2.5 months after infusion [69]. Widespread gene delivery to the canine skeletal musculature was also achieved with rAAV9, though with relatively poor delivery to the heart [70, 71].
AAV vectors are also being used as a vehicle to deliver antisense oligonucleotides (AONs), since AONs by themselves have not worked well in the heart [72]. Embedding the antisense DNA sequence into an AAV vector carrying a small nuclear RNA (snRNA) particle protects it from degradation and improves the efficiency of pre-mRNA splice site modulation. A single administration of a rAAV expressing an ASO gene restored dystrophin expression and ameliorated pathology in muscles of dystrophic mice [73, 74]. Although the level of dystrophin restoration will decline over time as AAV vectors are lost, thus requiring repeat injections, recent studies in mice showed sustained exon skipping in the heart after 1 year [75]. Based on these promising results, AAV-mediated exon skipping is currently in preclinical stages to treat DMD patients (http://clinicaltrials.gov/ct2/show/NCT01385917).
Surrogate Genes
Delivery of surrogate genes, such as utrophin, could also potentially circumvent an immune response against dystrophin in DMD patients. Utrophin is a close evolutionary relative (paralog) of dystrophin, sharing 80 % amino acid sequence identity, and is normally produced in muscle at the neuromuscular and myotendinous junctions. Utrophin has also been found to partially compensate for the loss of dystrophin in DMD patients and animal models. rAAV-mediated delivery of microutrophin performed favorably relative to microdystrophin in dystrophic mdx:utrophin−/− (double knock out, dko) mice [76]. Upregulation of utrophin by treatment with SMT C1100, an orally bioavailable small molecule utrophin modulator, partially reversed dystrophic pathology in mdx mice [77•]. A phase 1B clinical trial of SMT C1100 commenced in December 2013 and is being carried out by Summit, a UK-based drug discovery company. In addition to utrophin, rAAV-mediated transfer of follistatin, a muscle-building gene, improved histology and increased muscle mass and strength in mdx mice and non-human primates [78]. It is in the phase 1 clinical trial for patients with BMD and Sporadic Inclusion Body Myositis (http://clinicaltrials.gov/ct2/show/NCT01519349). Recently, rAAV-mediated co-delivery of microdystrophin and follistatin was shown to fully restore force and resistance to eccentric contraction-induced injury in aged mdx mice, thus presenting a viable treatment option for DMD patients with advanced pathology [79•].
Cell Therapies
One caveat for dystrophin replacement therapies is that effectiveness in patients is dependent on both the number of remaining myofibers and the degree to which fibrotic connective tissue might limit the accessibility of myofibers to AAV vectors or small molecules. For patients in advanced stages of the disease, the use of gene replacement/correction in conjunction with cell therapies would likely have the greatest potential for an effective therapy.
While gene replacement therapies typically aim to stop the progression of muscle degeneration, cell therapies carry possibilities of both halting disease progression and restoring lost muscle mass. Resident muscle stem cells, termed satellite cells [80], are responsible for normal muscle maintenance and essential for muscle regeneration following injury [81–83]. Upon injury, satellite cells are activated to both self-renew and give rise to pools of proliferating myogenic progenitor cells (myoblasts), which fuse to repair damaged myofibers [83]. Throughout constant bouts of injury and repair in DMD patients, the capacity of resident satellite cells to self-renew and replenish injured muscle eventually diminishes, contributing to significant loss of muscle mass and function. A major feature of DMD is therefore a loss of regenerative capacity, and hence it could be amenable to stem cell therapy.
Based on promising results in animal models [84], cell therapies for DMD initially focused on local intramuscular injections of myoblasts derived from healthy donors to re-introduce dystrophin expression in muscles of DMD patients [85–87]. Unfortunately myoblast transfer strategies have thus far failed to produce adequate therapeutic effects, in part because of issues of poor survivability, limited migration from the injection site and immune rejection of transplanted cells [85, 88–90]. Expectations have been further tempered by challenges in expanding sufficient numbers of myogenic stem/progenitor cells in culture while preserving their ability to self-renew and efficiently engraft in vivo [91]. However, these early observations helped identify hurdles that needed to be overcome to improve treatment outcomes.
These findings led to the exploration of alternative cell sources with myogenic potential that could be more easily grown in culture while also being able to distribute to a wider range of affected muscles following systemic, intravascular delivery. Bone marrow-derived stem cells as well as muscle blood vessel-associated cells such as pericytes and mesoangioblasts (MABs) were shown capable of contributing to muscle regeneration, albeit with lesser efficiency than traditional myogenic stem/progenitor cells [92, 93]. Promising cell candidates eventually emerged, including MABs for which phase 1/2 clinical trials relying on transplantations of MABs derived from healthy HLA-matched donors are in progress (EudraCT no. 2011-000176-33). The next goal was to generate MABs from a dystrophic patient, correct the genetic mutation during cell expansion in culture, then transplant the corrected cells back into the patient (an approach known as ex vivo gene therapy). However, a recent report showed a drastic reduction of MABs in dystrophic patients, thereby limiting the ability to obtain sufficient number of cells for transplantation [94].
Induced pluripotent stem (iPS) cells have recently gained attention in re-programming strategies aimed at “manufacturing” ideal cell candidates that fulfill many of the identified needs for successful stem cell therapies of DMD. These iPS cells can be “induced” to a pluripotent, stem cell state by addition of several combinations of transcription factors important during early embryonic development [94]. iPS cells harbor virtually limitless expansion potential during which dystrophin vectors can be re-introduced to correct or bypass the genetic mutation [95, 96]. Corrected iPS cells can be subsequently directed toward adopting myogenic fates using various techniques, such as gene transfer of myogenic determination factors, and serve as a patient-specific cell source for autologous transplantation. Successful re-programming of iPS cells into stem/progenitor cells capable myogenic differentiation has been reported following gene-transfer of the regulatory genes encoding MyoD, Pax7 and Pax3 [97, 98••, 99].
MyoD is an extremely important regulator of gene transcription in muscle, which plays a key role during muscle regeneration where its expression in muscle stem (satellite) cells precedes myogenic differentiation [100]. The potency of MyoD for inducing myogenic fates in both muscle and non-muscle cell types has been well documented [101, 102]. A recent study aimed at stimulating muscle differentiation in human iPS cells modified with a MyoD gene that could be turned on or off reported up to 90 % myogenic reprogramming efficiency in vitro with the ability of reprogrammed cells to engraft into dystrophic muscles [97]. A possible drawback of using MyoD is that its expression leads to irreversible muscle differentiation, potentially reducing the chance for transplanted cells to become muscle stem cells (rather than myofibers) within transplanted muscles. However, recent data have indicated that subsets of iPS cell-derived MyoD-expressing cells are capable of forming muscle stem cells and engrafting into regenerating myofibers [103].
Other transcription factors that control the decision of stem cells to become myogenic cells are also attractive gene-transfer candidates to generate new muscle stem cells. Pax7 plays a key part in the function of satellite cells and is indispensible for adult skeletal muscle regeneration following injury [82]. The transcription factor Pax7 has also been used to convert human iPS cells into a muscle fate, and those cells were also able to partially restore dystrophin expression, generate new satellite (stem) cells and improve muscle function following transplantation into dystrophic mice [104]. Pax3, like Pax7, is expressed during myogenesis as well as embryonic development of non-muscle lineages such as those giving rise to the peripheral nervous system and melanocytes [105]. This necessitates careful sorting of cells derived via forced Pax3/7 gene expression in order to separate non-myogenic and possibly tumorigenic cells from myogenic cells prior to transplantation. Similarly, a version of the Pax3 transcription factor was used to generate myogenic stem cells from iPS cells genetically corrected with a micro-utrophin gene [98••]. Following activation of the Pax3 gene, cells were allowed to differentiate in culture prior to cell sorting for markers normally found on mesodermal cells destined to form muscle. The resulting stem/progenitor cells exhibited typical myogenic differentiation characteristics in culture and were able to generate functional microutrophin-expressing myofibers, migrate to stem cell harboring sites in muscle and improve muscle function following intramuscular injection into dystrophic mice [98••]. Importantly, these cells were also able to disseminate to several affected muscle groups following intravenous delivery.
iPS cells could potentially also solve the issue of obtaining sufficient MABs from dystrophic patients for autologous cell transplantation therapies. In two recent reports, iPS cells derived from both mice and humans with limb-girdle muscular dystrophy (LGMD) or DMD were used to generate α-sarcoglycan- or microdystrophin-corrected MABs for transplantation studies in immunodeficient mouse models of each condition [94, 103]. Following intramuscular and intraarterial administration, gene-corrected MABs were stimulated to adopt myogenic fates using an inducible version of MyoD [102]. Induced MABs engrafted a majority of affected muscles where they both fused with myofibers and formed new satellite/stem cells. Engrafted muscles exhibited a restoration of up to 25 % of normal expression levels of the mutant gene as well as significant morphological and physiological improvements. While promising, further studies are warranted to determine the safety and efficacy of these cell therapy approaches before translation into patients.
Conclusions
Development of therapeutics for the muscular dystrophies had progressed from basic studies in mice to ongoing early phase human clinical trials. While none of the methods being tested has yet risen to the level of a cure or major therapy, multiple approaches are showing significant promise, and it is becoming clear that treatments can and will be developed. It will be important to continue refining current approaches in animal models while expanding testing in patients. Recent clinical results also serve as a reminder that early encouraging success is no guarantee of long-term benefit, and it will be important to seek alternative strategies while refining those approaches already in advanced stages of testing.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Rodino-Klapac LR, Mendell JR, Sahenk Z. Update on the treatment of Duchenne muscular dystrophy. Curr Neurol Neurosci Rep. 2013;13(3):332.
Eagle M, Baudouin SV, Chandler C, et al. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord. 2002;12(10):926–9.
England SB, Nicholson LV, Johnson MA, et al. Very mild muscular dystrophy associated with the deletion of 46 % of dystrophin. Nature. 1990;343(6254):180–2.
Neri M, Torelli S, Brown S, et al. Dystrophin levels as low as 30 % are sufficient to avoid muscular dystrophy in the human. Neuromuscul Disord. 2007;17(11–12):913–8.
Chamberlain JS. Dystrophin levels required for correction of Duchenne muscular dystrophy. Basic Appl Myol. 1997;7:251–5.
Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34(2):135–44.
Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6(7):1044–55 PMCID: 1369979.
Welch EM, Barton ER, Zhuo J, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87–91.
Auld DS, Lovell S, Thorne N, et al. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc Natl Acad Sci USA. 2010;107(11):4878–83 PMCID: 2841876.
Auld DS, Thorne N, Maguire WF, Inglese J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci USA. 2009;106(9):3585–90 PMCID: 2638738.
McElroy SP, Nomura T, Torrie LS, et al. A lack of premature termination codon read-through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biol. 2013;11(6):e1001593 PMCID: 3692445.
Hirawat S, Welch EM, Elfring GL, et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharm. 2007;47(4):430–44.
Aartsma-Rus A, Fokkema I, Verschuuren J, et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. 2009;30(3):293–9.
Goyenvalle A, Babbs A, Powell D, et al. Prevention of dystrophic pathology in severely affected dystrophin/utrophin-deficient mice by morpholino-oligomer-mediated exon-skipping. Mol Ther. 2010;18(1):198–205 PMCID: 2839228.
Mann CJ, Honeyman K, Cheng AJ, et al. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc Natl Acad Sci USA. 2001;98(1):42–7 PMCID: 14541.
Yokota T, Lu QL, Partridge T, et al. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. 2009;65(6):667–76.
Yokota T, Nakamura A, Nagata T, et al. Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic Acid Ther. 2012;22(5):306–15 PMCID: 3464409.
Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8(10):918–28 PMCID: 2755039.
van Deutekom JC, Janson AA, Ginjaar IB, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. New Engl J Med. 2007;357(26):2677–86.
•• Goemans NM, Tulinius M, van den Akker JT, et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. New Engl J Med. 2011;364(16):1513–22. A human clinical study showing that ASOs can generate widesrpead dystrophin production using a systemic, rather than a localized delivery method.
Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378(9791):595–605 PMCID: 3156980.
•• Cirak S, Feng L, Anthony K, et al. Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol Ther. 2012;20(2):462–7. PMCID: 3277241. The ability of morpholino ASOs to generate new dystrophin was demonstrated in DMD patients. This study provides the ratinale for larger scale studies of therapy.
Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74(5):637–47.
McDonald CM, Henricson EK, Abresch RT, et al. The 6-min walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve. 2013;48(3):343–56 PMCID: 3824082.
McDonald CM, Henricson EK, Abresch RT, et al. The 6-minute walk test and other clinical endpoints in Duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve. 2013;48(3):357–68 PMCID: 3826053.
Hoffman EP, Connor EM. Orphan drug development in muscular dystrophy: update on two large clinical trials of dystrophin rescue therapies. Discov Med. 2013;16(89):233–9.
Wright JF. Manufacturing and characterizing AAV-based vectors for use in clinical studies. Gene Ther. 2008;15(11):840–8.
Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16(6):1073–80.
Schultz BR, Chamberlain JS. Recombinant adeno-associated virus transduction and integration. Mol Ther. 2008;16(7):1189–99.
Abmayr S, Chamberlain J. The structure and function of dystrophin. In: Winder SJ, editor. The molecular mechanisms of muscular dystrophies. Georgetown: Landes Biosciences; 2006. p. 14–34.
Harper SQ, Hauser MA, DelloRusso C, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nature Med. 2002;8(3):253–61.
Gregorevic P, Allen JM, Minami E, et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nature Med. 2006;12(7):787–9.
Gregorevic P, Blankinship MJ, Allen JM, Chamberlain JS. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol Ther. 2008;16(4):657–64 PMCID: 2650831.
Banks GB, Chamberlain JS, Froehner SC. Truncated dystrophins can influence neuromuscular synapse structure. Mol Cell Neurosci. 2009;40(4):433–41 PMCID: 2826111.
Banks GB, Combs AC, Chamberlain JR, Chamberlain JS. Molecular and cellular adaptations to chronic myotendinous strain injury in mdx mice expressing a truncated dystrophin. Hum Mol Genet. 2008;17(24):3975–86 PMCID: 2638580.
Banks GB, Judge LM, Allen JM, Chamberlain JS. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet. 2010;6(5):e1000958 PMCID: 2873924.
Li J, Sun W, Wang B, Xiao X, Liu XQ. Protein trans-splicing as a means for viral vector-mediated in vivo gene therapy. Hum Gene Ther. 2008;19(9):958–64 PMCID: 2940629.
•• Odom GL, Gregorevic P, Allen JM, Chamberlain JS. Gene therapy of mdx mice with large truncated dystrophins generated by recombination using rAAV6. Mol Ther. 2011;19(1):36–45. PMCID: 3017440. Demonstrates the potential for rAAV vectors to deliver larger dystrophin expression cassettes than can be transferred using a single AAV vector. Larger casettes may allow for increased functional correction of the dystrophic pathophysiology.
Zhang Y, Duan D. Novel mini-dystrophin gene dual adeno-associated virus vectors restore neuronal nitric oxide synthase expression at the sarcolemma. Hum Gene Ther. 2012;23(1):98–103 PMCID: 3260444.
Zhang Y, Yue Y, Li L, et al. Dual AAV therapy ameliorates exercise-induced muscle injury and functional ischemia in murine models of Duchenne muscular dystrophy. Hum Mol Genet. 2013;22(18):3720–9 PMCID: 3749861.
Koo T, Popplewell L, Athanasopoulos T, Dickson G. Triple trans-splicing adeno-associated virus vectors capable of transferring the coding sequence for full-length dystrophin protein into dystrophic mice. Hum Gene Ther. 2013. doi:10.1089/hum.2008.058.
Ohshima S, Shin JH, Yuasa K, et al. Transduction efficiency and immune response associated with the administration of AAV8 vector into dog skeletal muscle. Mol Ther. 2009;17(1):73–80 PMCID: 2834993.
Wang Z, Allen JM, Riddell SR, et al. Immunity to adeno-associated virus-mediated gene transfer in a random-bred canine model of Duchenne muscular dystrophy. Hum Gene Ther. 2007;18(1):18–26.
Yuasa K, Yoshimura M, Urasawa N, et al. Injection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene products. Gene Ther. 2007;14(17):1249–60.
Mendell JR, Campbell K, Rodino-Klapac L, et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med. 2010;363(15):1429–37 PMCID: 3014106.
Bowles DE, McPhee SW, Li C, et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol Ther. 2012;20(2):443–55 PMCID: 3277234.
•• Flanigan KM, Campbell K, Viollet L, et al. Anti-dystrophin T cell responses in Duchenne muscular dystrophy: prevalence and a glucocorticoid treatment effect. Hum Gene Ther. 2013;24(9):797-806. PMCID: 3768239. This study showed that immune effector cells that recognize dystrophin are common in DMD patients. The work is important to guide safe development of gene thearpy for DMD.
Gregorevic P, Schultz BR, Allen JM, et al. Evaluation of vascular delivery methodologies to enhance rAAV6-mediated gene transfer to canine striated musculature. Mol Ther. 2009;17(8):1427–33 PMCID: 2788962.
Wang Z, Kuhr CS, Allen JM, et al. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol Ther. 2007;15(6):1160–6.
Rapti K, Louis-Jeune V, Kohlbrenner E, et al. Neutralizing antibodies against AAV serotypes 1, 2, 6, and 9 in sera of commonly used animal models. Mol Ther. 2012;20(1):73–83 PMCID: 3255603.
• Shin JH, Pan X, Hakim CH, et al. Microdystrophin ameliorates muscular dystrophy in the canine model of Duchenne muscular dystrophy. Mol Ther. 2013;21(4):750–7. PMCID: 3616540. Clear demonstration of the effectiveness of microdystrophin constructs in a large animal model for DMD.
Wang L, Calcedo R, Wang H, et al. The pleiotropic effects of natural AAV infections on liver-directed gene transfer in macaques. Mol Ther. 2010;18(1):126–34 PMCID: 2839221.
Halbert CL, Madtes DK, Vaughan AE, et al. Expression of human alpha1-antitrypsin in mice and dogs following AAV6 vector-mediated gene transfer to the lungs. Mol Ther. 2010;18(6):1165–72 PMCID: 2889746.
Bish LT, Sleeper MM, Sweeney HL. Percutaneous transendocardial delivery of self-complementary adeno-associated virus 6 in the canine. Methods Mol Biol. 2011;709:369–78.
Halbert CL, Miller AD, McNamara S, et al. Prevalence of neutralizing antibodies against adeno-associated virus (AAV) types 2, 5, and 6 in cystic fibrosis and normal populations: implications for gene therapy using AAV vectors. Hum Gene Ther. 2006;17(4):440–7.
Mingozzi F, Anguela XM, Pavani G, et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci Transl Med. 2013;5(194):194ra92.
• Chicoine L, Montgomery C, Bremer W, et al. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol Ther. 2013. doi:10.1038/mt.2013.244. Important approach that could limit problems from preexisiting immunity to AAV vectors during gene therapy.
Cordier L, Gao GP, Hack AA, et al. Muscle-specific promoters may be necessary for adeno-associated virus-mediated gene transfer in the treatment of muscular dystrophies. Hum Gene Ther. 2001;12:205–15.
Salva MZ, Himeda CL, Tai PW, et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol Ther. 2007;15(2):320–9.
Hartigan-O’Connor D, Kirk CJ, Crawford R, Mule JJ, Chamberlain JS. Immune evasion by muscle-specific gene expression in dystrophic muscle. Mol Ther. 2001;4(6):525–33.
Foster H, Sharp PS, Athanasopoulos T, et al. Codon and mRNA sequence optimization of microdystrophin transgenes improves expression and physiological outcome in dystrophic mdx mice following AAV2/8 gene transfer. Mol Ther. 2008;16(11):1825–32.
Chicoine LG, Rodino-Klapac LR, Shao G, et al. Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin alpha2 surrogates. Mol Ther. 2013. doi:10.1038/mt.2013.246.
Rodino-Klapac LR, Montgomery CL, Bremer WG, et al. Persistent expression of FLAG-tagged micro dystrophin in nonhuman primates following intramuscular and vascular delivery. Mol Ther. 2010;18(1):109–17 PMCID: 2839222.
Greelish JP, Su LT, Lankford EB, et al. Stable restoration of the sarcoglycan complex in dystrophic muscle perfused with histamine and a recombinant adeno-associated viral vector. Nat Med. 1999;5(4):439–43.
Gregorevic P, Blankinship MJ, Allen JM, et al. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med. 2004;10(8):828–34.
Cho WK, Ebihara S, Nalbantoglu J, et al. Modulation of Starling forces and muscle fiber maturity permits adenovirus-mediated gene transfer to adult dystrophic (mdx) mice by the intravascular route. Hum Gene Ther. 2000;11(5):701–14.
Su LT, Gopal K, Wang Z, et al. Uniform scale-independent gene transfer to striated muscle after transvenular extravasation of vector. Circulation. 2005;112(12):1780–8.
• Gao G, Bish LT, Sleeper MM, et al. Transendocardial delivery of AAV6 results in highly efficient and global cardiac gene transfer in rhesus macaques. Hum Gene Ther. 2011;22(8):979–84. Data supporting the use of rAAV6 vectors for cardiac gene therapy in a non-human primate model.
Pan X, Yue Y, Zhang K, et al. Long-term robust myocardial transduction of the dog heart from a peripheral vein by adeno-associated virus serotype-8. Hum Gene Ther. 2013;24(6):584–94 PMCID: 3689160.
Kornegay JN, Li J, Bogan JR, et al. Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol Ther. 2010;18(8):1501–8 PMCID: 2927072.
Yue Y, Ghosh A, Long C, et al. A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol Ther. 2008;16(12):1944–52.
Alter J, Lou F, Rabinowitz A, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. 2006;12(2):175–7.
Goyenvalle A, Vulin A, Fougerousse F, et al. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science. 2004;306(5702):1796–9.
Goyenvalle A, Babbs A, Wright J, et al. Rescue of severely affected dystrophin/utrophin-deficient mice through scAAV-U7snRNA-mediated exon skipping. Hum Mol Genet. 2012;21(11):2559–71 PMCID: 3349427.
Le Hir M, Goyenvalle A, Peccate C, et al. AAV genome loss from dystrophic mouse muscles during AAV-U7 snRNA-mediated exon-skipping therapy. Mol Ther. 2013;21(8):1551–8 PMCID: 3734654.
Odom GL, Gregorevic P, Allen JM, Finn E, Chamberlain JS. Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol Ther. 2008;16(9):1539–45 PMCID: 2643133.
• Tinsley JM, Fairclough RJ, Storer R, et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One. 2011;6(5):e19189. PMCID: 3089598. Best demostration to date that utrophin upregulation might be achieved using small molecules.
Kota J, Handy CR, Haidet AM, et al. Follistatin gene delivery enhances muscle growth and strength in nonhuman primates. Sci Transl Med. 2009;1(6):6ra15 PMCID: 2852878.
• Rodino-Klapac LR, Janssen PM, Shontz KM, et al. Micro-dystrophin and follistatin co-delivery restores muscle function in aged DMD model. Human Mol Genet. 2013;22(24):4929–37. A combinatorial approach to DMD therapy that augments muscle mass while restoring dystrophin production. Such an aproach could be especially important for older patients.
Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;94:93–5 PMCID: 2225012.
Montarras D, L’Honore A, Buckingham M. Lying low but ready for action: the quiescent muscle satellite cell. FEBS J. 2013;280(17):4036–50.
Lepper C, Partridge TA, Fan CM. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development. 2011;138(17):3639–46 PMCID: 3152922.
Relaix F, Zammit PS. Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development. 2012;139(16):2845–56.
Partridge TA, Morgan JE, Coulton GR, Hoffman EP, Kunkel LM. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature. 1989;337(6203):176–9.
Skuk D, Goulet M, Roy B, et al. Dystrophin expression in muscles of Duchenne muscular dystrophy patients after high-density injections of normal myogenic cells. J Neuropathol Exp Neurol. 2006;65(4):371–86.
Skuk D, Roy B, Goulet M, et al. Dystrophin expression in myofibers of Duchenne muscular dystrophy patients following intramuscular injections of normal myogenic cells. Mol Ther. 2004;9(3):475–82.
Mendell JR, Kissel JT, Amato AA, et al. Myoblast transfer in the treatment of Duchenne’s muscular dystrophy. N Engl J Med. 1995;333(13):832–8.
Fan Y, Maley M, Beilharz M, Grounds M. Rapid death of injected myoblasts in myoblast transfer therapy. Muscle Nerve. 1996;19(7):853–60.
Skuk D, Paradis M, Goulet M, Tremblay JP. Ischemic central necrosis in pockets of transplanted myoblasts in nonhuman primates: implications for cell-transplantation strategies. Transplantation. 2007;84(10):1307–15.
Skuk D, Goulet M, Tremblay JP. Transplanted myoblasts can migrate several millimeters to fuse with damaged myofibers in nonhuman primate skeletal muscle. J Neuropathol Exp Neurol. 2011;70(9):770–8.
Montarras D, Morgan J, Collins C, et al. Direct isolation of satellite cells for skeletal muscle regeneration. Science. 2005;309(5743):2064–7.
Peault B, Rudnicki M, Torrente Y, et al. Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol Ther. 2007;15(5):867–77.
Farini A, Razini P, Erratico S, Torrente Y, Meregalli M. Cell based therapy for Duchenne muscular dystrophy. J Cell Physiol. 2009;221(3):526–34.
Tedesco FS, Gerli MF, Perani L, et al. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci Transl Med. 2012;4(140):140ra89.
Seto JT, Ramos JN, Muir L, Chamberlain JS, Odom GL. Gene replacement therapies for Duchenne muscular dystrophy using adeno-associated viral vectors. Curr Gene Ther. 2012;12(3):139–51.
Konieczny P, Swiderski K, Chamberlain JS. Gene and cell-mediated therapies for muscular dystrophy. Muscle Nerve. 2013;47(5):649–63.
Tanaka A, Woltjen K, Miyake K, et al. Efficient and reproducible myogenic differentiation from human iPS cells: prospects for modeling Miyoshi Myopathy in vitro. PLoS ONE. 2013;8(4):e61540 PMCID: 3633995.
•• Filareto A, Parker S, Darabi R, et al. An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat Commun. 2013;41549. PMCID: 3595133. Most promising study to date showing the potential for iPS cells to be used in cell transplantation in an animal model for DMD. Also showed that autologous iPS cells could be used for ex vivo gene therapy.
Darabi R, Pan W, Bosnakovski D, et al. Functional myogenic engraftment from mouse iPS cells. Stem Cell Rev. 2011;7(4):948–57.
Ishibashi J, Perry RL, Asakura A, Rudnicki MA. MyoD induces myogenic differentiation through cooperation of its NH2- and COOH-terminal regions. J Cell Biol. 2005;171(3):471–82 PMCID: 2171269.
Weintraub H, Tapscott SJ, Davis RL, et al. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci USA. 1989;86(14):5434–8 PMCID: 297637.
Kimura E, Han JJ, Li S, et al. Cell-lineage regulated myogenesis for dystrophin replacement: a novel therapeutic approach for treatment of muscular dystrophy. Hum Mol Genet. 2008;17(16):2507–17 PMCID: 2574879.
Tedesco FS, Hoshiya H, D’Antona G, et al. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci Transl Med. 2011;3(96):96ra78.
Darabi R, Arpke RW, Irion S, et al. Human ES- and iPS-derived myogenic progenitors restore dystrophin and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell. 2012;10(5):610–9 PMCID: 3348507.
Buckingham M, Relaix F. The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu Rev Cell Dev Biol. 2007;23:645–73.
Acknowledgments
We thank John Hall for critical reading of the manuscript. Supported by NIH grants R37AR40864 and R01AG033610 (to JSC). JTS was supported by an Early Career Fellowship from the NHMRC of Australia (Grant No. 1036656).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Seto, J.T., Bengtsson, N.E. & Chamberlain, J.S. Therapy of Genetic Disorders: Novel Therapies for Duchenne Muscular Dystrophy. Curr Pediatr Rep 2, 102–112 (2014). https://doi.org/10.1007/s40124-014-0044-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40124-014-0044-x