
Abstract
Objective
To assess the pharmacokinetics and safety of liquid palivizumab compared with lyophilized palivizumab.
Methods
This phase 2, randomized, double-blind crossover study included premature infants aged ≤6 months born ≤35 weeks gestational age. Patients were randomized in a 1:1 ratio to receive a single 15 mg/kg intramuscular dose of liquid (sequence A group) or lyophilized (sequence B group) palivizumab on Day 0. Patients crossed over to receive the alternate formulation on Day 30. Serum palivizumab and antidrug antibody (ADA) levels were measured on Day 0 (predose), Day 30 (before the dose of alternate formulation), and Day 60 (30 days after the dose of alternate formulation). Patients were followed for safety through Day 60 (30 days after the dose of alternate formulation).
Results
A total of 153 infants were randomized into the study (sequence A 75; sequence B 78). Sequence A and sequence B trough serum palivizumab levels were similar on Day 30 (51.7 and 49.1 µg/mL, respectively) and Day 60 (84.8 and 87.2 µg/mL, respectively). The ratio of the geometric means using both Day 30 and Day 60 serum concentrations was 1.040 (90% CI 0.998–1.083), which was within the prespecified bioequivalence range of 0.8–1.25. Adverse events (AEs) were similar between the palivizumab liquid and lyophilized groups and within each treatment sequence. Serious AEs (SAEs) were experienced by 3% of infants in both liquid palivizumab and lyophilized palivizumab groups. None of the SAEs were determined to be related to study drug. Among the 124 infants (81% of total) evaluated for ADA, 2 (1.6%) tested positive for ADA at Day 60 (1 in each of sequence A and B).
Conclusion
Liquid and lyophilized formulations of palivizumab were bioequivalent with similar safety profiles in infants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Respiratory syncytial virus (RSV) is a viral pathogen that is a common cause of acute lower respiratory infection in infants and children worldwide [1]. By 2 years of age, most children have been infected with RSV at least once [2]. The risk for severe RSV is particularly high in children who were born prematurely, or who have congenital heart disease (CHD), or chronic lung disease of prematurity (CLDP) [2]. In 2005, it was estimated that there were 33.8 million cases of RSV-associated acute lower respiratory infection worldwide in children <5 years of age [1]. Of these, 3.4 million were severe and required hospitalization, and 66,000–199,000 cases resulted in death [1].
Respiratory syncytial virus immune globulin (RespiGam, MedImmune, Gaithersburg, MD, USA) was the first RSV-specific prevention used in high-risk infants [3]. Although this therapy was effective, it required several hours of an intravenous (IV) infusion [3]. Palivizumab (MedImmune, Gaithersburg, MD, USA), an F-protein–specific humanized monoclonal antibody, was the first monoclonal antibody developed to prevent an infectious disease and represented an advance in RSV prevention. Palivizumab was approved by the Food and Drug Administration (FDA) in 1998 and is indicated for the prevention of serious lower respiratory tract disease caused by RSV in children at high risk for RSV disease [3]. In addition, palivizumab offers a more convenient, intramuscular (IM), route of administration [4]. Palivizumab was first manufactured as a lyophilized powder [5]. Lyophilized palivizumab is administered at a dose of 15 mg/kg IM monthly throughout the RSV season [4]. The efficacy of lyophilized palivizumab is well established; results from three randomized, placebo-controlled studies have shown that palivizumab resulted in a 45–82% reduction in RSV hospitalizations [6–8]. However, lyophilized palivizumab requires reconstitution with sterile water, which takes approximately 20 min, and the drug must be used within 6 h of reconstitution [5]. A liquid formulation of palivizumab bypasses the need for reconstitution and simplifies the administration of palivizumab. This liquid formulation was approved by the FDA in 2004 based, in part, on the results of this study [5], and has been the only formulation distributed in the United States since 2006. The reduced time required for administration of liquid versus lyophilized palivizumab has additional clinical relevance in that it enables high-risk infants to spend less time in a healthcare provider’s waiting room, thereby decreasing their exposure to other sick children visiting the pediatrician. Single-dose pharmacokinetics (PK) and safety profile of the liquid formulation of palivizumab administered IV and IM were first tested and compared with the lyophilized formulation in 53 healthy adult volunteers. In this parallel cohort study, liquid palivizumab demonstrated PK and safety profiles similar to those of lyophilized palivizumab [9].
The goal of the current study was to establish bioequivalence and safety of the liquid formulation in a pediatric population for whom palivizumab is approved. This randomized, crossover study assessed the PK, safety, and tolerability of the liquid formulation compared with the lyophilized formulation of palivizumab in infants ≤6 months of age who were born ≤35 weeks gestational age.
Methods
Patients
Infants aged ≤6 months who were born ≤35 weeks gestational age were included in the study. Infants were excluded if they were hospitalized or receiving mechanical ventilation at the time of study entry; had CLDP, CHD, renal impairment, hepatic dysfunction, chronic seizure disorder, or immunodeficiency; had acute illness or progressive clinical disorder; had a history of recent difficult venous access; had an active infection; or had ever received palivizumab.
Study approval was obtained by appropriate institutional review board(s) (Supplementary Appendix) before initiation. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Written informed consent was obtained from each study patient’s parent or legal guardian before study entry or any study procedures were performed.
Study Drug
Liquid palivizumab was provided in 3-mL vials containing 100 mg/mL of sterile liquid product in a volume of 1.2 mL (25 mM histidine, 1.6 mM glycine, at pH 6.0). Lyophilized palivizumab was provided in 5-mL vials containing 100 mg of sterile lyophilized product (25 mM histidine, 1.6 mM glycine, and 3% w/v mannitol at pH 6.0, before lyophilization) that yielded palivizumab at a concentration of 100 mg/mL after reconstitution with 1 mL of sterile water for injection. Study medications were administered at a dose of 15 mg/kg IM.
Study Design
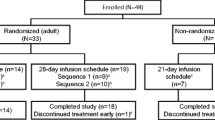
This phase 2, randomized, double-blind, 2-period crossover study (NCT00240929) was conducted at 21 sites in the United States. Patients were randomized from November 2002 through February 2003 in a 1:1 ratio to either the sequence A group or the sequence B group (Fig. 1). Study medication was supplied to the pharmacy as open-labeled lyophilized or liquid palivizumab. The pharmacist prepared the study medication with the appropriate formulation, and the medication was dispensed from the pharmacy in identical syringes. The investigators, study nurses, and patient’s parents were blinded to the study medication. Patients received a single 15 mg/kg IM dose of liquid (sequence A) or lyophilized (sequence B) palivizumab on Day 0. Patients crossed over to receive the alternate formulation on Day 30. The design and endpoints of the trial were finalized following regulatory input. After study completion (i.e., after patients had received 2 doses of study drug), investigators were free to give patients additional doses of commercially available lyophilized palivizumab for the remainder of the RSV season. Liquid palivizumab was not commercially available at the time of the study and was used only in the experimental setting.

Study design. PK pharmacokinetics
Pharmacokinetics
Bioequivalence was assessed by comparing the relative bioavailability of the two palivizumab formulations. Because the indicated patient population for palivizumab consists of preterm infants from whom only limited numbers of blood samples and total blood volume could be collected, the standard bioequivalence parameters of maximum concentration (C max) and area under the curve (AUC), which require a full PK profile, could not be used to establish bioequivalence. Serum samples for determining palivizumab concentrations were collected at baseline, on study Day 30 (before the dose of alternate formulation), and study Day 60 (30 days after the dose of alternate formulation). Serum samples were analyzed at a central laboratory (PPD Development, Richmond, VA, USA) to determine palivizumab concentration using a validated enzyme-linked immunosorbent assay (ELISA) [10]. The ELISA used for measuring serum palivizumab concentration was developed by MedImmune and provides linear results in the range 10–750 µg/mL. Patient samples with concentrations below the lower limit of quantitation (LLOQ) of the assay of 10 µg/mL were reported as <LOQ. Results were analyzed using Softmax Pro 2.2.1 software (Molecular Devices Corporation, Sunnyvale, CA, USA).
Safety
Adverse events (AEs) were monitored for 30 days after each study drug injection. AEs and serious AEs (SAEs) were graded by severity (level 1–4) and relationship to study drug (none, remote, possible, probable, or definite) by the study investigator. AEs were considered related to the study medication (as determined by the blinded site investigator) if they were reported as possibly, probably, or definitely related. SAEs included those that resulted in death; were life-threatening; or resulted in inpatient hospitalization or prolongation of existing hospitalization [11].
Antidrug Antibodies
Serum samples for determining antidrug antibodies (ADA) to palivizumab from patients who had previously consented to additional testing were collected at baseline and on study Days 30 and 60. ADA assays were performed with the qualified assay described in previous studies [10]. Patient serum samples and negative and positive controls were diluted 1:10 before performing the assay. Solutions were incubated for 1 h, and anti-palivizumab immunoglobulin was detected with horseradish peroxidase-conjugated palivizumab. A titer >1:10 was considered positive for ADA. Results were analyzed using Softmax Pro 3.2.1 software (Molecular Device Corporation).
Statistical Analyses
The PK analysis included all patients receiving two full doses of study drug, without major protocol violations, and for whom the study visits occurred within the specified visit windows. All patients who received study drug were included in the safety summaries.
An enrollment goal of 60 per sequence group was initially calculated based on estimates of between-patient and within-patient variability obtained from previous studies, and was calculated to yield an 80% chance that the 90% confidence interval (CI) of the within-patient standard deviations (SD) of the log trough concentration would be between 0.80 and 1.25. The sample size was increased to 75 because of potential difficulties in obtaining adequate blood samples from infants, and to allow for withdrawals and dropouts in the study.
An analysis of variance (ANOVA) was performed on log-transformed 30-day postdose serum concentrations, which included both Day 30 and Day 60 trough serum concentrations due to the crossover design. The model accounted for all sources of variation that could be reasonably assumed to have an effect on the serum concentration, namely, sequence, patient, formulation, and period. The geometric means of the trough concentration (C trough) for each formulation, the ratio of the geometric means (with the lyophilized formulation as the reference group) and the associated 90% CI were estimated. If the 90% CI for the ratio of the geometric means of the C trough was within 0.8–1.25 of the statistical interval, the two formulations would be considered bioequivalent. The CI was to be constructed only if a significant sequence effect was not demonstrated. If a significant sequence effect was demonstrated, a 90% CI using only the Day 30 data was to be constructed. This additional CI is reported here for convenience.
The incidence of AEs was summarized separately for liquid and lyophilized palivizumab during each of the 3 study periods: study Days 0–30 (i.e., from after the first injection through 30 days or up to the date of the second injection), study Days 30–60 (i.e., from after the second injection through 30 days), and study Days 0–60 (i.e., through the respective 30-day follow-up periods for patients receiving study drug at study Day 0, study Day 30, or both).
Results
Patients
A total of 153 patients were randomized into the study (75 to sequence A, 78 to sequence B), and 152 completed the study through 30 days after the second injection of the study drug. One infant in the sequence B group did not complete the study as their parents withdrew consent after the first study drug injection. Generally, demographic and baseline characteristics were similar between the sequence A and sequence B groups (Table 1).
Pharmacokinetics
The trough serum palivizumab levels on study Days 30 and 60 were similar in both sequence groups. The geometric mean trough serum palivizumab levels obtained on study Day 30 were 51.7 µg/mL in the sequence A group (30 days after receipt of liquid palivizumab) and 49.1 µg/mL in the sequence B group (30 days after receipt of lyophilized palivizumab), reflecting similarity in trough concentrations following liquid and lyophilized formulations, respectively (Table 2). The arithmetic mean (SD) levels obtained on study Day 30 were 54.8 (20.1) µg/mL in the sequence A group and 51.6 (17.5) µg/mL in the sequence B group. On study Day 60, the geometric mean trough serum palivizumab levels were 84.8 µg/mL with sequence A and 87.2 µg/mL with sequence B, and the arithmetic mean (SD) levels were 88.6 (27.0) µg/mL with sequence A and 90.9 (26.7) µg/mL with sequence B. The trough levels increased for all patients in sequence A, and in all but three patients in sequence B, from study Day 30 to study Day 60; mean (SD) trough levels are shown in Fig. 2. Using an ANOVA model, the ratio of the geometric means of the C trough for each formulation was 1.040 and its associated 90% CI was 0.998–1.083, which was within the bioequivalence limits of 0.8–1.25 (Table 2). The results of the ANOVA detected a significant period effect, as expected, due to accumulation of concentrations after the second dose; however, the ANOVA did not detect a statistically significant formulation or sequence effect, indicating no evidence of a differential carryover effect between the 2 sequences. Analyzed as a parallel study (Table 2), the ratio of the geometric means of the Day 30 C trough was 1.051 (90% CI 0.963–1.148), which was also within the bioequivalence limits of 0.8–1.25.

Mean (SD) trough serum palivizumab concentrations on Day 30 and Day 60 (30 days postdose)
Safety
Overall, 50.0% (76/152) of infants receiving liquid palivizumab and 49.0% (75/153) receiving lyophilized palivizumab experienced ≥1 AE from study Days 0–60. The AEs were generally similar between the two groups, and the most common AEs overall were typical for the population of infants ≤6 months of age with a history of prematurity (Table 3). The proportion of infants with ≥1 AE reported during each of the Day 0–30 and 30–60 study periods was similar between the liquid and lyophilized palivizumab groups (Table 4) and within each treatment sequence. The number of infants with ≥1 AE was slightly higher after the first injection than after the second injection for both sequence A and sequence B. Most AEs were level 1 or 2 in severity. Only 4 AEs in the liquid palivizumab group (fever, injection site reaction; n = 2 each) and 2 AEs in the lyophilized palivizumab group (injection site reaction, pneumonia) were considered to be related to study medication. No deaths occurred during the study. From study Days 0–60, the number of SAEs was the same for each group (n = 5), representing 5 (3.3%) and 4 (2.6%) infants in the liquid and lyophilized palivizumab groups, respectively (Table 5). Generally, SAEs were similar between the liquid and lyophilized palivizumab groups and were also similar after each injection in both the sequence A and B groups (Table 5). None of the SAEs were considered to be related to palivizumab.
Antidrug Antibodies
ADAs were assessed in 81.3% (61/75) of infants in sequence A and 83.3% (65/78) of infants in sequence B. Of these, 1.6% (2/126) tested positive for ADA (defined as a titer ≥1:10). One infant in sequence A (1/61; 1.6%) had a titer of 1:40 at study Day 60 and another infant in sequence B (1/65; 1.5%) had a titer of 1:10 at study Day 60.
Discussion
Similarity in PK and safety of liquid and lyophilized formulations after 3 mg/kg administered IM and 15 mg/kg administered IV was first assessed in healthy adult volunteers in a double-blinded fashion [9]. Liquid and lyophilized palivizumab had similar safety profiles, with no serious AEs reported [9]. Both formulations of palivizumab also exhibited similar PK profiles, with mean [standard error (SE)] C max of 32.6 (2.35) µg/mL and 29.6 (2.84) µg/mL, and mean (SE) AUC of 890 (111.5) µg × day/mL and 844 (119.9) µg × day/mL in the 3 mg/kg IM liquid and lyophilized groups, respectively, and mean (SE) C max of 502.5 (35.89) µg/mL and 585 (32.40) µg/mL, and mean (SE) AUC of 6,673 (749.2) µg × day/mL and 6,310 (414.2) µg × day/mL in the 15 mg/kg IV liquid and lyophilized groups, respectively [9].
Based on the similarity in palivizumab PK between the two formulations in the exploratory study in adults, our current study was designed to evaluate bioequivalence with respect to serum concentrations and to assess similarity in safety between liquid and lyophilized formulations in children at high risk for RSV disease. A crossover design was used since this allows for the assessment of bioequivalence while minimizing the impact of subject-to-subject variability on the results of the analysis. Given that palivizumab is administered monthly in infants, serum trough concentrations were used to assess similarity in palivizumab exposures between lyophilized and liquid formulations in a crossover fashion.
An analysis of palivizumab PK across 22 studies (including data from the current study) [12] showed that body weight impacts palivizumab exposure, and gestational age only has a minimal effect on palivizumab concentrations. Across a range of gestational and postnatal ages, palivizumab serum concentrations were similar, suggesting the appropriateness of body weight dosing [12]. Moreover, palivizumab 15 mg/kg demonstrated comparable therapeutic concentrations in patients with different demographic characteristics [12].
Findings from this study of infants aged ≤6 months with a history of prematurity of ≤35 weeks gestation showed that liquid and lyophilized palivizumab formulations exhibit comparable PK and similar safety profiles. Similar trough palivizumab levels were observed between the two sequence groups and the 90% CIs for the ratios of least squares geometric means of trough levels for the two formulations were within the 90% confidence limits for bioequivalence.
The half-life of palivizumab is approximately 20 days; palivizumab accumulation is expected upon redosing on Day 30 based on the frequency of dosing compared with the half-life. The trough serum palivizumab levels in the present study were generally consistent with the levels previously reported from the randomized, double-blind, placebo-controlled efficacy study in infants ≤35 weeks gestation and <6 months of age or ≤2 years of age with CLDP, in which mean trough serum levels through 30 days after the first and second doses were 37 and 57 µg/mL, respectively, after 15 mg/kg of lyophilized palivizumab [8]. Serum levels from a phase 2, randomized, double-blind study of liquid palivizumab in premature infants with and without CLDP showed a mean trough serum palivizumab level of 87.4 µg/mL on Day 60 [13]. It is difficult to compare absolute levels between studies, given the subtle differences in assays or study conduct, which could lead to different results. However, all studies have shown higher values at Day 60 relative to Day 30 due to accumulation of palivizumab with repeated dosing.
Liquid palivizumab has been used in the United States since its FDA approval in 2004 and has demonstrated a similar safety profile to lyophilized palivizumab in the postmarketing setting. In the current study, a similar incidence of AEs was observed regardless of palivizumab formulation or the sequence of palivizumab administration. The incidence of SAEs was also similar between the liquid and lyophilized palivizumab groups. The AEs considered to be related to the study medications in the present study were consistent with those reported in a larger study with lyophilized palivizumab, including fever (2.8%), nervousness (2.5%), and injection site reaction (2.3%) [8]. Additionally, the AEs reported in the present study were generally consistent with those reported in a large (n = 3,306) study of infants who received liquid palivizumab [14]. The most commonly reported AEs in that study were upper respiratory tract infection (30.1%), pyrexia (16.9%), and rhinitis (13.5%) [14]. Overall, the reported AEs and SAEs were generally as expected for the studied population of infants ≤6 months of age with a history of prematurity.
The crossover study design, a strength of this study, allowed for the evaluation of the safety of the sequential exposure of one palivizumab formulation to another. One of the limitations of the study is that there was no washout period. A drug washout period of approximately 3 months would have potentially placed these infants at risk during the RSV season. This crossover design, with the alternative formulation administered at the next scheduled monthly dosing period, was also felt to mimic a potential clinical situation where infants receiving lyophilized palivizumab could be switched to the liquid formulation within a single RSV season. The ANOVA demonstrated no significant differential carryover effect. Moreover, the geometric mean ratio and associated 90% CI constructed after a single dose using Day 30 C trough data alone were within the bioequivalence limits, supporting the bioequivalence conclusion in the absence of a washout period in this study.
In conclusion, findings from this study show comparable PK profiles of the liquid and lyophilized formulations of palivizumab in infants, supporting the bioequivalence of the two formulations. The safety profiles were also similar between the liquid and lyophilized formulations of palivizumab in the studied pediatric population.
References
Nair H, Nokes DJ, Gessner BD, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet. 2010;375:1545–55.
Committee on Infectious Diseases. From the American Academy of Pediatrics: policy statements–modified recommendations for use of palivizumab for prevention of respiratory syncytial virus infections. Pediatrics. 2009;124:1694–701.
Meissner HC, Long SS, American Academy of Pediatrics Committee on Infectious Diseases and Committee on Fetus and Newborn. Revised indications for the use of palivizumab and respiratory syncytial virus immune globulin intravenous for the prevention of respiratory syncytial virus infections. Pediatrics. 2003;112:1447–52.
American Academy of Pediatrics Committee on Infectious Diseases and Committee on Fetus and Newborn. Revised indications for the use of palivizumab and respiratory syncytial virus immune globulin intravenous for the prevention of respiratory syncytial virus infections. Pediatrics. 2003;112:1442–6.
Mejias A, Chavez-Bueno S, Sanchez PJ. Respiratory syncytial virus prophylaxis. Neoreviews. 2005;6:e26–31.
Blanken MO, Rovers MM, Molenaar JM, et al. Respiratory syncytial virus and recurrent wheeze in healthy preterm infants. N Engl J Med. 2013;368:1791–9.
Feltes TF, Cabalka AK, Meissner HC, et al. Palivizumab prophylaxis reduces hospitalization due to respiratory syncytial virus in young children with hemodynamically significant congenital heart disease. J Pediatr. 2003;143:532–40.
The IMpact-RSV Study Group. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics. 1998;102:531–7.
Data on File—Study MI-CP080. MedImmune, LLC. Gaithersburg.
Subramanian KN, Weisman LE, Rhodes T, et al. Safety, tolerance and pharmacokinetics of a humanized monoclonal antibody to respiratory syncytial virus in premature infants and infants with bronchopulmonary dysplasia. MEDI-493 Study Group. Pediatr Infect Dis J. 1998;17:110–5.
Abarca K, Jung E, Fernandez P, et al. Safety, tolerability, pharmacokinetics, and immunogenicity of motavizumab, a humanized, enhanced-potency monoclonal antibody for the prevention of respiratory syncytial virus infection in at-risk children. Pediatr Infect Dis J. 2009;28:267–72.
Robbie GJ, Zhao L, Mondick J, Losonsky G, Roskos LK. Population pharmacokinetics of palivizumab, a humanized anti-respiratory syncytial virus monoclonal antibody, in adults and children. Antimicrob Agents Chemother. 2012;56:4927–36.
Fernandez P, Trenholme A, Abarca K, et al. A phase 2, randomized, double-blind safety and pharmacokinetic assessment of respiratory syncytial virus (RSV) prophylaxis with motavizumab and palivizumab administered in the same season. BMC Pediatr. 2010;10:38.
Carbonell-Estrany X, Simoes EA, Dagan R, et al. Motavizumab for prophylaxis of respiratory syncytial virus in high-risk children: a noninferiority trial. Pediatrics. 2010;125:e35–51.
Acknowledgments
This study and article publication charges were supported by MedImmune (Gaithersburg, MD, USA). All named authors meet the ICMJE criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published. Medical writing assistance was provided by John E. Fincke, PhD, and Anny Wu, PharmD, of Complete Healthcare Communications, Inc., (Chadds Ford, PA, USA) and was funded by MedImmune.
Conflict of interest
Gabriel Robbie is an employee of MedImmune. Doris Makari is an employee of MedImmune. Brian Harris is an employee of MedImmune. Hasan Jafri is an employee of MedImmune. MedImmune employees may hold stock or stock options in AstraZeneca. Genevieve Losonsky was an employee of MedImmune at the time of study conduct and analysis.
Compliance with ethics guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Written informed consent was obtained from each study patient’s parent or legal guardian before study entry or any study procedures were performed.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Trial registration: ClinicalTrials.gov #NCT00240929.
Genevieve A. Losonsky: Formerly affiliated with MedImmune
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Robbie, G.J., Makari, D., Harris, B. et al. Randomized, Double-Blind Study of the Pharmacokinetics and Safety of Palivizumab Liquid Formulation Compared with Lyophilized Formulation. Infect Dis Ther 3, 203–214 (2014). https://doi.org/10.1007/s40121-014-0042-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-014-0042-x