
Abstract
Introduction
Bamlanivimab and etesevimab (BAM + ETE) are monoclonal antibodies (mAbs) effective in reducing COVID-19-related hospitalizations and all-cause mortality in adult participants at increased risk for severe disease. We present pharmacokinetic (PK), efficacy, and safety results from pediatric participants (< 18 years of age) with COVID-19 who were treated with BAM + ETE.
Methods
In an addendum to the phase 2/3 BLAZE-1 clinical trial (NCT04427501), pediatric participants received open-label weight-based dosing (WBD, n = 94) based on exposure-matching to the authorized dose of BAM + ETE in adult participants. For efficacy and safety assessments, placebo (n = 14) and BAM + ETE (n = 20)-treated adolescent participants (> 12 to < 18 years of age) from the BLAZE-1 trial were included in the overall pediatric population (N = 128). All participants had mild to moderate COVID-19 upon enrollment and ≥ 1 risk factor for severe COVID-19. The primary objective was to characterize the PK of BAM and ETE in the WBD population.
Results
The median age of the participants was 11.2 years, 46.1% were female, 57.9% were Black/African American, and 19.7% were Hispanic/Latino. The area under the curve for BAM and ETE in the WBD population was similar to that previously observed in adults. There were no COVID-19-related hospitalizations or deaths. All adverse events (AE) except one were mild or moderate, with one participant reporting a serious AE.
Conclusion
WBD in pediatric participants achieved similar drug exposures compared to adult participants that received the authorized BAM + ETE dose. The pediatric efficacy and safety data were consistent with adults receiving mAbs for COVID-19.
Trial Registration Number
NCT04427501.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
One in four children hospitalized with COVID-19 requires intensive care. |
There is an unmet need for pediatric COVID-19 treatment options during mild to moderate disease presentation for high-risk patients. |
This study investigates weight-based dosing of bamlanivimab and etesevimab administered together in pediatric, ambulatory, high-risk participants with COVID-19. |
What was learned from the study? |
Our findings reveal similar pharmacokinetics, safety, and clinical status profiles between weight-based dosing of bamlanivimab and etesevimab in pediatrics to adults administered the authorized doses of bamlanivimab and etesevimab. |
These results support the first FDA authorization of a monoclonal antibody treatment in pediatric participants aged < 12 years and reveal distinct approaches to dosing selection and to testing COVID-19 therapeutics in pediatric populations. |
Introduction
SARS-CoV-2 infections in pediatric populations (< 18 years of age) have been rising in the US, constituting 18% of positive cases, as of January 12, 2023 [1]. Compared to adults, pediatric populations present with fewer cases, milder symptoms, and reduced hospitalizations [2, 3]; however, one in four hospitalized children requires intensive care [2]. Prevention of severe COVID-19 in pediatric populations remains a primary approach to treatment and suggests the need for treatment options in pediatric population during mild to moderate disease presentation.
Prior to the Omicron variant surge, the neutralizing monoclonal antibodies bamlanivimab (BAM) and etesevimab (ETE) administered together were authorized for treatment of mild to moderate COVID-19 in adults and adolescents ≥ 12 years of age, weighing at least 40 kg and at high-risk for progression to severe disease [4, 5]. The phase 2/3 BLAZE-1 clinical trial demonstrated that the combination of BAM + ETE reduced COVID-19-related hospitalization and all-cause mortality in adolescent and adult participants with mild to moderate COVID-19 [4, 6, 7]. To address treatment gaps in children < 12 years of age with COVID-19, the original BLAZE-1 study was expanded with a pediatric addendum to investigate the pharmacokinetics (PK), efficacy, and safety of BAM + ETE in pediatric and adolescent participants (< 18 years of age).
Guidelines for pediatric clinical trial design require purposeful reassessment of adult dosing to a pediatric population to minimize potential risk for the participants. Trial design should also consider the timely access of potentially critical therapeutics to participants. Thus, development of the BLAZE-1 pediatric addendum required distinct approaches. First, efficacy outcomes for pediatric patients were extrapolated from adult clinical data if exposure-matching was achieved between our proposed pediatric dosing and the authorized BAM + ETE treatments for COVID-19. Complete extrapolation of efficacy from adult clinical data to a pediatric population based on PK exposure-matching aligned with FDA draft guidance [8,9,10], thereby reducing the number of participants necessary to demonstrate efficacy and the need for dedicated pediatric pharmacokinetic clinical trials. Second, previously enrolled adolescents (12 to < 18 years of age) from the phase 2/3 BLAZE-1 clinical trial program (some of whom were reported on previously [4, 6]) were included in efficacy and safety analyses, thereby reducing the number of participants needed for enrollment.
The BLAZE-1 pediatric addendum was an open-label, phase 2/3 study performed across 14 clinical sites within the US. The adolescent participants (12 to < 18 years of age) in previous phase 2/3 BLAZE-1 cohorts were included in the efficacy and safety analyses. All participants had mild to moderate COVID-19 upon enrollment, at least one risk factor for severe COVID-19, and received treatment within 3 days of a positive SARS-CoV-2 test. The primary objective of the current study was to characterize the pharmacokinetics of weight-based dosing (WBD) of BAM + ETE in pediatric participants and to assess the safety and efficacy (i.e., viral dynamics, clinical status, and symptom resolution) of BAM + ETE in pediatric participants. The results reported here represent final analyses of all pediatric participants in BLAZE-1.
Methods
Study Design
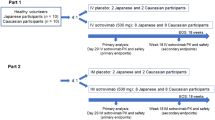
A total of 128 pediatric participants (< 18 years of age) were evaluated. Of the 128 participants, 94 pediatric participants < 18 years old were enrolled in an open-label pediatric addendum of the BLAZE-1 study (NCT04427501), 33 adolescent participants (ages 12 to < 18 years old) were enrolled in earlier randomized, double-blind, placebo (Pbo)-controlled arms (arms 7, 8, 9, 13, 14) of the BLAZE-1 study, and 1 adolescent participant in an earlier open-label arm investigating faster drug administration times of BAM + ETE (arm 21) [4, 6, 7]. Pediatric addendum participants (arm 22) received WBD to match the exposures observed in adults (> 18 years of age) and adolescents (12 to < 18 years of age) who received the authorized dose of 700 mg BAM + 1400 mg ETE (700/1400 mg BAM + ETE) and were included in pharmacokinetic, safety, and efficacy analyses. Of the 34 adolescent participants from prior BLAZE-1 treatment arms, 20 received BAM + ETE and 14 received Pbo and were included in safety analyses (Figure S1); 1 participant (arm 22) was excluded from efficacy analyses as they were not assessed for viral dynamics or symptom resolution. The first and last participant visit in the pediatric addendum were March 31, 2021, and December 14, 2021, respectively. The first and last participant visit in the other treatment arms were September 4, 2020, and July 2, 2021, respectively.
WBD was informed by PK exposure-matching to the authorized BAM + ETE adult dose. BAM + ETE was given as 700 mg + 1400 mg to those WBD participants who were ≥ 40 kg in body weight, 350 mg + 700 mg to those > 20 kg to < 40 kg, 175 mg + 350 mg to those > 12 to 20 kg, and 15 mg/kg + 30 mg/kg to those ≥ 1.5 kg to 12 kg. Of adolescent participants from prior BLAZE-1 treatment arms, BAM + ETE doses were 2800 mg + 2800 mg (2800/2800 mg), 700 mg + 1400 mg (700/1400 mg), or 350 mg + 700 mg (350/700 mg) depending on the study arm. Participants assigned to the Pbo arm received a 0.9% sodium chloride injection. Each participant received a single dose of study intervention, and participants were followed for 85 days post-treatment.
The trial complied with the Declaration of Helsinki, the International Conference on Harmonization Guidelines for Good Clinical Practice, and applicable local regulations. The protocol was reviewed and approved by the institutional review board - WCG IRB (20201599), the ethics committees of all participating centers, and all participants or legally authorized representatives gave written informed consistent and/or assent prior to study entry.
Enrollment Criteria
For a full list of inclusion and exclusion criteria, see Supplemental material. In brief, participants had to be 0 (≥ 32 weeks gestational age and ≥ 1.5 kg) to 17 years of age, have mild or moderate COVID-19 with at least one symptom, have at least one risk factor for developing severe COVID-19, and have had sample collection for the first positive SARS-CoV-2 viral infection determination ≤ 3 days prior to the start of the infusion. The most predominant risk factor for severe COVID-19 was participants meeting the body mass index (BMI) requirement (BMI percentile ≥ 85%). Risk factors affecting more than one participant included chronic respiratory disease, immunosuppressant therapy, and diabetes. Participants were excluded from the study if they had a diagnosis of multisystem inflammatory syndrome in children (MIS-C), had a history of a positive SARS-CoV-2 test prior to the one serving as eligibility for this study, or were hospitalized for COVID-19. COVID-19 severity was determined based on FDA guidance documents [8, 9]. Participants met criteria for mild or moderate COVID-19 severity based on shortness of breath at rest, respiratory rate, and heart rate. Participants with severe COVID-19 were excluded from the study based on pre-determined inclusion and exclusion criteria.
Pharmacokinetic Exposure-Matching
Efficacy data of BAM + ETE from clinical studies in adults with COVID-19 were extrapolated to pediatric participants based on PK exposure- matching. Justification for complete extrapolation was based on regulatory guidance for pediatric drug development [8,9,10] that specified conditions for appropriate extrapolation to pediatric populations such as common initial stage of mild or moderate COVID-19 in adults and pediatric participants [11, 12], similar course of SARS-CoV-2 infection in pediatric participants compared to adults, albeit usually less severe in pediatric participants, and similar BAM + ETE mechanism of action, regardless of host.
SARS-CoV-2 Spike Genotypic Analysis
Nasopharyngeal samples were obtained at study enrollment (baseline sample) and subsequently at days 3, 5, 7, 11, and 29. To assess SARS-CoV-2 variant lineage and the prevalence of treatment-emergent resistant variants, viral nucleic acid was extracted from the nasal samples and next-generation sequencing was performed on qPCR-positive samples as described in Gottlieb et al. [7]. Baseline viral lineage was determined using Pangolin (Version 3.1.11 [13]) using the assignment algorithm pangoLEARN (24 August 2021 release with pango-designation version 1.2.66). High-level strain definitions were as assigned by the embedded Scorpio software (version 0.3.12, constellations version 0.0.15), which uses defined constellations of mutations to attribute a summary viral strain classification to a sequence. Presence of a treatment-emergent protein substitution was determined by comparing the sequencing results from each study participant’s baseline sample with the post-treatment samples. Treatment-emergent substitutions that resulted in an observed pseudovirus neutralization reduction of ≥ 5-fold for BAM, ETE, or BAM + ETE were considered treatment-emergent resistant substitutions.
Statistical Analysis
Change from baseline (CFB) in SARS-CoV-2 viral load was calculated as the visit value of interest minus the baseline value. Baseline was defined as the last non-missing assessment recorded on or prior to the date of first study drug infusion. Mean CFB to day 3, day 5, and day 7 in SARS-CoV-2 viral load was estimated using descriptive statistics. Persistent high viral load (PHVL) was defined as a (log10) viral load > 5.27 on day 7 [7]. The proportions of participants with PHVL at Day 7 and those with COVID-19-related hospitalization or death were provided.
Participants in the Pbo-controlled arms (arms 7–9 and 13–14) were rated for the severity of eight COVID-19 symptoms daily through day 11, on day 22, and day 29 on a scale of none/absent = 0; mild = 1; moderate = 2; and severe = 3. The symptom questionnaire for the Pbo-controlled arms accounted for shortness of breath, feeling feverish, body aches and pains, sore throat, chills, headache, cough, and fatigue symptoms. Participants in the pediatric addendum (arm 22) reported the presence or absence of the eight symptoms in addition to the following five symptoms: diarrhea, nasal congestion, nausea, stomachache, and vomiting. Complete symptom resolution was defined as the first day all eight symptoms for fixed dosing and Pbo arms and all 13 symptoms for the WBD pediatric arm were absent. Sustained complete symptom resolution was defined as the first of 2 consecutive days that all symptoms were absent. The proportion of participants with complete symptom resolution was summarized for baseline and post-baseline on day 3, 5, 7, and 11. The time to sustained symptom resolution was analyzed using Kaplan-Meier estimates through day 29.
The proportion of participants experiencing COVID-19-related hospitalization (defined as ≥ 24 h of acute care) or death from any cause by day 29 was reported. The proportion of participants experiencing treatment-emergent adverse events (TEAEs), defined as an event that first occurred or worsened in severity after baseline, and serious adverse events (SAE), was also examined.
Sample size needed to achieve adequate trial power to meet PK objective was estimated using Monte Carlo simulations. To accomplish this, first, a validated PK model of the monoclonal antibody with established allometric (weight-based) function was used to simulate PK profile of trial participants over the wide range of weight distribution. PK model-based estimations were conducted to support sample size needed to achieve reasonable precision and bias of the estimates in pediatric patients across the weight categories. BAM + ETE concentration data were analyzed using a population PK approach via nonlinear mixed-effects modeling with NONMEM software. For safety and efficacy, descriptive data were provided. For continuous measures, number of participants, mean, standard deviation, median, minimum, and maximum were included. For categorical measures, frequency counts and percentages were included. Dropouts and missing data were accommodated following data imputation methods defined per analysis model, except for data imputation for a mixed effects model for repeated measures analysis because the addendum did not include inferential analyses. The final database lock occurred on January 25, 2022.
Results
A total of 128 pediatric participants were enrolled into the BLAZE-1 clinical trial, of which 94 were enrolled in the pediatric open-label addendum and received WBD BAM + ETE, and 34 were randomized to receive 350/700 mg, 700/1400 mg, or 2800/2800 mg BAM + ETE or Pbo in other BLAZE-1 treatment arms. The mean (± SD) age of all pediatric participants (N = 128) was 11.2 ± 4.7 years. Participants were distributed into four age categories: 0 to < 2 years (n = 8), 2 to < 6 years (n = 10), 6 to < 12 years (n = 36), and 12 to ≤ 17 years (n = 74). Of all pediatric participants, approximately 46.1% were identified as female, 57.9% as black or African American, and 19.7% as Hispanic. The BMI mean for all pediatric participants was 25.6 ± 8.6. Three participants, all in the WBD treatment group, received a COVID-19 mRNA vaccination prior to study drug treatment. These participants, aged 12, 16, and 17 years, received the first vaccine dose 37, 12, and 164 days prior to study drug treatment, respectively.
In total, pediatric participants in the study were infected primarily with Delta (B.1.617.2; 50.4%) and Alpha (B.1.1.7; 10.5%) variant lineages; a sizeable percentage of participants were also infected with non-WHO defined variants (23.8%). In participants receiving WBD (n = 94), the majority were infected with the Delta (69.7%) and Alpha (14.5%) COVID-19 variants.
PK parameters were similar for adults assigned to the authorized 700/1400 mg BAM + ETE (arm 9) and pediatric participants assigned to WBD (arm 22) that were selected based on exposure-matching to adults. Body weight was the only clinically meaningful patient factor identified that impacted the PK of pediatric participants, and PK clearances and volumes were adjusted to account for body weight effects. Concentration-time profiles and area under the concentration versus time curve (AUC) from zero to infinity for each WBD category were overlaid with 90% prediction intervals for adults receiving the authorized dose of 700/1400 mg BAM + ETE (Fig. 1, S2). Pediatric participants that received WBD displayed similar exposures for AUC and serum concentrations compared to adult participants who received the authorized 700/1400 mg BAM + ETE. Observed BAM + ETE serum concentrations on day 29 (Table 1) were also consistent with model-predicted BAM + ETE PK, and mean concentrations were within the 5th and 95th percentile range of adult 700/1400 mg BAM + ETE (Fig. 2).

Pediatric serum concentration profiles by weight category for BAM and ETE and exposure-matched adults. Pediatric serum concentration profiles of BLAZE-1 pediatric PK population by weight category-matched adult concentration profiles (PK model-estimated 5th and 95th percentiles) administered BAM 700 mg (left) and ETE 1400 mg (right)

PK profile simulations of BAM + ETE. PK profile simulations of BAM + ETE were conducted in a simulation dataset of 518 virtual participants weighing ≤ 12 kg, weights ranging from 3.2 to 12 kg, and ages ranging from 0.5 to 53 months. Datasets display 90% prediction intervals for BAM (top) and ETE (bottom) and concentration-time profiles in pediatric participants weighing ≤ 12 kg following administration of 12/24 mg/kg BAM + ETE (left panels) and 15/30 mg/kg BAM + ETE (right panels) compared with exposures in adult participants at the authorized dose of 700 mg/1400 mg BAM + ETE
The change in viral load from baseline was measured at 3, 5, and 7 days after BAM + ETE or Pbo infusion (Table S1). Regardless of study intervention (including placebo), mean viral clearance continually declined at each post-baseline visit. In all pediatric participants, the viral load change on day 7 was – 4.0 from a baseline value of 6.3. In pediatric participants receiving WBD, the viral load change on day 7 was – 4.4 from a baseline value of 6.5. All age group categories that received WBD, including 0 to < 2, 2 to < 6, 6 to < 12, and 12 to ≤ 17 years of age, displayed reductions in viral load from baseline to day 7. At day 7, 13 (10.2%) pediatric participants had PHVL.
In all pediatric participants, the number of participants with complete symptom resolution (Table S2) increased from 20 (15.7%) at day 3 to 96 (75.6%) at day 11. In pediatric participants that received WBD, the number of participants with complete symptom resolution increased from 17 (18.1%) at day 3 to 72 (76.6%) at day 11. Proportion of participants with sustained complete symptom resolution was 111 (87.4%) of total participants and 81 (86.2%) for WBD-treated participants by day 29. The safety data from pediatric participants receiving BAM + ETE (n = 114) were consistent with previous reports of adults receiving BAM + ETE. TEAEs occurred in 14.8% (n = 19) of participants administered BAM + ETE (Table 2). TEAEs were balanced across arms in the Pbo-controlled portions of the study. All but one TEAE were mild or moderate in severity. The reported severe TEAE was an increase in blood creatine phosphokinase on day 1 in a participant receiving WBD. The increase was reduced by 50% on day 3 and had normalized by day 29; the event was not associated with any clinical outcome. The reported mild and moderate TEAEs were primarily blood creatine phosphokinase increases and white blood cell count decreases, which resolved and were considered unrelated to open-label study drug. There was one SAE of diabetic ketoacidosis in a participant with type 1 diabetes mellitus receiving WBD. The SAE was considered moderate in severity and resulted in a 2-day hospitalization approximately 1 month after the study drug infusion. This SAE was not considered related to open-label study drug or COVID-19. A TEAE of moderate infusion site extravasation was reported in a pediatric participant receiving 350/700 mg BAM + ETE over a 3-min rapid IV infusion. No immediate hypersensitivity reactions were reported. One potential non-immediate hypersensitivity event (rash of moderate severity) was reported in a participant receiving 700/1400 mg BAM + ETE. The event occurred more than 24 h after infusion and was considered not related to blinded study drug. Throughout the 85-day study duration, there were no discontinuations from the study due to AE or deaths.
The incidence of participants with any treatment-emergent variant with neutralization impact of five-fold or greater on either BAM or ETE was 10% (1/10) for Pbo and 10.2% (10/98) for pooled BAM + ETE-treated participants. Six of these participants (all in BAM + ETE) did not have baseline sequences and were imputed to be treatment emergent. The remaining four participants that were treated with BAM + ETE had variants with known ETE neutralization impact at D420A and N460T/Y that were not present at baseline. Although the treatment-emergent variants D420A and N460T/Y should only have a neutralization impact on ETE and not BAM, all four of the participants had a primary infection with the Delta virus, which shows reduced neutralization to BAM [5]; therefore, the treatment-emergent ETE variants will likely result in antiviral resistance to both BAM and ETE. Of these four participants, three had evidence of delayed viral clearance (defined by PHVL at Day 7). None of the ten participants with suspected treatment-emergent resistance was hospitalized or experienced severe outcomes.
Discussion
Here we report the pharmacokinetic, efficacy, and safety results from an open-label addendum to the ongoing Blaze-1 phase 2/3 clinical trial for treatment of SARS-CoV-2 (NCT04427501) investigating WBD of BAM + ETE in pediatric participants at increased risk for severe COVID-19. The PK analyses confirmed that the WBD selected for pediatric participants (0 to < 18 years) provided comparable exposures to those observed in adults (≥ 18 years) and adolescent participants (12 to < 18 years), weighing at least 40 kg, and supported extrapolation of efficacy from adults based on exposure-matching for authorization. The safety profile of BAM + ETE in pediatric participants identified no new safety risks compared to the established safety profile. In contrast to adults, no COVID-19-related hospitalizations or deaths were reported in our pediatric populations. Furthermore, the observed 10% frequency of treatment-emergent substitutions of known neutralization impact was comparable to the rate seen in adults and was considered minimal.
These findings expand on similar results reported in a September 2021 interim analysis that supported BAM + ETE as the first authorized recombinant anti-SARS-CoV-2 monoclonal antibody therapy for high-risk participants < 12 years of age presenting with mild to moderate COVID-19 (authorized December 3, 2021 [5]). Soon after authorization, the Omicron variant became predominant in the US and globally. BAM + ETE displayed attenuated neutralization against the Omicron variant [5, 14], and this ultimately led to a pause of the Emergency Use Authorization (EUA) on January 24, 2022, for participants of all ages. Nevertheless, these results reveal distinct approaches to dose selection and to testing COVID-19 therapeutics in pediatric populations and suggest that PK, efficacy, and safety are comparable to adults for treatment of BAM + ETE-sensitive SARS-CoV2 variants.
The PK modeling approach was successful in predicting pediatric doses to match adult and adolescent exposures at the previously authorized 700/1400 mg BAM + ETE dose. The PK parameters were similar between adult and pediatric participants after accounting for body weight effects. Other than body weight effect, there were no clinically meaningful covariates identified that impacted the PK of pediatric participants across the age and weight ranges evaluated. The WBD-based dosing administered to pediatric participants provided similar exposures, based on AUC and serum concentrations at day 29, compared to adult participants who received the previously authorized 700/1400 mg BAM + ETE dose. These analyses met all pre-specified PK exposure-matching to support full extrapolation of efficacy data from adults to pediatric participants for the authorized use of BAM + ETE.
Interim PK, safety, and efficacy analysis results were submitted to the FDA for BAM + ETE EUA for pediatrics in November 2021. As body weight alone may not adequately predict PK changes due to maturation of organs or tissues in neonates and infants [15], FDA suggested the inclusion of age as a maturation function in the evaluation of the proposed dosing regimen in neonates and infants (weighing ≤ 12 kg) from the proposed 15 mg/kg and 30 mg/kg BAM + ETE to 12 mg/kg and 24 mg/kg BAM + ETE. The PK model was updated to include a maturation function published by Robbie et al. [16] that described the further reduction in drug clearance in the younger pediatric population in addition to the effect of low body weight on clearance. Simulations were conducted that incorporated the effect of maturation in infants and neonates on BAM + ETE clearance. Although there were no safety concerns with the original 15 mg/kg and 30 mg/kg BAM + ETE evaluated in the study, simulations demonstrated that the lower doses of BAM + ETE in participants ≤ 12 kg improved the precision of exposure-matching to adults at the authorized doses (Fig. 2). These simulations supported the reduced doses in neonates from the previous protocol-specified doses evaluated for BAM + ETE and were used to update the health care provider factsheet [5].
The overall safety profile, including low incidence of infusion-related reactions and hypersensitivity events, was consistent with that observed in adults. The rates of SAE in pediatric participants administered BAM + ETE (0.9%) were similar to those reported in adults (1.4% [4] and 1.2% [6]), as was the rate of TEAEs (14.9%, compared to 12.5% [4] and 9.2% [6]). The most frequently reported TEAEs within pediatric participants administered BAM + ETE were increases in blood creatine kinase and decreases in white blood cell count. There were no symptomatic AEs associated with these; thus, these lab abnormalities did not result in clinically meaningful findings.
Some children mount a strong inflammatory response associated with SARS-CoV-2 infection 2–6 weeks after infection, resulting in life-threatening MIS-C. In the US, MIS-C has a reported incidence of 5.1 per 1,000,000 individuals, with a higher incidence in Black and Hispanic/Latino children. There were no reported cases of MIS-C throughout the study. Given our total sample size of 124 pediatric participants, the lack of reported cases of MIS-C throughout this study is consistent with the reported incidence ratio of MIS-C in the US [17].
Complete symptom resolution in pediatric participants receiving WBD BAM + ETE increased from day 3 to 11 and aligns with an increase in viral load reduction from baseline to day 7. Compared to adults, pediatric participants present with fewer symptoms [18] but have similar levels of viral load. Similarly, the reduction in viral load from baseline to day 7 was approximately 19-fold in participants receiving WBD BAM + ETE compared to previously reported 16-fold reduction from baseline to day 7 in adult participants receiving the authorized 700/1400 mg BAM + ETE [4].
This study describes PK, efficacy, and safety outcomes in pediatric participants treated with BAM + ETE or Pbo; however, treatment in pediatric populations remains a challenge because of a lack of clinical data supporting the efficacy of monoclonal antibody therapy for COVID-19 in pediatrics and differences in the clinical course [19] and post-acute COVID-19 [20] in pediatric participants compared to adults. Although the current study addresses PK, efficacy, and safety of BAM + ETE in pediatric participants, the lack of a Pbo-controlled cohort limits the utility of the data found herein. Furthermore, previous EUA guidelines for administration of BAM + ETE following exposure to SARS-CoV-2 were based on limited preliminary data, mostly in adult patients, and highlights the need for dedicated clinical trials in pediatrics to facilitate health care providers with a better understanding of the appropriate conditions to treat COVID-19 infected pediatric patients with monoclonal antibody.
Study limitations include the small sample size of the pediatric population and the lack of placebo comparison within the WBD population. Pediatric populations were exclusively investigated in the US, where COVD-19 mortality rates can vary widely compared to other countries [21], suggesting potential differences in pediatric population characteristics outside of the US.
Conclusion
This is the first study conducted and completed in a high-risk pediatric population with mild to moderate COVID-19 treated with neutralizing monoclonal antibodies. The results of this study provide preliminary data and bridge the gap for future monoclonal antibody studies that are urgently needed in a disease that can potentially be serious in pediatric participants.
References
Children and COVID-19: state-level data report. American Academy of Pediatrics, 2023. https://www.aap.org/en/pages/2019-novel-coronavirus-covid-19-infections/children-and-covid-19-state-level-data-report/. Accessed Jan 19, 2023.
Delahoy MJ, Ujamaa D, Whitaker M, et al. Hospitalizations associated with COVID-19 among children and adolescents — COVID-NET, 14 states March 1, 2020–August 14, 2021. MMWR Morb Mortal Wkly Rep. 2021;70:1255–60.
Laboratory-confirmed COVID-19-associated hospitalizations. 2022. https://gis.cdc.gov/grasp/covidnet/covid19_3.html. Accessed Feb 3, 2022.
Dougan M, Nirula A, Azizad M, et al. Bamlanivimab plus Etesevimab in mild or moderate COVID-19. N Engl J Med. 2021;385:1382–92.
Fact sheet for health care providers emergency use authorization (EUA) of Bamlanivimab and Etesevimab. Food and drug administration, 2021. https://www.fda.gov/media/145802/download. Accessed Feb 22, 2022.
Dougan M, Azizad M, Mocherla B, et al. A randomized, placebo-controlled clinical trial of Bamlanivimab and Etesevimab together in high-risk ambulatory patients with COVID-19 and validation of the prognostic value of persistently high viral load. Clin Infect Dis. 2022;75:e440–9.
Gottlieb RL, Nirula A, Chen P, et al. Effect of Bamlanivimab as monotherapy or in combination with Etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. JAMA. 2021;325:632–44.
Guidance for industry. COVID-19: developing drugs and biological products for treatment or prevention. Food and Drug Administration, 2020. https://www.fda.gov/media/137926/download. Accessed Oct 3, 2021.
Development of anti-infective drug products for the pediatric population. Food and Drug Administration 2020. https://www.fda.gov/media/139586/download. Accessed Oct 3, 2021.
Guidance for industry. Pediatric study plans: content of and process for submitting initial pediatric study plans and amended initial pediatric study plans. Food and Drug Administration 2020. https://www.fda.gov/media/86340/download. Accessed Oct 3, 2021.
Zimmermann P, Curtis N. Coronavirus infections in children including COVID-19: an overview of the epidemiology, clinical features, diagnosis, treatment and prevention options in children. Pediatr Infect Dis J. 2020;39:355–68.
Siddiqi HK, Mehra MR. COVID-19 illness in native and immunosuppressed states: a clinical-therapeutic staging proposal. J Heart Lung Transplant. 2020;39:405–7.
O’Toole A, Scher E, Underwood A, et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021;7:veab064.
Hoffmann M, Kruger N, Schulz S, et al. The Omicron variant is highly resistant against antibody-mediated neutralization: implications for control of the COVID-19 pandemic. Cell. 2022;185:447–56.e11.
Holford N, Heo YA, Anderson B. A pharmacokinetic standard for babies and adults. J Pharm Sci. 2013;102:2941–52.
Robbie GJ, Zhao L, Mondick J, Losonsky G, Roskos LK. Population pharmacokinetics of palivizumab, a humanized anti-respiratory syncytial virus monoclonal antibody, in adults and children. Antimicrob Agents Chemother. 2012;56:4927–36.
Payne AB, Gilani Z, Godfred-Cato S, et al. Incidence of multisystem inflammatory syndrome in children among US persons infected with SARS-CoV-2. JAMA Netw Open. 2021;4: e2116420.
Chung E, Chow EJ, Wilcox NC, et al. Comparison of symptoms and RNA levels in children and adults with SARS-CoV-2 infection in the community setting. JAMA Pediatr. 2021;175: e212025.
Hoang A, Chorath K, Moreira A, et al. COVID-19 in 7780 pediatric patients: a systematic review. EClinicalMedicine. 2020;24: 100433.
Smane L, Roge I, Pucuka Z, Pavare J. Clinical features of pediatric post-acute COVID-19: a descriptive retrospective follow-up study. Ital J Pediatr. 2021;47:177.
Mortality analyses. Johns Hopkins University of Medicine: Coronavirus Resource Center, 2022. https://coronavirus.jhu.edu/data/mortality. Accessed Mar 16, 2022.
Acknowledgements
We are grateful for the participation of the children and their parents/guardian without whom this research could not have been done.
Funding
This study and the journal’s fee were funded by Eli Lilly and Company.
Medical Writing and/or Editorial Assistance
We thank Carmen Deveau, PhD, and Meena Ravuri, PhD, of Eli Lilly and Company for providing medical writing and editorial support throughout the manuscript. We thank Lisa Ferguson-Sells, BS, Lisa O-Brien, BS, and Emmanuel Chigutsa, PhD, of Eli Lilly and Company for assistance with PK/PD analysis. We thank Timothy R. Holzer, PhD, Philip J. Ebert, PhD, Richard E. Higgs, PhD, John Calley, PhD, Gary Heady, PhD, Ling Zhang, PhD, Angie Fulford, PhD, Drew Nedderman, PhD, Erin Wray, PhD, and Leslie O’Neil Reising, MS, of Eli Lilly and Company for assistance with viral sequencing for the BLAZE-1 study. We thank Honglu Liu, MSc, Christopher Kaiser, PhD, and the BLAZE-1 statistical analyst team of Eli Lilly and Company for their support with statistical analyses. All medical writing and editorial assistance were funded by Eli Lilly and Company.
Author Contributions
Conceptualization: Jenny Y. Chien, Mark Williams, Design: Jenny Y. Chien, Dipak R. Patel, Constance J. Krull; Data analysis: Jenny Y. Chien, Amanda J. Long, Lisa F. Macpherson, William J. Muller; Acquisition of data: Himanshu P. Upadhyaya, Dipak R. Patel, Matthew M. Hufford, Jocelyn Y. Ang, Peter Chen, Jeffrey A. Potts, Timothy Quinn; Interpretation of data: Himanshu P. Upadhyaya, Amanda J. Long, Martin S. Bohm, Lisa F. Macpherson, Matthew M. Hufford, Constance J. Krull, Jocelyn Y. Ang, William J. Muller; Manuscript drafting: Himanshu P. Upadhyaya, Jenny Y. Chien, Nicole L. Kallewaard, Lisa F. Macpherson; Manuscript review and revision: All authors.
List of Investigators
We thank the Blaze-1 investigators (listed in supplementary material) and their support staff for their dedication, invaluable insights, and participant-focused care throughout this program.
Prior Publication
The content was previously presented as a poster at 29th Annual Conference on Retroviruses and Opportunistic Infections, February 12–16, 2022. The corresponding abstract was published in the journal Topics in Antiviral Medicine. Citation: Conference on Retroviruses and Opportunistic Infections, CROI 2022. Virtual. 30(1 SUPPL) (pp 296), 2022. Date of Publication: March 2022.
Disclosures
Himanshu P. Upadhyaya, Jenny Y. Chien, Amanda J. Long, Martin S. Bohm, Lisa F. Macpherson, Dipak R. Patel, Matthew M. Hufford, Constance J. Krull and Mark Williams are employees and minor shareholders of Eli Lilly and Company. Nicole L. Kallewaard is an employee and minor shareholder of Eli Lilly and Company, and a former employee and minor stockholder of AstraZeneca. Jocelyn Y. Ang reports receiving funding for institutional research from Eli Lilly and Company. Peter Chen reports receiving personal fees from Eli Lilly and Company. William J. Muller reports receiving funding from Eli Lilly and Company; grants or contracts for institutional research from Ansun, Astellas, AstraZeneca, Enanta Pharmaceuticals, Gilead, Janssen, Karius, Melinta, Merck, Moderna, Nabriva, Paratek, Pfizer, Roche and Tetraphase; consulting fees from AstraZeneca, DiaSorin, Finley Law Firm, P.C., Sanofi and Seqirus; honoraria from Contemporary Pediatrics; travel support from Merck to attend meetings, and serving as advisory board for Adagio Therapeutics and ProventionBio. Jeffrey A. Potts and Timothy Quinn declare no conflict of interest.
Compliance with Ethics Guidelines
The trial complied with the Declaration of Helsinki, the International Conference on Harmonization Guidelines for Good Clinical Practice, and applicable local regulations. The protocol was reviewed and approved by the institutional review board—WCG IRB (20201599), the ethics committees of all participating centers, and all participants or legally authorized representatives gave written informed consistent and/or assent prior to study entry. Lilly voluntarily asked the FDA to revoke the EUA for bamlanivimab 700 mg alone in April 2021. This request was not due to any new safety concerns and was due to the evolving variant landscape in the US and to complete transition to bamlanivimab and etesevimab together for treatment of COVID-19 in the US.
Data Availability
All data generated or analyzed during this study are included in this published article/as supplementary information files.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Upadhyaya, H.P., Chien, J.Y., Long, A.J. et al. Pharmacokinetics, Efficacy, and Safety of a SARS-CoV-2 Antibody Treatment in Pediatric Participants: An Open-Label Addendum of a Placebo-Controlled, Randomized Phase 2/3 Trial. Infect Dis Ther 12, 1861–1873 (2023). https://doi.org/10.1007/s40121-023-00832-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-023-00832-y