
Abstract
Introduction
Clinical metagenomic next-generation sequencing (mNGS) has proven to be a powerful diagnostic tool in pathogen detection. However, its clinical utility has not been thoroughly evaluated.
Methods
In this single-center prospective study at the First Affiliated Hospital of Soochow University, a total of 228 samples from 215 patients suspected of having acute or chronic infections between June 2018 and December 2018 were studied. Samples that met the mNGS quality control (QC) criteria (N = 201) were simultaneously analyzed using conventional tests (CTs), including multiple clinical microbiological tests and real-time PCR (if applicable).
Results
Pathogen detection results of mNGS in the 201 QC-passed samples were compared to CTs and exhibited a sensitivity of 98.8%, specificity of 38.5%, and accuracy of 87.1%. Specifically, 109 out of 160 (68.1%) CT+/mNGS+ samples exhibited concordant results at the species/genus level, 25 samples (15.6%) showed overlapping results, while the remaining 26 samples (16.3%) had discordant results between the CT and mNGS assays. In addition, mNGS could identify pathogens at the species level, whereas only the genera of some pathogens could be identified by CT. In this cohort, mNGS results were used to guide treatment plans in 24 out of 41 cases that had available follow-up information, and the symptoms were improved in over 70% (17/24) of them.
Conclusion
Our data demonstrated the analytic performance of our mNGS pipeline for pathogen detection using a large clinical cohort and strongly supports the notion that in clinical practice, mNGS represents a valuable supplementary tool to CTs to rapidly determine etiological factors of various types of infection and to guide treatment decision-making.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Why carry out this study? | |
Rapid and broad detection of pathogens in infectious diseases is critical for diagnosis and timely treatment, especially in severe conditions. | |
The application value of metagenomic next-generation sequencing (mNGS) in real-world clinical practice was investigated in a large cohort with diverse pathogens and multiple sample types. | |
What was learned from the study? | |
The pathogen detection results of mNGS were comprehensively compared with conventional tests and exhibited a sensitivity of 98.8%, specificity of 38.5%, and accuracy of 87.1%. | |
The value of mNGS results in guiding treatment plans was also demonstrated in this study, which emphasized that mNGS represents a powerful supplementary tool not only in diagnosis but also in treatment decision-making. |
Introduction
Metagenomic next-generation sequencing (mNGS) is a rapid and universal pathogen detection method for infectious disease diagnostics. Since the first diagnosis of an infected patient using mNGS in 2014, mNGS has contributed significantly to the diagnosis of infections in recent years [1,2,3]. All types of infectious pathogens, such as bacteria, fungi, viruses, and parasites, can be detected using mNGS, with no need for preclinical information [4]. Additionally, mNGS can be performed within 48 h, offering opportunities to save lives. To date, some in-house mNGS methods have been built for pathogen detection. The application of mNGS to different infected body fluids and tissue samples provides a faster sample-to-answer time for pathogen detection and presents high accuracy compared to conventional tests (CT) [5,6,7,8,9,10]. CTs have been the foundation of microbial infection detection in clinical practice for many decades. They include diverse clinical microbiological tests (CMTs) such as culture, Gram staining, acid-fast staining, and serological assays as well as real-time PCR. While bacterial culture remains the gold standard test for infection diagnosis, it is often time consuming and exhibits a relatively low sensitivity. Serological assays, such as (1–3)-β-d-glucan (G) and Aspergillus galactomannan (GM), are commonly used for diagnosing fungal infections. Real-time PCR has emerged as a rapid and sensitive method for pathogen detection by quantifying microbial DNA or RNA, but it may still require the pre-identification of specific pathogen targets. Thus, none of these CTs can comprehensively detect all potential pathogens in a single run, unlike mNGS.
Despite the significant advantages of mNGS over CTs in detecting pathogens, there are multiple technical and regulatory obstacles preventing this technology from being used widely, and it remains in the early stage of clinical adoption [11, 12]. A limited number of large-scale validation studies of the clinical performance of mNGS have been reported [13,14,15]. Furthermore, whether mNGS diagnostic results can inform clinical decision-making remains to be answered. Here, we utilized an mNGS pipeline to detect pathogens from multiple sample types and compared the results with those from CTs, demonstrating the clinical utility of mNGS in real-world clinical practice.
Methods
Patient Enrollment and Study Design
We prospectively collected 228 samples from a total of 215 patients suspected of having acute or chronic infections of the lower respiratory tract, bloodstream, central nervous system, etc., who were admitted to the First Affiliated Hospital of Soochow University in Suzhou, China, between June 2018 and December 2018. The samples were analyzed by the Department of Pathology using CMTs (accompanied by real-time PCR for CMT-negative samples) and simultaneously using mNGS. Patients over 18 years old and with characteristic clinical signs, symptoms, and laboratory tests suggesting infections were included in this study. Exclusion criteria included sample unavailability, an infection with manifest symptoms indicative of a known pathogen, and other ineligible conditions determined by investigators. This study was approved by the ethics committee of the First Affiliated Hospital of Soochow University (no. 2018-189) and was conducted according to the principles of the Helsinki Declaration. Written informed consent was collected from each patient for their participation in and the publication of this study.
CMT and Real-Time PCR
Sputum and bronchoalveolar lavage fluid (BALF) samples were inoculated onto sheep blood or chocolate agar plates. Positive blood culture bottles were used to prepare Gram stains and were subcultured on sheep blood or chocolate agar plates. The sheep blood and chocolate agar plates were incubated at 35 °C in 5% CO2. Brucella agar plates were incubated anaerobically and were used to subculture bacteria from positive anaerobic blood culture bottles. Colonies on the plates were recovered using a 10-μL inoculating loop and spotted onto a target slide prepared according to the manufacturer's instructions for analysis using the Vitek MS system and the accompanying software (version 2.0; BioMérieux, France).
The diagnosis of tuberculosis infection was performed using the Kinyoun cold Ziehl–Neelsen stain (Baso, China). Plasma cytomegalovirus and Epstein–Barr virus DNA levels were assessed using real-time PCR (DaAnGene, China). Serological fungus (1–3)-β-d-glucan (G) and Aspergillus galactomannan (GM) assays were used for the serological diagnosis of fungal infections (Dynamiker Biotechnology, China).
mNGS Assay, Sample Processing, and Nucleic Acid Extraction
Clinical samples were pre-processed for mNGS as follows. Peripheral blood samples were centrifuged at 1800g for 10 min at 4 °C, and sputum/BALF samples were liquefied by 0.1% dithiothreitol for 1 h at 60 °C. Then an in-house host DNA depletion pipeline was performed, followed by DNA extraction using the QIAamp® Circulating Nucleic Acid Kit (Qiagen) or the Tiangen Magnetic DNA Kit (Tiangen) according to the manufacturer’s instructions. A no-template control (NTC) for each batch was processed in parallel with the clinical samples. The quantity, quality, and purity of extracted DNA were evaluated for each sample using Qubit, agarose gel electrophoresis, and NanoDrop, respectively.
Library Construction and Sequencing
Plasma cell-free DNA was used directly for library preparation, while DNA extracted from sputum and BALF was sonicated into 150–300 bp fragments with ultrasonication parameters of 30 s on and 30 s off for 10 cycles (Bioruptor Plus, Diagenode). All DNA libraries were constructed using the KAPA Hyper Prep Kit (KAPA Biosystems) or the Hieff NGS OnePot II DNA Library Prep Kit for Illumina (Yeasen), following the manufacturer’s protocols. Sequencing was performed on an Illumina NextSeq550Dx (Illumina) sequencing system. The samples with more than 25 ng of DNA after library construction and over eight million raw reads passed the quality control (QC) process.
mNGS Database Construction and Data Analysis
The mNGS database included the reference genomes of human and microorganisms as well as plasmid/cloning vector sequences. The human reference genome, hs37d5, was downloaded from the UCSC Genome Browser. The microorganism genome database, consisting of genomes or scaffolds of 12,000+ bacteria, 18,000+ fungi, 4600+ viruses, and 100+ parasites, was downloaded from National Center for Biotechnology Information (NCBI: ftp://ftp.ncbi.nlm.nih.gov/genomes/genbank/ and ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/), with the assembly level matching either the complete genome or chromosome criteria. Plasmid, plastid, mitochondrion, and UniVec sequences from NCBI RefSeq and human clone sequences from the NCBI Nucleotide database were downloaded to construct the plasmid and cloning vector databases.
Raw sequencing data were split using bcl2fastq2, according to the sample sequence index, and high-quality sequencing data were generated by removing low-quality reads, adapter contamination, and duplicated and short reads (length < 36 bp) using Trimmomatic software. Human host sequences were identified by mapping to the human reference genome (hs37d5) using bowtie2. Reads that could not be mapped to the human genome were remapped to the plasmid and cloning vector database. The retained unmapped reads were aligned to the microorganism genome database for microbial identification using Kraken2 [16, 17].
mNGS Interpretation and Reporting
The mNGS pathogen detection pipeline was described in previous studies [18,19,20], and the criteria for detection positivity were as follows: (i) at least one species-specific read for Mycobacterium, Nocardia, and Legionella pneumophila detection; (ii) for other bacteria, fungi, virus, and parasites, at least three unique reads were needed; (iii) pathogens were excluded if the ratio of microorganism reads per million of a given sample versus NTC was < 10. A clinical adjudication committee composed of three physicians from the Departments of Pneumology and Hematology and the Clinical Laboratory determined the final clinical diagnosis by reviewing the etiological results, radiological testing results, and other relevant records.
mNGS Analytical Performance Characteristics
The limits of detection (LoDs) of the mNGS assay were determined using 11 representative microorganisms, including two DNA viruses, five tough-to-lyse Gram-positive (G+) bacteria, two easy-to-lyse Gram-negative (G−) bacteria, and two tough-to-lyse yeast (Table 1). The selected strains were spiked into clinically negative plasma in five tenfold serial dilutions, with 20 replicates per dilution. LoD tests were performed and calculated for each organism using the bioinformatic pipeline, as described in previous studies [18,19,20]. With 20 replicates performed for each tested concentration in each organism, we accepted the lowest concentration at which ≥ 95% detection was achieved as the LoD.
Statistics
Comparative analyses were conducted using McNemar's chi-squared test and Fisher's exact test in the R statistical software package (version 3.6.2). P values < 0.05 were considered statistically significant.
Results
Patient Overview
A total of 228 samples, including 34 peripheral blood specimens, 147 sputum specimens, and 47 BALF specimens, were collected from 215 patients. Twenty-seven patients had two or more samples. Notably, a total of 201 samples from 188 patients passed the QC process of the mNGS pipeline and were included in the following analyses (Fig. 1A). Therein, the median age of all cases was 58 years; 9 cases (4.8%) were 18 years of age or younger and 21 cases (11.2%) were older than 80 years of age. Approximately 67.6% (127/188) of the cases were male. Eighty cases (42.6%) had lower respiratory tract infections, 7 had infections at other sites, including bloodstream infections, while the remaining 101 had infections at unknown sites (Supplementary Information: Table S1).

Metagenomic analysis of clinical samples and quality control (QC) data. A Overview of the study design and sample filtering in this study. B Scatter plot showing the distribution of quality metrics of all samples (N = 228) in the total amount of DNA after library construction (x-axis) and raw reads (y-axis). The gray dashed line indicates the filter criteria (25 ng and 8 million). C mNGS QC process. The stacked column chart shows the percentages of the samples that passed and failed for each sample type
Performance Evaluation of the mNGS Assay
The LoDs for 11 representative species are shown in Table 1, and range from 10 to 16,000 copies/mL (Table 1). Notably, the genome size of a given pathogen could be a significant factor affecting the LoD. For instance, the LoD of Cryptococcus neoformans, with a 19-Mb genome, was 10 copies/mL, while adenovirus type 35, with a much smaller genome (0.035 Mb), could be detected at 16,000 copies/mL.
In silico data simulation was performed to evaluate the performance of the mNGS analytical pipeline as follows. One thousand species were randomly selected from the microorganism genome database and 50 sequencing reads from each selected organism were computationally generated, which were then mixed into the whole-genome sequencing results of a healthy donor to generate a simulated dataset of eight million reads. The pathogen analytical pipeline was repeated 10 times and the detection rates were all over 82%, demonstrating the reliability of this analytic pipeline (Supplementary Information: Fig. S1).
Comparison of the mNGS and CT Results for the Clinical Samples
All clinical samples underwent CMT analysis, and 12 CMT-negative samples had sufficient remaining volume for a real-time PCR test (positive rate: 7/12, 58.3%). Either CMT+ or PCR+ samples were considered CT+. As shown in Fig. 1, 201 out of 228 (88.2%) samples passed the QC process of the mNGS analysis. The pass rates for different sample types were comparable (blood: 85.3%, BALF: 87.2%, sputum: 89.1%). Compared to the CT results, mNGS exhibited 98.8% sensitivity in all 201 samples, with 100% for blood, 96.6% for BALF, and 99.2% for sputum samples, respectively (Fig. 2A). The overall specificity was 38.5% and the accuracy was 87.1%. A total of 160 samples (79.6%, 160/201) were positive for both mNGS and CT, and 15 (7.5%) were double negative (Fig. 2B). 109 out of 160 double-positive samples exhibited concordant results between mNGS and CT, of which 77 were concordant at the species level and 32 were concordant at the genus level (Fig. 2B). An additional 25 samples showed overlapping results between mNGS and CT, with the same pathogen detected at least once. The remaining 26 samples had discordant results between mNGS and CT. The pathogen composition analysis of the 109 samples with concordant results of mNGS and CT showed that most of them were infected by single bacteria, including 38.5% G− bacteria, 20.2% G+ bacteria, and 10.1% Mycobacterium tuberculosis (MTB), followed by 24.8% of samples with polymicrobial infections (multiple bacteria, 20.2%; bacteria plus fungi, 3.7%; fungi plus viruses, 0.9%). Only a small proportion of the samples had viral (1.8%) and fungal (4.6%) infections (Fig. 2B).

Validation of the mNGS assay and performance analytics. A 2 × 2 contingency tables comparing the performance of mNGS relative to CTs for 201 samples that passed the mNGS QC threshold. Performance analytics including sensitivity, specificity, and accuracy are shown below each table. The comparison was performed using Fisher’s exact test. B Comparison of the mNGS and CT results
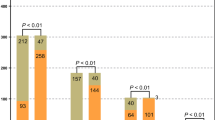
The number of samples that were positive for each pathogen is summarized in Fig. 3A, and the positive rates for bacterial pathogens (p < 0.05) and viruses (p < 0.001) were found to be significantly higher when using mNGS than CTs (Fig. 3B). However, more fungi-infected samples were identified by CTs. Notably, for some pathogens, the species level could only be identified by mNGS, while CTs could identify pathogens at the genus level, such as Neisseria and Candida. Fourteen bacteria, two fungi, and four viral species were only detected by mNGS.

Pathogens detected by mNGS and/or CT assays. A Pathogens that tested positive either by mNGS or CTs are grouped into three categories: bacteria, fungi, and viruses. B The total number of positive samples for each category is summarized in the bar plot. *p < 0.05; ***p < 0.001. 1Viridans streptococci include Streptococcus oralis, Streptococcus anginosus, Streptococcus mitis, and Streptococcus pseudopneumoniae. 2For the samples which are positive in both CTs and mNGS, 8/14 samples of Mycobacterium tuberculosis, 13/13 samples of Neisseria subflava, 1/9 samples of Corynebacterium striatum, 4/4 samples of Neisseria elongate, 1/1 samples of Neisseria flavescens, and 4/7 samples of Candida albicans were only detected at genus level by CT
Application of mNGS Results in Clinical Diagnosis and Treatment Decision-Making
Among the samples whose results were not concordant between mNGS and conventional tests, 41 cases had follow-up medication information. The results of mNGS led to the modification of treatment in 58.5% (24/41) of cases (Fig. 4), including 14 with overlapping (N = 7) or discordant (N = 7) pathogens between mNGS and CTs and 10 that were only mNGS+ (Supplementary Information: Table S2). Specifically, 16 patients received antibiotic therapies with mNGS-detected Klebsiella pneumoniae, P. aeruginosa, or L. pneumophila infections, and the antiviral treatments oseltamivir and ganciclovir were given to 3 patients. Five patients who had Cryptococcus neoformans, Pneumocystis jirovecii, or Candida glabrata infections, as indicated by the mNGS results, were administered antifungal treatments. Over 70% (17/24) of the patients who received mNGS-guided therapies showed significant improvements in their infectious symptoms (Supplementary Information: Table S2). However, the mNGS results did not assist treatment decision-making in the remaining 17 cases. For instance, the original therapies were maintained in 11 cases, and the pathogens detected by mNGS were not deemed the etiology in 4 cases where antibiotic treatments were given to patients with mNGS-detected fungal infections. One patient whose sample was positive in both mNGS (Haemophilus parainfluenzae) and CTs (viridans streptococci) was later diagnosed with lung cancer and received chemotherapy. One mNGS−/CT+ patient was treated with antibiotics.

The application of mNGS results in clinical diagnosis and treatment decision-making. The mNGS results were adopted to build and improve treatment plans in 24 out of 41 cases that had available follow-up treatment information
Three representative CT-negative patients were cured by the mNGS-advised treatment. P19 was a 54-year-old male who was admitted to the hospital after 5 days of fever and cough. Legionella pneumophila was detected by mNGS in his BALF sample. After the completion of a 10-day course of moxifloxacin (BAYER Avelox), significant resolution of the clinical symptoms was observed. In the second case, an 83-year-old male patient (P38) with fever and chest pain for 2 weeks was CT-negative but Cryptococcus neoformans infection was identified by mNGS in his blood sample. The patient was then treated with an 18-day course of voriconazole, and his symptoms were resolved before discharge. P1, an 80-year-old male, experienced foamy urine for over 6 months and was admitted to the hospital after 10 days of chest pain and shortness of breath. The original CT result was also negative in this case and Pneumocystis jeroveci was detected in blood, sputum, and BALF samples using mNGS. A 12-day course of ganciclovir, tigecycline, meropenem, caspofungin (Cancidas), and sulfamethoxazole tablet resolved his clinical symptoms.
Discussion
The CMT is the most basic tool for growing most microorganisms, but it requires considerable amounts of laboratory equipment, consumables, and time to detect pathogens, which delays the targeted treatment for infections [21]. Considering the rapid turnaround and high sensitivity of mNGS, it may hasten clinical decision-making and guide clinical laboratories to adjust the culture conditions for fastidious or specific microorganisms, which may increase diagnostic and prognostic accuracy and improve treatment efficacy. In our study, all viruses (6/6) and approximately half of the bacteria (29/44) and fungi (5/10) were only detected by mNGS in at least one sample (Fig. 3A), as the culture incubation of some organisms is difficult or even impossible using existing cultivation approaches. In our cohort, a total of 24 samples only presented positive pathogen detection by mNGS, including Legionella pneumophila, Cryptococcus neoformans, Pneumocystis jirovecii, and Aspergillus fumigatus, which were difficult to detect using traditional methods.
We demonstrated a 98.8% sensitivity of mNGS, whereas that of CTs ranged from 47.9% [22] to 92.9% [23] in previous studies. Background interference that is generally from human host DNA is a major factor limiting the sensitivity of mNGS, as it is unbiasedly sequenced with pathogens. Given the influx of immune cells during the infection, the human-to-microbial DNA ratio may be even higher in samples from inflamed or infected sites than in those from healthy sites. The overabundance of human sequencing reads in clinical samples can surpass 90% in sputum [24] and 99.9% in cerebrospinal fluid [25].
The overall rate of pathogen detection using mNGS was significantly higher than the rates achieved using other methods [26], especially for respiratory samples such as BALF and sputum. In previous studies, the positive detection rate using mNGS in BALF was 65%, which was much higher than that of microbiological tests (35%) [27]. For severe and critically ill patients, the positive detection rate obtained using mNGS was 92.3% in BALF and 66.7% in sputum samples [28]. A previous study of pneumocystis pneumonia diagnosis demonstrated that Pneumocystis jirovecii was detected in all BALF, sputum, and blood samples using mNGS, while only 38% (5/13) of the samples were positive using conventional methods [29]. Furthermore, mNGS exhibited a high sensitivity for pathogen detection in BALF samples and can be used to guide clinical practice [30]. However, the difference between the mNGS and CT results for sputum samples was not significant in this study (p > 0.05), which might be due to the small number of negative samples.
Previous studies suggested that mNGS was less likely to be affected by prior antibiotic usage than culture and susceptibility testing [13, 31, 32], as cell-free DNA may remain stable during the first week of treatment and is eliminated from the liver within 2–3 weeks if a favorable treatment outcome is observed [33, 34]. A retrospective review showed that inappropriate initial antimicrobial therapies for the treatment of septic shock occur in about 20% of patients and are associated with a fivefold reduction in survival [35]. The overall concordance rate between mNGS and CT in this study proved that mNGS could be an effective method for clinical pathogen detection, which has been discussed in multiple clinical mNGS studies [13, 36,37,38].
The clinical impacts of mNGS on the diagnosis and treatment of infections were evaluated in this study. Notably, mNGS and CTs showed identical detection results for 124 cases (109 samples with concordant results and 15 double-negative samples), which indicates that mNGS can play an important role in determining the etiologies of infectious diseases. We also observed 15 double-negative cases whose clinical condition was improving without symptoms of an active infection. Such observations can thus help clinicians to appropriately manage antibiotic treatments for patients.
This study exhibits several limitations that warrant consideration. First, the dearth of available samples constrained result validation by real-time PCR for some cases. Secondly, the study predominantly focused on sputum and BALF samples, which leads to a bias towards respiratory infection in the cohort. The inadequate representation of other sites of infection and sample types deserves further attention. Lastly, as it is a single-center study, regional bias and a restricted sample size are unavoidable. Thus, it is imperative that future studies employ larger cohorts and conduct more comprehensive assessments to address these limitations.
Conclusions
In conclusion, we utilized the mNGS assay to detect pathogens in multiple types of clinical samples and compared the results with those from parallel-performed CTs. The results of this comparison suggest that the application of mNGS as a supplementary method for pathogen detection and treatment decision-making should be promoted in clinical practice, although it is necessary to be aware of its additional costs.
References
Wilson MR, Naccache SN, Samayoa E, Biagtan M, Bashir H, Yu G, et al. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N Engl J Med. 2014;370(25):2408–17.
Wilson MR, Suan D, Duggins A, Schubert RD, Khan LM, Sample HA, et al. A novel cause of chronic viral meningoencephalitis: Cache Valley virus. Ann Neurol. 2017;82(1):105–14.
Grumaz S, Stevens P, Grumaz C, Decker SO, Weigand MA, Hofer S, et al. Next-generation sequencing diagnostics of bacteremia in septic patients. Genome Med. 2016;8(1):73.
Simner PJ, Miller S, Carroll KC. Understanding the promises and hurdles of metagenomic next-generation sequencing as a diagnostic tool for infectious diseases. Clin Infect Dis. 2018;66(5):778–88.
Blauwkamp TA, Thair S, Rosen MJ, Blair L, Lindner MS, Vilfan ID, et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat Microbiol. 2019;4(4):663–74.
Schlaberg R, Chiu CY, Miller S, Procop GW, Weinstock G, et al. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Arch Pathol Lab Med. 2017;141(6):776–86.
Miller S, Naccache SN, Samayoa E, Messacar K, Arevalo S, Federman S, et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019;29(5):831–42.
Ivy MI, Thoendel MJ, Jeraldo PR, Greenwood-Quaintance KE, Hanssen AD, Abdel MP, et al. Direct detection and identification of prosthetic joint infection pathogens in synovial fluid by metagenomic shotgun sequencing. J Clin Microbiol. 2018;56(9):e00402-18.
Schlaberg R, Queen K, Simmon K, Tardif K, Stockmann C, Flygare S, et al. Viral pathogen detection by metagenomics and pan-viral group polymerase chain reaction in children with pneumonia lacking identifiable etiology. J Infect Dis. 2017;215(9):1407–15.
Gu W, Deng X, Lee M, Sucu YD, Arevalo S, Stryke D, et al. Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat Med. 2021;27(1):115–24.
Jia X, Hu L, Wu M, Ling Y, Wang W, Lu H, et al. A streamlined clinical metagenomic sequencing protocol for rapid pathogen identification. Sci Rep. 2021;11(1):1–10.
de Vries JJ, Brown JR, Couto N, Beer M, Le Mercier P, Sidorov I, et al. Recommendations for the introduction of metagenomic next-generation sequencing in clinical virology, part ii: bioinformatic analysis and reporting. J Clin Virol. 2021;138:104812.
Miao Q, Ma Y, Wang Q, Pan J, Zhang Y, Jin W, et al. Microbiological diagnostic performance of metagenomic next-generation sequencing when applied to clinical practice. Clin Infect Dis. 2018;67(Suppl 2):S231–40.
Grumaz S, Grumaz C, Vainshtein Y, Stevens P, Glanz K, Decker SO, et al. Enhanced performance of next-generation sequencing diagnostics compared with standard of care microbiological diagnostics in patients suffering from septic shock. Crit Care Med. 2019;47(5):e394–402.
Xing XW, Zhang JT, Ma YB, He MW, Yao GE, Wang W, et al. Metagenomic next-generation sequencing for diagnosis of infectious encephalitis and meningitis: a large, prospective case series of 213 patients. Front Cell Infect Microbiol. 2020;10:88.
Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15(3):R46.
Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20(1):257.
Zeng X, Wu J, Li X, Xiong W, Tang L, Li X, et al. Application of metagenomic next-generation sequencing in the etiological diagnosis of infective endocarditis during the perioperative period of cardiac surgery: a prospective cohort study. Front Cardiovasc Med. 2022;9: 811492.
Liu L, Sun B, Ying W, Liu D, Wang Y, Sun J, et al. Rapid diagnosis of Talaromyces marneffei infection by metagenomic next-generation sequencing technology in a Chinese cohort of inborn errors of immunity. Front Cell Infect Microbiol. 2022;12: 987692.
Qian L, Shi Y, Li F, Wang Y, Ma M, Zhang Y, et al. Metagenomic next-generation sequencing of cerebrospinal fluid for the diagnosis of external ventricular and lumbar drainage-associated ventriculitis and meningitis. Front Microbiol. 2020;11: 596175.
Rajapaksha P, Elbourne A, Gangadoo S, Brown R, Cozzolino D, Chapman J. A review of methods for the detection of pathogenic microorganisms. Analyst. 2019;144(2):396–411.
Shi CL, Han P, Tang PJ, Chen MM, Ye ZJ, Wu MY, et al. Clinical metagenomic sequencing for diagnosis of pulmonary tuberculosis. J Infect. 2020;81(4):567–74.
Blauwkamp TA, Thair S, Rosen MJ, Blair L, Lindner MS, Vilfan ID, et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. 2019;4(4):663–74
Feigelman R, Kahlert CR, Baty F, Rassouli F, Kleiner RL, Kohler P, et al. Sputum DNA sequencing in cystic fibrosis: non-invasive access to the lung microbiome and to pathogen details. Microbiome. 2017;5(1):20.
Salzberg SL, Breitwieser FP, Kumar A, Hao H, Burger P, Rodriguez FJ, et al. Next-generation sequencing in neuropathologic diagnosis of infections of the nervous system. Neurol Neuroimmunol Neuroinflamm. 2016;3(4): e251.
Wu X, Li Y, Zhang M, Li M, Zhang R, Lu X, et al. Etiology of severe community-acquired pneumonia in adults based on metagenomic next-generation sequencing: a prospective multicenter study. Infect Dis Ther. 2020;9(4):1003–15.
Chen H, Yin Y, Gao H, Guo Y, Dong Z, Wang X, et al. Clinical utility of in-house metagenomic next-generation sequencing for the diagnosis of lower respiratory tract infections and analysis of the host immune response. Clin Infect Dis. 2020;71(Suppl 4):S416–26.
Chen S, Zhu Q, Xiao Y, Wu C, Jiang Z, Liu L, et al. Clinical and etiological analysis of co‐infections and secondary infections in COVID‐19 patients: an observational study. Clin Respir J. 2021:15(7):815–25.
Zhang Y, Ai J-W, Cui P, Zhang W-H, Wu H-L, Ye M-Z. A cluster of cases of pneumocystis pneumonia identified by shotgun metagenomics approach. J Infect. 2019;78(2):158–69.
Li Y, Sun B, Tang X, Liu Y-l, He H-Y, Li X-Y, et al. Application of metagenomic next-generation sequencing for bronchoalveolar lavage diagnostics in critically ill patients. Eur J Clin Microbiol Infect Dis. 2020;39(2):369–74.
Rhodes J, Hyder JA, Peruski LF, Fisher C, Jorakate P, Kaewpan A, et al. Antibiotic use in Thailand: quantifying impact on blood culture yield and estimates of pneumococcal bacteremia incidence. Am J Trop Med Hyg. 2010;83(2):301–6.
Zhang XX, Guo LY, Liu LL, Shen A, Feng WY, Huang WH, et al. The diagnostic value of metagenomic next-generation sequencing for identifying Streptococcus pneumoniae in paediatric bacterial meningitis. BMC Infect Dis. 2019;19(1):495.
Hong DK, Blauwkamp TA, Kertesz M, Bercovici S, Truong C, Banaei N. Liquid biopsy for infectious diseases: sequencing of cell-free plasma to detect pathogen DNA in patients with invasive fungal disease. Diagn Microbiol Infect Dis. 2018;92(3):210–3.
Imbert S, Gauthier L, Joly I, Brossas J-Y, Uzunov M, Touafek F, et al. Aspergillus PCR in serum for the diagnosis, follow-up and prognosis of invasive aspergillosis in neutropenic and nonneutropenic patients. Clin Microbiol Infect. 2016;22(6):562.e1–e8.
Kumar A, Ellis P, Arabi Y, Roberts D, Light B, Parrillo JE, et al. Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest. 2009;136(5):1237–48.
Thoendel MJ, Jeraldo PR, Greenwood-Quaintance KE, Yao JZ, Chia N, Hanssen AD, et al. Identification of prosthetic joint infection pathogens using a shotgun metagenomics approach. Clin Infect Dis. 2018;67(9):1333–8.
Zhang HC, Ai JW, Cui P, Zhu YM, Hong-Long W, Li YJ, et al. Incremental value of metagenomic next generation sequencing for the diagnosis of suspected focal infection in adults. J Infect. 2019;79(5):419–25.
Jiang X, Liu Z, Xia Y, Luo J, Xu J, He X, et al. Low FAT4 expression is associated with a poor prognosis in gastric cancer patients. Oncotarget. 2018;9(4):5137–54.
Acknowledgements
Funding
The study was supported by the National Natural Science Foundation of China [No. 82070180], the Jiangsu Provincial Key Research and Development Program [BE2019656], and the Suzhou Science and Technology Planning Project [SZM2021011]. The Rapid Service Fee was funded by the National Natural Science Foundation of China [No. 82070180].
Author Contributions
Jun He designed the study. Jun He and Lin Wang revised the manuscript. Jie Xu, Peng Zhou, and Hailong Fu contributed to the investigation and formal analysis. Jia Liu, Weiwei Wu, Qiuxiang Ou, and Yutong Ma visualized the results and wrote the manuscript. Lina Zhao and Qingzhen Han contributed to data curation and validation. All authors read and approved the final manuscript.
Disclosures
Jia Liu, Weiwei Wu, Qiuxiang Ou, and Yutong Ma are employees of Dinfectome Inc., Nanjing, Jiangsu, China. Jie Xu, Peng Zhou, Lina Zhao, Hailong Fu, Qingzhen Han, Lin Wang, and Jun He have nothing to disclose.
Compliance with Ethics Guidelines
This study was approved by the ethics committee of the First Affiliated Hospital of Soochow University (No. 2018–189) and conducted according to the principles of the Helsinki Declaration. All participants signed informed consent forms for participation and publication.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Xu, J., Zhou, P., Liu, J. et al. Utilizing Metagenomic Next-Generation Sequencing (mNGS) for Rapid Pathogen Identification and to Inform Clinical Decision-Making: Results from a Large Real-World Cohort. Infect Dis Ther 12, 1175–1187 (2023). https://doi.org/10.1007/s40121-023-00790-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-023-00790-5