
Abstract
Introduction
Shigellosis is a major health concern among children < 5 years of age from developing countries, and there are no widely available vaccines to prevent it. The GMMA-based 1790GAHB investigational vaccine against Shigella sonnei was well tolerated and immunogenic in phase 1 and 2 studies conducted in healthy adults from Shigella endemic and non-endemic populations. Based on pooled data of five individual trials, we assessed the association between vaccine administration and the risk of neutropenia as well as the overall safety profile of 1790GAHB.
Methods
The risk ratio (RR) of neutropenia was evaluated between participants receiving 1790GAHB (vaccinees) and active comparator/placebo (controls) using different ethnicity-specific absolute neutrophil count (ANC) thresholds established to define neutropenia. Safety was assessed in terms of solicited, unsolicited, and serious adverse events (AEs).
Results
Of the 279 participants, 11 (5.5%) vaccinees and 4 (5.0%) controls had ANC below the appropriate threshold within 7 days post-vaccination. RR was 0.96 [95% confidence interval (CI) 0.54–1.70]. When neutrophil counts of participants of African descent were measured against an ethnicity non-specific threshold, they resulted in neutropenia episodes in 30 (37.0%) vaccinees and 16 (30.2%) controls, while only 2 (2.5%) vaccinees and 1 (1.9%) control had neutropenia when the ethnicity-specific threshold was applied. RRs were 0.98 (95% CI 0.75–1.28) and 1.30 (95% CI 0.1–17.6), respectively. Solicited and unsolicited AEs were slightly more frequent among vaccinees than controls. No serious AEs, other than neutropenia cases, were recorded in the vaccine group.
Conclusion
By applying the appropriate threshold, no increased risk of neutropenia was identified in vaccinees compared with the controls. The frequency of neutropenia events varied drastically when ethnicity-appropriate thresholds were applied. This observation highlights the importance of selecting appropriate cut-off values according to the correct population reference. Overall, the 1790GAHB vaccine demonstrated an acceptable safety profile.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This is a meta-analysis to assess the overall safety profile of 1790GAHB and its association with risk of neutropenia |
The overall safety profile of 1790GAHB was acceptable, without any safety concern |
Overall, no increased risk of neutropenia was identified for the 1790GAHB vaccine compared with comparator vaccine/placebo within 7 days post-vaccination |
The frequency of neutropenia events depends on the threshold used for the neutropenia definition |
Neutropenia cut-off values need to be selected considering the geographical ancestry of the target population |
Introduction
Shigella is the second leading cause of diarrhea-related mortality globally, after rotavirus, with the highest burden in children < 5 years of age in low- and middle-income countries (LMIC) [1, 2]. There is no widely available vaccine against Shigella, though several candidate vaccines are currently under development [3,4,5,6]. While numerous vaccine types (animal cell culture-based vaccines, whole-cell or subunit vaccines, and recombinant vaccines) and manufacturing processes are well established and widely used, many efforts have recently been devoted to developing new versatile technologies that are able to produce a wide range of low-cost and high-yield vaccines in a relatively short period of time [7]. This would be particularly important for the immunization of pediatric populations from LMIC against several high-burden infectious diseases and to effectively control outbreaks. Besides efficacy and cost-effectiveness, the safety profile of vaccines is a key aspect [8]. Characterization of vaccine safety in clinical development is typically based on the collection of a predefined set of local and systemic solicited adverse events (AEs) for 7 days post-vaccination, unsolicited AEs for approximately 1 month post-vaccination, and serious AEs (SAEs) throughout the study duration. Particularly in the initial trials of a clinical development plan, safety assessment also includes monitoring of hematology/biochemistry parameters. Evaluation of the laboratory safety profile in early development studies is critical to assess whether vaccination has the potential to induce adverse effects on major organ systems in exposed individuals. Additionally, laboratory results from early phase development may also help predicting, appropriately evaluating, and addressing potential early safety signals occurring after licensure, when standardized laboratory testing outside of a controlled clinical trial setting may be less feasible. In fact, the collection of samples and evaluation of laboratory test results are usually challenging in post-marketing settings, particularly in communities with limited access to adequate healthcare or with limited healthcare systems. Neutrophil count is one of the laboratory parameters routinely tested during early vaccine studies. Post-vaccination neutropenia (reduced neutrophil count) has been identified as a common but generally transient and clinically insignificant occurrence in clinical trials conducted with both investigational and licensed vaccines. [9]. Nevertheless, it should be carefully monitored as it is considered a potential warning sign associated with an increased risk for infection. Absolute neutrophil counts (ANCs) are commonly used to define neutropenia and grade its severity. However, ANCs are known to vary by age, sex, and ethnicity [10]. Thus, specific clinical laboratory reference intervals, adapted to the trial setting and population, should be used to avoid misclassification of neutropenia and related safety signals.
The Generalized Modules for Membrane Antigens (GMMA) technology is one of the potential new technologies to design effective and affordable vaccines [7, 11]. The investigational vaccine against Shigella sonnei (1790GAHB) is the first GMMA-based vaccine that has been demonstrated to be well tolerated and immunogenic in healthy adults from both Shigella endemic and non-endemic regions [12,13,14,15]. Although the vaccine was considered to have an acceptable safety profile in terms of local and systemic solicited AEs, unsolicited AEs, and SAEs, eight neutropenia events were recorded during the two initial, parallel, phase 1 studies [12]. Thus, in subsequent studies, neutropenia was monitored as an AE of special interest (AESI) [13,14,15]. To ensure a systematic approach for evaluation of neutropenia events observed during the 1790GAHB vaccine development, we conducted a more comprehensive safety assessment based on a meta-analysis of individual patient safety data.
This article presents an overall evaluation of the safety profile of the 1790GAHB vaccine based on data collected in the five studies performed in healthy adults from endemic and non-endemic populations [12,13,14,15]. A special emphasis was placed on neutropenia and on the implications of applying ethnicity-specific laboratory thresholds for the evaluation of potential laboratory findings in early clinical studies and for safety signal evaluation.
Methods
Study Design and Participants
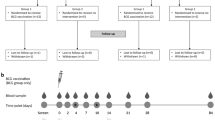
The 1790GAHB vaccine was tested in phase 1 dose escalation studies conducted in France (Study 1) and the UK (Study 2) [12]. In an extension study conducted 2–3 years later, a booster dose was given to some of the participants who completed the three-dose schedule during Study 1 and was compared to a single dose administered to unprimed healthy individuals (Study 3) [13]. The safety and immunogenicity of two doses of 1790GAHB were also evaluated in a phase 2a study in Kenya, where Shigella is endemic (Study 4) [14], and in a phase 2b controlled human infection model study in the US, in which efficacy against moderate to severe diarrhea was also assessed (Study 5) [15]. More details about the study design and number of participants in each trial are presented in Fig. 1. GSK clinical data for each trial were reviewed and combined by pooling the required study data tabulation model (SDTM) domains to create the analysis data model (ADaM) domains. The different versions of SDTMs were harmonized. Participants who received any dosage of 1790GAHB vaccine intramuscularly, intradermally, or intranasally were eligible to be included in the 1790GAHB group. The control group comprised participants who received placebo or active comparator vaccines (the quadrivalent A, C, W, and Y meningococcal vaccine conjugated to a nontoxic mutant of diphtheria toxin [Menveo, GSK] followed by the reduced-antigen-content diphtheria toxoid, tetanus toxoid, and acellular pertussis vaccine [Tdap; Boostrix, GSK]) during the respective studies. Seven participants of Study 1 who also participated in Study 3 were counted twice and analyzed as independent entities because the time interval between the two studies (2–3 years) was sufficiently long to assume that post-booster safety endpoints were associated with booster vaccination and were independent from primary vaccination.

Study design and participants of clinical trials with the S. sonnei 1790GAHB candidate vaccine included in the current analysis (exposed set). OAg O-Antigen. Population set for each analysis was defined according to the evaluated endpoints: neutropenia events were analyzed in the neutropenia safety set, solicited/unsolicited adverse events were analyzed in the solicited/unsolicited safety set, and serious adverse events were analyzed in the exposed set. 1790GAHB study vaccine was administered by intramuscular route. aParticipants were randomized to receive the study vaccine by intradermal, intranasal, or intramuscular route
Approvals from ethics committees and local authorities were obtained for all five studies from which data were used. The present analysis was a post hoc analysis of anonymized data collected in completed trials; thus, ethics or regulatory approvals were not necessary.
Study Objectives
The primary objective of this meta-analysis was to evaluate the impact of different thresholds established to define neutropenia on the association between vaccine administration and the risk of neutropenia. Neutropenia in adults is defined by the US Food and Drug Administration (FDA) as ANC < 2000 cells/µl and by the Division of Acquired Immunodeficiency Syndrome (DAIDS) as ANC < 1000 cells/µl. The secondary objective was to assess the pooled safety and reactogenicity of the 1790GAHB vaccine in terms of solicited AEs, unsolicited AEs, and SAEs.
Statistical Analysis
The primary objective was evaluated in the neutropenia safety set, which included all participants exposed to study interventions and with valid laboratory data on ANC. The number and percentage of participants with neutropenia for 7- and 28-day periods post-each dose and overall were calculated. Neutropenia was defined using what we label as ‘the most appropriate threshold’ according to the geographic ancestry of participants: DAIDS threshold for Black/African American participants and FDA threshold for other than Black/African American participants [16, 17]. The DAIDS grading scale was selected for Black/African American participants because it more appropriately reflects the normal range of ANC in healthy Black populations compared to the FDA scale. It was selected based on the data safety monitoring board (DSMB) recommendation of Study 4 and observations from Karita et al. [18]. Neutropenia events were graded on a 4-grade scale (Table 1). The kappa statistic (κ) was used to quantify the degree of agreement between the two thresholds evaluating neutropenia events/non-events and the two neutropenia grading scales, along with their p-values and confidence intervals (CIs). Risk ratios (RRs) of vaccinated participants reporting neutropenia at least once against participants receiving control were calculated. A generalized linear model was used with the binary distribution and link function log to compute the log-transformed RRs and their corresponding 95% CIs. Treatment group was modeled as fixed effect and study as random effect. Then, values were exponentiated to obtain the RRs and their corresponding 95% CIs. When zero events were observed in any treatment group, the RR was not calculated.
The measures used for continuous endpoints were geometric mean (GM) and geometric mean ratio (GMR, post-vaccination versus baseline) of ANCs, ratio of GM, and ratio of GMR of participants receiving the vaccine versus controls. The ANC values were logarithmically transformed (base 10) to fulfill the normal distribution assumption. GMs and ratio of GMs along with their 95% CIs were obtained from a two-way analysis of variance with factors for treatment group, baseline measurement, interaction of treatment group, and baseline measurement (fixed effect) and study (random effect). Between-group ratio of GMs and ratio of GMRs along with their 95% CIs were obtained from a two-way analysis of covariance with factors for treatment group (fixed effect) and study (random effect). The analysis was performed by dose and overall. For each participant, we accounted for repeated measures with autoregressive correlation structure for the different time points. Measures and their 95% CIs estimated from the models were then exponentiated.
Solicited AEs collected within 7 days after each dose were evaluated in the solicited safety set, which comprised all participants exposed to study interventions and with valid data on solicited AEs. Unsolicited AEs collected within 28 days after each dose were assessed in the unsolicited safety set including all exposed participants who did not withdraw before the subsequent visit and were not lost to follow-up on the subsequent visit. Safety sets were defined on vaccination visit level, i.e., participants who did not receive a vaccine dose at a certain visit were excluded from the corresponding neutropenia/solicited/unsolicited safety set. SAEs within 28 days after each dose were evaluated in all participants who received a study vaccination (exposed set). The number and percentage of participants experiencing solicited or unsolicited AEs were summarized overall and after each dose with their 95% CIs. RRs of vaccinated participants reporting an AE at least once against participants receiving control were calculated using the generalized linear model, as described earlier in this section. The original verbatim terms used by investigators to identify unsolicited AEs were mapped to Preferred Terms (PT) using the Medical Dictionary for Regulatory Activities (MedDRA) version as valid at the time of each separate study. All reported unsolicited AEs, as well as AEs judged by the investigator as causally related to the study vaccine/control, were summarized according to PT within the System Organ Class (SOC). When an AE occurred more than once for a participant, the maximal severity and strongest relationship to the vaccine/control were accounted.
Statistical analyses were performed using SAS (version 9.4) statistical software (SAS Institute Inc., Cary, NC, USA).
Results
In total, 280 participants were included in the current meta-analysis: 199 participants received the 1790GAHB vaccine, 24 received active comparator, and 57 received placebo during the studies (Fig. 1). All participants in Study 1, Study 3, Study 4, and Study 5 were vaccinated intramuscularly. In Study 2, intramuscular route of administration was used in 6 vaccinees and 1 control, intradermal route in 16 vaccinees and 6 controls, and intranasal route in 16 vaccinees and 7 controls. ANC data were available for 279 participants (199 in the 1790GAHB group and 80 in the control group). Demographic characteristics are presented in Table 2.
A total of 11 (5.5%) participants in the 1790GAHB group and 4 (5.0%) participants in the control group had an ANC below the appropriate threshold (DAIDS threshold for Black/African American participants and FDA for other than Black/African American participants) within 7 days after vaccination, the overall RR being 0.96 (95% CI 0.54–1.70) (Fig. 2A). Similarly, no statistically significant differences were observed between groups for the GM and GMR of ANC values (Fig. 2B, C). All neutropenia events were reported at grade 1 intensity. When extending the observational period to 28 days, 16 (8.0%) vaccinees and 4 (5.0%) controls reported ANC below the appropriate threshold, resulting in an RR of 1.44 (95% CI 0.73–2.82). Participants reported all neutropenia events at grade 1 intensity, except for three participants in the 1790GAHB group for whom intensity was grade 2. When ANC thresholds were not adjusted to ethnicity (applying the FDA threshold to all participants), differences between groups were not statistically significant during the 7-day (RR of 0.88 [95% CI 0.59–1.31]) and statistically significant during the 28-day (RR of 0.82 [95% CI 0.71–0.94]) post-vaccination periods (Supplementary Table 1).

Percentage of participants with ANC below the DAIDS threshold for Black/African American and the FDA threshold for other than Black/African American participants (a) and geometric mean (b) and geometric mean ratio (c) of ANC within 7 days after each dose and overall (neutropenia safety set). ANC absolute neutrophil count, RR risk ratio, DAIDS Division of Acquired Immunodeficiency Syndrome, FDA US Food and Drug Administration. aUpper limit is 26.5. Error bars represent 95% confidence intervals. The area of the orange circles is proportional to the values of risk ratios/between-group ratios
Among Black/African American participants, at least one neutropenia event was recorded for 30 (37.0%) vaccinees and 16 (30.2%) controls within 7 days post-vaccination when applying the FDA threshold (Figs. 3A, 4). Based on the FDA definition, grade 3 events were reported by two (2.5%) vaccinees and one (1.9%) control (Fig. 4). When applying the DAIDS threshold, only two (2.5%) vaccinees and one (1.9%) control had ANC below the threshold, all events being of grade 1 intensity (Figs. 3B, 4). Between-group RRs were 0.98 (95% CI: 0.75–1.28) for FDA and 1.30 (95% CI 0.10–17.6) for the DAIDS definition (Fig. 3A, B). Similar results were observed when extending the post-vaccination period to 28 days: 36 (44.4%) vaccinees and 23 (43.4%) controls had at least one neutropenia event by the FDA threshold, which translated to 3 (3.7%) and 1 (1.9%), respectively, by applying the DAIDS threshold. The kappa coefficient (κ = 0.06, p < 0.001) indicated that there was no to slight agreement between the two thresholds on identifying neutropenia events in Black/African American participants.

Percentage of Black/African American participants with ANC below the FDA (a) and DAIDS (b) thresholds and geometric mean (c) and geometric mean ratio (d) of ANC within 7 days after each dose and overall (neutropenia safety set). ANC absolute neutrophil count, RR risk ratio, DAIDS Division of Acquired Immunodeficiency Syndrome, FDA US Food and Drug Administration. *Upper limit is 97.5. **Upper limits are 52.2 (1790GAHB group) and 97.5 (control group). #Upper limits are > 999.9 in both groups. §Upper limits are 17.1 (1790GAHB group) and 6.7 (control group). The area of the orange circles is proportional to the values of risk ratios/between-group ratios

Percentage of Black/African American participants with neutropenia as defined by DAIDS and FDA, within 7 days after each dose and overall (neutropenia safety set). DAIDS Division of Acquired Immunodeficiency Syndrome, FDA US Food and Drug Administration, RR risk ratio of vaccinated participants reporting neutropenia at least once against participants receiving control. The area of the orange circles is proportional to the values of risk ratios/between-group ratios
Within 7 days after any dose, at least one solicited (local and systemic) AE was reported by 182 (91.5%) participants in the 1790GAHB group and 58 (71.6%) participants in the control group (Table 3), the RR being 1.25 (95% CI 1.06–1.47). Solicited AEs of grade 3 intensity were reported by 13 (6.5%) vaccinees and 1 (1.2%) control (Fig. 5). Overall, fatigue was the most frequent solicited systemic AE, reported by 78 (39.2%) participants in the 1790GAHB group and 32 (39.5%) participants in the control group (Supplementary Table 2). Among participants in the vaccine group, the most frequent grade 3 solicited systemic AEs were fatigue (six participants; 3.0%) and headache (four participants; 2.0%). Overall, local pain within 7 days after vaccination was reported by 159 (79.9%) vaccinees and 41 (50.6%) controls. Grade 3 local pain was reported by four (2.0%) participants in the vaccine group.

Summary of solicited adverse events reported within 7 days after each dose and overall (solicited safety set). AE adverse event, RR risk ratio. Solicited AEs collected in the studies were pain, erythema, induration, swelling, facial edema, nasal pain, rhinorrhea (local), and arthralgia, chills, fatigue, headache, malaise, myalgia, and fever (systemic). The area of the orange circles is proportional to the values of risk ratios/between-group ratios
At least one unsolicited AE within 28 days after any dose was reported by 141 (70.9%) participants in the 1790GAHB group and 51 (63.0%) participants in the control group, the RR being 1.17 (95% CI 1.04–1.30). The numbers of participants reporting at least one unsolicited AE considered related or possibly related to study interventions were 71 (35.7%) and 22 (27.2%) in the 1790GAHB and control groups, respectively. Seven (3.5%) vaccinees and 1 (1.2%) control reported an AE at grade 3 intensity (Table 3). Grade 3 AEs considered related or possibly related to study interventions in the 1790GAHB group were two cases of neutropenia, one case each of injection site pain, headache, and myalgia; one case of neutropenia was reported in the control group. During the 28-day post-vaccination period, five (2.5%) participants in the 1790GAHB group and one (1.2%) in the control group reported neutropenia events which were classified as SAEs: these events were considered AESI as per study protocols, and, as such, they were reported as SAEs even if not meeting SAE definition. No other SAEs or fatalities were reported during the 28-day post-vaccination period.
Discussion
Abnormalities in hematological parameters, including ANC, are used to evaluate the eligibility of volunteers to participate in clinical trials and to assess any emerging safety signals during the conduct of the trial. Misclassification of ANC values may affect enrollment and might also lead to incorrect interpretation of the safety profile of a pharmaceutical product [19]. During the conduct of the S. sonnei 1790GAHB vaccine studies, transient and clinically asymptomatic neutropenia events have been observed. These events were mainly reported in participants of African descent. In the phase 2a study (Study 4) conducted in Kenya, the first 18 participants were enrolled using an ANC threshold of 1800 cells/µl (first cohort), while upon recommendation of the DSMB, the remaining 54 participants were screened using the DAIDS threshold of 1000 cells/µl (second cohort). Five participants reported a total of eight neutropenia events in the first cohort compared to one participant with two neutropenia events in the second cohort [14], clearly demonstrating the impact of using ethnicity-specific reference ranges for the definition of neutropenia. Our meta-analysis on pooled individual patient data confirmed this observation, indicating that the number of study participants reporting neutropenia varies drastically when ethnicity-specific thresholds are applied. Thus, neutropenia cut-off values need to be selected according to the correct population. A recent systematic review of literature data showed that individuals developing neutropenia post-vaccination had a significantly lower baseline ANC than those who did not develop neutropenia and were mainly of African descent, who are known to have lower average baseline ANC than other ethnicities [9]. This is in line with our observation and suggests that the definition of normal laboratory ranges for neutropenia in this setting should also consider ethnicity [9, 18]. Ranges derived from other than Black/African American populations to evaluate the safety profile of vaccines in clinical trials performed in people of African descent are suboptimal and may have relevant implications on safety assessments.
By applying the appropriate threshold for each population (i.e., DAIDS for Black/African American and FDA for other than Black/African American), the adjusted RR estimation indicated no statistically significant increased risk of experiencing neutropenia amongst participants who received the S. sonnei 1790GAHB candidate vaccine compared with participants receiving the placebo or the comparators during both the 7- and 28-day post-vaccination periods. The ratio of ANC GM also confirmed this trend.
Solicited and unsolicited AEs were slightly more frequent in the 1790GAHB group than in the control group. However, most participants in the control group received the placebo formulation instead of a marketed comparator vaccine (used only in Study 4); therefore, an increased risk of AE occurrence in vaccinees can be expected. Additionally, based on previous observations from the phase 2a study (Study 4), the reactogenicity profile of the 1790GAHB vaccine tended to be milder than that of Tdap administered as active comparator [14]. More data would be needed to appropriately compare the safety profile of 1790GAHB and marketed vaccines. Overall, the number of participants reporting grade 3 events (both solicited and unsolicited) in the 1790GAHB group was limited, confirming the vaccine’s tolerability.
Except for events of neutropenia that had been predefined as SAEs during the studies due to AESI reporting procedure specified in the protocol, no other SAEs occurred within the 28-day post-vaccination period.
The relatively small sample size is a potential limitation of this analysis. It is acknowledged that, due to their small sample size, early studies can only identify common and acute AEs, while uncommon or rare AEs, or those with delayed onset, can typically only be detected in subsequent larger clinical trials or in post-marketing settings. Another limitation is the difference between 1790GAHB vaccine formulations administered in the individual clinical trials (e.g., different dose levels or routes of administration), which was not considered in this analysis. Nevertheless, only a small proportion of the pooled population received intranasal or intradermal injection [12]. Moreover, the DAIDS threshold is primarily designed for people with human immunodeficiency infection; thus, validation of this definition in the broader population might be warranted. It should also be noted that use of the FDA threshold for some participants other than Black or African American may be more conservative compared to local laboratory normal ranges proposed by study sites (e.g., lower limit of the normal range in France during Study 1 and Study 3 was 1700 cells/µl). Thus, reference ranges should be adjusted to the targeted population.
Despite these limitations, this study used robust data, available from phase 1 and 2 clinical trials, and provided a systematic evaluation of the neutropenia-related events. Results can stimulate the expansion of this methodological approach in future studies to the development of other vaccines and to other biochemical parameters.
Conclusions
No increased risk of neutropenia was detected in adults vaccinated with the investigational S. sonnei 1790GAHB vaccine compared to placebo or other licensed vaccines. Compared to the DAIDS definition, the use of the FDA definition overestimated the occurrence of neutropenia in the Black/African American adult population in all treatment groups. Any cut-off used to establish abnormal values, including neutropenia cut-off, need to be selected according to the correct trial population reference. Standard guidance on hematological and biochemical toxicity grading scales (considering region-specific variability) for trials conducted in specific populations would facilitate vaccine signal detection, safety profile comparisons, and consistency in AE reporting. The safety profile of the GMMA technology and the 1790GAHB vaccine was acceptable in healthy adults from Shigella endemic and non-endemic regions; no major concerns have been identified during early clinical development.
Change history
18 April 2022
A Correction to this paper has been published: https://doi.org/10.1007/s40121-022-00634-8
References
GBD 2016 Diarrhoeal Disease Collaborators. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect Dis. 2018;18:1211–28. https://doi.org/10.1016/S1473-3099(18)30362-1.
Kotloff KL, Riddle MS, Platts-Mills JA, Pavlinac P, Zaidi AKM. Shigellosis. Lancet. 2018;391:801–12. https://doi.org/10.1016/s0140-6736(17)33296-8.
Cohen D, Atsmon J, Artaud C, Meron-Sudai S, Gougeon M-L, Bialik A, et al. Safety and immunogenicity of a synthetic carbohydrate conjugate vaccine against Shigella flexneri 2a in healthy adult volunteers: a phase 1, dose-escalating, single-blind, randomised, placebo-controlled study. Lancet Infect Dis. 2021;21:546–58. https://doi.org/10.1016/S1473-3099(20)30488-6.
Frenck RW Jr, Baqar S, Alexander W, Dickey M, McNeal M, El-Khorazaty J, et al. A Phase I trial to evaluate the safety and immunogenicity of WRSs2 and WRSs3; two live oral candidate vaccines against Shigella sonnei. Vaccine. 2018;36:4880–9. https://doi.org/10.1016/j.vaccine.2018.06.063.
Riddle MS, Kaminski RW, Di Paolo C, Porter CK, Gutierrez RL, Clarkson KA, et al. Safety and immunogenicity of a candidate bioconjugate vaccine against Shigella flexneri 2a administered to healthy adults: a single-blind, randomized phase I study. Clin Vaccine Immunol. 2016;23:908–17. https://doi.org/10.1128/cvi.00224-16.
van der Put RMF, Kim TH, Guerreiro C, Thouron F, Hoogerhout P, Sansonetti PJ, et al. A synthetic carbohydrate conjugate vaccine candidate against shigellosis: improved bioconjugation and impact of alum on immunogenicity. Bioconjug Chem. 2016;27:883–92. https://doi.org/10.1021/acs.bioconjchem.5b00617.
Kis Z, Shattock R, Shah N, Kontoravdi C. Emerging technologies for low-cost, rapid vaccine manufacture. Biotechnol J. 2019;14: e1800376. https://doi.org/10.1002/biot.201800376.
Scholtz M, Duclos P. Immunization safety: a global priority. Bull World Health Organ. 2000;78:153–4.
Muturi-Kioi V, Lewis D, Launay O, Leroux-Roels G, Anemona A, Loulergue P, et al. Neutropenia as an adverse event following vaccination: results from randomized clinical trials in healthy adults and systematic review. PLoS ONE. 2016;11: e0157385. https://doi.org/10.1371/journal.pone.0157385.
Denic S, Narchi H, Al Mekaini LA, Al-Hammadi S, Al Jabri ON, Souid AK. Prevalence of neutropenia in children by nationality. BMC Hematol. 2016;16:15. https://doi.org/10.1186/s12878-016-0054-8.
Micoli F, Alfini R, Di Benedetto R, Necchi F, Schiavo F, Mancini F, et al. GMMA Is a versatile platform to design effective multivalent combination vaccines. Vaccines (Basel). 2020;8:540. https://doi.org/10.3390/vaccines8030540.
Launay O, Lewis DJM, Anemona A, Loulergue P, Leahy J, Sciré AS, et al. Safety profile and immunologic responses of a novel vaccine against Shigella sonnei administered intramuscularly, intradermally and intranasally: results from two parallel randomized phase 1 clinical studies in healthy adult volunteers in Europe. EBioMedicine. 2017;22:164–72. https://doi.org/10.1016/j.ebiom.2017.07.013.
Launay O, Ndiaye AGW, Conti V, Loulergue P, Scire AS, Landre AM, et al. Booster vaccination with GVGH Shigella sonnei 1790GAHB GMMA vaccine compared to single vaccination in unvaccinated healthy European adults: results from a phase 1 clinical trial. Front Immunol. 2019;10:335. https://doi.org/10.3389/fimmu.2019.00335.
Obiero CW, Ndiaye AGW, Sciré AS, Kaunyangi BM, Marchetti E, Gone AM, et al. A phase 2a randomized study to evaluate the safety and immunogenicity of the 1790GAHB generalized modules for membrane antigen vaccine against Shigella sonnei administered intramuscularly to adults from a shigellosis-endemic country. Front Immunol. 2017;8:1884. https://doi.org/10.3389/fimmu.2017.01884.
Frenck RW, Conti V, Ferruzzi P, Ndiaye AGW, Parker S, McNeal MM, et al. Efficacy, safety, and immunogenicity of the Shigella sonnei 1790GAHB GMMA candidate vaccine: results from a phase 2b randomized, placebo-controlled challenge study in adults. EClinicalMedicine. 2021;39: 101076. https://doi.org/10.1016/j.eclinm.2021.101076.
US Food and Drug Administration Guidance for industry: toxicity grading scale for healthy adult and adolescent volunteers enrolled in preventive vaccine clinical trials. 2007 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/toxicity-grading-scale-healthy-adult-and-adolescent-volunteers-enrolled-preventive-vaccine-clinical. Accessed 18 Aug 2021.
US Department of Health and Human Services NIoH, National Institute of Allergy and Infectious Diseases, Division of AIDS, Division of AIDS (DAIDS) Table for grading the severity of adult and pediatric adverse events, Corrected Version 2.1. 2017 https://rsc.niaid.nih.gov/sites/default/files/daidsgradingcorrectedv21.pdf. Accessed 18 Aug 2021.
Karita E, Ketter N, Price MA, Kayitenkore K, Kaleebu P, Nanvubya A, et al. CLSI-derived hematology and biochemistry reference intervals for healthy adults in eastern and southern Africa. PLoS ONE. 2009;4: e4401. https://doi.org/10.1371/journal.pone.0004401.
Koen A, Jose L, Madhi SA, Fix A, Cryz S, Groome MJ. Neutrophil counts in healthy South African infants: implications for enrollment and adverse event grading in clinical trials in an African setting. J Pediatr X. 2019;1: 100005. https://doi.org/10.1016/j.ympdx.2019.100005.
Acknowledgements
The contribution of study participants and the current and former GVGH staff members is gratefully acknowledged.
Funding
This analysis was funded by GlaxoSmithKline Biologicals SA. GlaxoSmithKline Biologicals SA also took responsibility for all costs associated with the development of the present manuscript, including the journal’s Rapid Service Fees.
Medical Writing and Editorial Assistance
The authors thank Kishor Mariyala, Sateesh Aravapalli, and Rob Mulder for providing statistical programming activities. The authors thank Modis for editorial assistance and manuscript coordination, on behalf of GSK. Botond Nagy provided medical writing support and Elise Verplancke coordinated the manuscript development.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Iris De Ryck, Eleanna Sarakinou, Usman Nakakana, Giulia Luna Cilio, Venanzio Vella, Joachim Auerbach, Valentino Conti, Audino Podda, and Augustin Ndiaye were involved in the study conception and design and in data analysis and data interpretation. Iris De Ryck, Eleanna Sarakinou, Usman Nakakana, Joachim Auerbach, and Audino Podda were involved in acquisition and generation of data. Eleanna Sarakinou and Audino Podda performed the study. All authors contributed substantially to the development of the manuscript and approved the final version.
Disclosures
Iris De Ryck, Eleanna Sarakinou, Usman Nakakana, Giulia Luna Cilio, Augustin Ndiaye, Valentino Conti, and Audino Podda are employees of GSK group of companies. Joachim Auerbach was an employee of the GSK group of companies at the time of this study and is now affiliated with Coalition for Epidemic Preparedness Innovations, London, UK. Iris De Ryck, Venanzio Vella, Joachim Auerbach, Valentino Conti, and Audino Podda hold shares in the GSK group of companies. The authors declare no other non-financial relationships and activities or conflicts of interest.
Compliance with Ethics Guidelines
Approvals from ethics committees and local authorities were obtained for all five studies from which data were used. The present analysis was a post hoc analysis of anonymized data collected in completed trials, thus ethics or regulatory approvals were not necessary.
Data Availability
GSK makes available the anonymized individual participant data and associated documents from interventional clinical studies which evaluate medicines, upon approval of proposals submitted to https://www.clinicalstudydatarequest.com. To access data for other types of GSK sponsored research, for study documents without patient-level data and for clinical studies not listed, please submit an enquiry via the website.
Trademark Statement
Menveo and Boostrix are trademarks owned by or licensed to the GSK group of companies.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original article was revised due to an error in copyright line
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
De Ryck, I., Sarakinou, E., Nakakana, U. et al. GMMA Technology for the Development of Safe Vaccines: Meta-Analysis of Individual Patient Data to Assess the Safety Profile of Shigella sonnei 1790GAHB Vaccine in Healthy Adults, with Special Focus on Neutropenia. Infect Dis Ther 11, 757–770 (2022). https://doi.org/10.1007/s40121-022-00596-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-022-00596-x