
Abstract
Introduction
The efficacy of natalizumab was evaluated in Japanese patients with relapsing-remitting multiple sclerosis (RRMS) in a 24-week, phase 2 bridging study. An open-label, 2-year extension study from this trial was conducted to assess the safety and efficacy of natalizumab treatment in Japanese patients.
Methods
A total of 97 patients (43 previously on placebo; 54 previously on natalizumab) who had completed the bridging study were treated with 300 mg natalizumab every 4 weeks. Multiple sclerosis relapses, changes in Expanded Disability Status Scale (EDSS) scores, and adverse events were assessed at regular intervals. Anti-natalizumab and anti-JC virus (JCV) antibodies were measured.
Results
After 2 years of natalizumab treatment, the mean adjusted annualized relapse rate was 0.30 (95% confidence interval [CI]: 0.18–0.52) among previously-on-placebo patients and 0.13 (95% CI: 0.05–0.29) among previously-on-natalizumab patients. The mean change in EDSS score from baseline to week 120 was −0.03 among previously-on-placebo patients and −0.18 among previously-on-natalizumab patients. In both groups, >90% of patients experienced ≥1 adverse event. Two previously-on-placebo patients developed persistently positive anti-natalizumab antibodies. Approximately 65% of all patients tested positive for anti-JCV antibodies at open-label treatment initiation. No deaths or progressive multifocal leukoencephalopathy cases were reported.
Conclusions
The efficacy and safety findings from this 2-year open-label extension study are comparable to and confirm the results of other clinical trials of natalizumab conducted in non-Asian patient populations, and provide longer-term evidence of efficacy and safety in Japanese patients.
Trial registration
ClinicalTrials.gov identifier NCT01416155.
Funding
Biogen.
Similar content being viewed by others


Introduction
Natalizumab, a humanized anti-α4 integrin monoclonal antibody, has demonstrated efficacy in reducing relapse rates and disability worsening over 2 years among patients with relapsing-remitting multiple sclerosis (RRMS) in phase 3 studies that enrolled predominantly non-Asian patient populations [1, 2]. Results from the observational open-label Safety of TYSABRI Re-dosing and Treatment (STRATA) study showed that RRMS patients from the phase 3 clinical trials experienced continued reductions in annualized relapse rate (ARR), stability of Expanded Disability Status Scale (EDSS) scores, and low rates of confirmed EDSS worsening with natalizumab treatment after 4–5 years of follow-up [3]. These results have been supported by results from studies conducted in the clinical practice setting [4,5,6,7].
More recently, the safety and efficacy of natalizumab in Japanese patients with RRMS was demonstrated in a 24-week phase 2 study [8]. After 24 weeks of double-blind treatment, the adjusted ARR was significantly lower in natalizumab- than placebo-treated patients (0.53 vs. 1.73, P < 0.001), and a significantly larger percentage of natalizumab- than placebo-treated patients were relapse-free (79% vs. 38%, P < 0.001). From baseline to week 24, the mean EDSS score improved in the natalizumab group and worsened in the placebo group (change of −0.22 vs. 0.19, P = 0.019).
Here, we report results from the 2-year open-label extension study of patients who completed the phase 2 bridging study. The primary objective of this study was to evaluate the 2-year safety and tolerability of natalizumab in Japanese patients with RRMS. The secondary objective was to evaluate the 2-year efficacy of natalizumab in this patient population.
Methods
Study Design and Patient Population
This study was a 2-year open-label extension study of patients who successfully completed part A or part B of the phase 2 bridging study [8]. Briefly, Japanese patients (aged 18–65 years) were eligible for the phase 2 bridging study if, prior to enrollment, they had a diagnosis of RRMS as defined by the revised McDonald criteria [9], ≥1 exacerbation within the previous year, and an EDSS score [10] between 0.0 and 6.0 (inclusive) for part A and between 0.0 and 5.5 (inclusive) for part B. The use of concomitant immunosuppressive or immunomodulatory treatment (including interferon beta and long-term systemic corticosteroids) was not permitted at any time during the bridging or extension study; however, steroids were permitted in each study to treat on-study relapses.
In part A of the open-label bridging study, 12 patients received 300 mg natalizumab administered intravenously (IV) every 4 weeks for 24 weeks to assess pharmacodynamics and pharmacokinetics. Part B was a placebo-controlled double-blind study in which 94 patients were randomized (1:1) to receive 300 mg IV natalizumab or placebo every 4 weeks for 24 weeks, and the ARR and proportion of relapse-free patients were assessed. EDSS scores and changes from baseline were analyzed, although these variables were not pre-specified endpoints. The current study focused on the longer-term follow-up of subjects enrolled in parts A and B. To avoid potential bias, the assessment of efficacy included only the population randomized to blinded treatment in part B, while safety assessments included patients enrolled in both parts A and B.
Compliance with Ethics Guidelines
The study protocol was approved by the ethics committees at each participating site, and all patients provided written informed consent. The study was conducted in accordance with the International Conference on Harmonisation Guideline for Good Clinical Practice and the ethical principles outlined in the Declaration of Helsinki of 1964, as revised in 2013.
Assessment of Efficacy
For the open-label extension, clinical relapses were defined as new or recurrent neurological symptoms not associated with fever or infection and lasting for ≥24 h. Relapses were assessed throughout the 2 years of the extension study. EDSS scores were recorded by the examining neurologist, and because the EDSS was assessed every 12 weeks, the EDSS scores presented in this analysis were collected at week 120 of the extension study.
Assessment of Safety and Tolerability
For the open-label extension, physical examinations were performed and vital signs were measured every 4 weeks, and neurological examinations and laboratory testing were conducted every 12 weeks. A 12-lead electrocardiogram was performed at week 24. Blood samples were taken at baseline and every 12 weeks thereafter to assess serum anti-natalizumab antibodies using a bridging enzyme-linked immunosorbent assay (ELISA) [11]. Anti-JC virus (JCV) antibody testing for progressive multifocal leukoencephalopathy (PML) risk assessment was conducted every 24 weeks. Anti-JCV antibodies were detected using the STRATIFY JCV® two-step ELISA (Focus Diagnostics, Cypress, CA, USA) [12, 13].
The immunogenicity population included all patients who received ≥1 infusion of study treatment, had a negative baseline antibody screening result, and had ≥1 post-baseline antibody assessment. Patients were considered persistently positive for anti-natalizumab antibodies if they had two positive results separated by ≥6–12 weeks. During the 24-week bridging study, the protocol was amended to include anti-JCV antibody testing at screening and week 24. Because this testing was begun when the study was already in progress, not all patients were tested for anti-JCV antibodies. During the open-label extension study, anti-JCV antibodies were tested at the open-label baseline (week 24 of the bridging study) and every 24 weeks thereafter.
Treatment-emergent adverse events (TEAEs) were monitored throughout the study. A TEAE was defined as any untoward medical occurrence regardless of its relationship to study medication. Severity (mild, moderate, or severe) and the possible relationship to study treatment (related or not related) were determined for each TEAE. A serious adverse event (SAE) was defined as any untoward medical occurrence that resulted in death, was considered by the investigator to be life-threatening, required hospitalization or prolonged existing hospitalization, resulted in persistent or significant disability or a congenital abnormality/birth defect, or was considered by the investigator to jeopardize the patient’s well-being.
Statistical Analysis
All variables were analyzed using the intent-to-treat population, which included all patients who received ≥1 infusion of study treatment and had ≥1 post-baseline assessment of the variable of interest. In the extension study, efficacy variables (ARR, the proportion of relapse-free patients, and the change in EDSS score from baseline) were analyzed only for patients who had participated in the randomized double-blind placebo-controlled portion of the bridging study (part B). ARR was calculated as the total number of relapses experienced in a treatment group divided by the total number of days in the study multiplied by 365. Using a Poisson regression model, ARRs were adjusted for baseline relapse rate (i.e., the number of relapses in the year prior to screening for the 24-week bridging study) and reported with their 95% confidence intervals (CIs). EDSS scores and changes from baseline were summarized using descriptive statistics. Safety variables were assessed in patients enrolled in the extension study who had participated in either part A or B in the bridging study and were summarized using descriptive statistics.
Results
Patient Disposition
Ninety-seven patients who completed the 24-week double-blind treatment period of the bridging study were enrolled in the open-label extension, including 43 patients who had received placebo and 54 (10 from part A and 44 from part B) who had received natalizumab (Fig. 1). Overall, 39 patients (40%) completed the open-label extension, including 16 patients (37%) who had previously received placebo and 23 (43%) who had previously received natalizumab. Fifty-eight patients withdrew from the study (27 from the previously-on-placebo group and 31 from the previously-on-natalizumab group). The most common reasons for study withdrawal were withdrawal of consent (33 of 58 patients), investigator decision (8 of 58 patients), and TEAEs (8 of 58 patients).

Study flow. PD pharmacodynamic, PK pharmacokinetic
Baseline characteristics were generally comparable between treatment groups; the mean (standard deviation [SD]) age of the patients was 37 (9) years, and 68% of patients were women. Overall, the mean (SD) time for participation in the open-label extension study (including patients who had withdrawn from the extension study) was 25.8 (11.0) months. Table 1 summarizes patient characteristics by treatment group. The mean (SD) number of doses of study treatment during the open-label extension was 24.9 (14.0), with all extension study patients receiving ≥1 dose of study treatment and >90% of patients receiving ≥6 doses (Table 1). In the overall population, the most frequently reported concomitant medications (excluding contrast medium) were loxoprofen (64%), PL® granules (caffeine, salicylamide, paracetamol, and promethazine methylene; 39%), rebamipide (34%), and methylprednisolone (used to treat on-study relapses; 31%) (Table 1).
Efficacy
Multiple sclerosis (MS) relapses and changes in disability were assessed in patients from part B of the bridging study. Relapse activity after 96 weeks in the extension study is summarized in Table 2. The mean adjusted ARR was 0.30 (95% CI: 0.18–0.52) in patients who had previously received placebo and 0.13 (95% CI: 0.05–0.29) in patients who had previously received natalizumab (Fig. 2). Throughout 96 weeks of open-label natalizumab treatment, the proportions of patients with known relapse status who were relapse-free were 46% (12 of 26 patients) in the previously-on-placebo group and 55% (12 of 22 patients) in the previously-on-natalizumab group. Relapse-free status was unknown for an additional 17 previously-on-placebo and 22 previously-on-natalizumab patients, which includes patients who withdrew from the study and did not experience a relapse prior to withdrawal.

Annualized relapse rate. *P < 0.001 vs. placebo (Poisson regression model). †Adjusted for baseline relapse rate. CI confidence interval
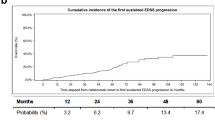
The mean change in EDSS score from baseline to week 120 was −0.03 in the previously-on-placebo group and −0.18 in the previously-on-natalizumab group.
Safety and Tolerability
Most patients in both treatment groups experienced ≥1 TEAE, including 39 of 43 patients (91%) in the previously-on-placebo group and 53 of 54 patients (98%) in the previously-on-natalizumab group. The most frequently reported TEAEs by MedDRA System Organ Class (those occurring in >30% of patients in both treatment arms) were infections and infestations, followed by nervous system and gastrointestinal disorders in both the previously-on-placebo group and the previously-on-natalizumab group (Table 3). Sixteen of 43 patients (37%) in the previously-on-placebo group and 17 of 54 patients (31%) in the previously-on-natalizumab group experienced TEAEs that were considered related to study treatment (Table 4).
SAEs were experienced by 12 of 43 patients (28%) and 17 of 54 patients (31%) in the previously-on-placebo and previously-on-natalizumab groups, respectively. The most frequently reported SAE was MS relapse (Table 5). Three previously-on-placebo patients (7%) and four previously-on-natalizumab patients (7%) experienced treatment-related SAEs. In the previously-on-placebo group, the three patients with treatment-related SAEs had meningitis and a mycoplasma infection, a skin rash, and an ectopic pregnancy, respectively. In the previously-on-natalizumab group, each of the following treatment-related SAEs was reported in one patient: esophageal ulcer and eosinophilia, interstitial lung disease, meningitis, and hypothyroidism. No deaths or cases of PML were reported during the open-label extension study.
Anti-Natalizumab Antibody Status
In the immunogenicity population, two patients in the previously-on-placebo group were persistently positive for anti-natalizumab antibodies. One of these patients experienced an infusion reaction, and both patients withdrew from the study. In addition, 1 patient was transiently positive for anti-natalizumab antibodies, with a positive result at week 12 and negative results at weeks 24, 36, and 48. In the previously-on-natalizumab group, no patients who were negative for anti-natalizumab antibodies during the 24-week bridging study became persistently positive for anti-natalizumab antibodies during the open-label extension study.
Anti-JCV Antibody Status
Overall, 69 of the 92 patients tested (75%) were anti-JCV antibody-positive at some point during the study. In the previously-on-natalizumab group, 17 of 25 patients (68%) and 37 of 51 patients (73%) were anti-JCV antibody-positive at baseline and overall, respectively. Similarly, 17 of 27 patients (63%) and 32 of 41 patients (78%) in the previously-on-placebo group were anti-JCV antibody-positive at baseline and overall, respectively.
Discussion
The findings of this open-label extension study of natalizumab in Japanese patients with RRMS support and extend the efficacy and safety results from the preceding two-part 24-week double-blind bridging study. During the 24 weeks in the bridging study, ARR decreased from 2.00 to 0.53 in the natalizumab group and from 1.90 to 1.73 in the placebo group [8]. During the 2-year extension study, ARR remained lower in the previously-on-natalizumab group than in the previously-on-placebo group (0.13 vs. 0.30), and the percentage of relapse-free patients appeared higher in the previously-on-natalizumab group than in the previously-on-placebo group (55% vs. 46%). Thus, slightly better efficacy was observed over the 2 years following the conclusion of the bridging study in patients who received continuous natalizumab treatment than in patients who initiated natalizumab in this extension phase, which suggests an association between earlier initiation of natalizumab and clinical benefits, similar to results seen in non-Asian populations [3]. However, statistical analysis of this potential relationship was not conducted, as it was considered inappropriate to compare 2 non-randomized groups both receiving natalizumab treatment in the open-label extension study.
Overall, changes in EDSS scores during the extension study were small in both the previously-on-placebo and previously-on-natalizumab groups, consistent with changes observed during the double-blind study [8]. The trend of EDSS score reduction in both groups and the slightly larger reduction observed in the previously-on-natalizumab group suggest that natalizumab, on average, also prevents disability progression in Japanese patients.
In terms of safety and tolerability, MS relapse and nasopharyngitis were the most frequently reported events in both groups. With MS relapse excluded from the incidence rate of TEAEs, 39 of 43 patients (91%) in the previously-on-placebo group and 52 of 54 patients (96%) in the previously-on-natalizumab group experienced a TEAE. These results were generally similar to the results that included MS relapse as a TEAE.
Although the bridging study was only 24 weeks in duration, similar results were observed: 87% of placebo patients and 72% of natalizumab patients reported ≥1 TEAE, with MS relapse and infections the most commonly reported TEAEs in both groups [8]. The incidence of testing persistently positive for anti-natalizumab antibodies was 2% (2 of 96 patients in the immunogenicity population) in the extension study, which is slightly lower than the 3–6% incidence that has been reported in non-Asian populations [1, 3, 11]. One of the two patients who were positive for anti-natalizumab antibodies experienced an infusion reaction, and both patients withdrew from the study. It is difficult to draw conclusions about the relationship between anti-natalizumab antibodies and efficacy or safety in this study, given the small number of patients who were anti-natalizumab antibody-positive. In general, the presence of antibodies against natalizumab has been associated with reduced clinical efficacy, likely due to accelerated clearance of natalizumab and more hypersensitivity and infusion-related reactions, including headache, urticaria, rigors, nausea, vomiting, and flushing [14, 15].
The risk of developing PML is increased in patients who test positive for anti-JCV antibodies, who have prior immunosuppressant use, and who use natalizumab for >2 years [16]. No cases of PML were reported during the 24-week placebo-controlled trial or during year 1 of the open-label extension study.
This study had several key limitations. As the study includes patients from a clinical trial population, the generalizability of these results to the Japanese RRMS patients being treated with natalizumab in real-world settings is unclear. In addition, because the treatment groups in this extension study were not randomized, statistical comparison of efficacy variables was not conducted.
Overall, results from this analysis of efficacy and safety data from patients enrolled in the 2-year open-label extension study of natalizumab were consistent with findings recently reported for patients who received natalizumab during the 24-week double-blind bridging study [8].
Conclusions
The efficacy and safety findings from the double-blind bridging study [8] and the open-label extension study are comparable to and confirm results reported from other clinical trials of natalizumab conducted in non-Asian patient populations [1, 2, 17], and provide longer-term evidence of the efficacy and safety of natalizumab treatment in Japanese patients with RRMS.
References
Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910.
Rudick RA, Stuart WH, Calabresi PA, Confavreux C, Galetta SL, Radue EW, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):911–23.
O’Connor P, Goodman A, Kappos L, Lublin F, Polman C, Rudick RA, et al. Long-term safety and effectiveness of natalizumab redosing and treatment in the STRATA MS Study. Neurology. 2014;83(1):78–86.
Butzkueven H, Kappos L, Pellegrini F, Trojano M, Wiendl H, Patel RN, et al. Efficacy and safety of natalizumab in multiple sclerosis: interim observational programme results. J Neurol Neurosurg Psychiatry. 2014;85(11):1190–7.
Kallweit U, Jelcic I, Braun N, Fischer H, Zörner B, Schreiner B, et al. Sustained efficacy of natalizumab in the treatment of relapsing-remitting multiple sclerosis independent of disease activity and disability at baseline: real-life data from a Swiss cohort. Clin Neuropharmacol. 2012;35(2):77–80.
Putzki N, Yaldizli O, Bühler R, Schwegler G, Curtius D, Tettenborn B. Natalizumab reduces clinical and MRI activity in multiple sclerosis patients with high disease activity: results from a multicenter study in Switzerland. Eur Neurol. 2010;63(2):101–6.
Sangalli F, Moiola L, Bucello S, Annovazzi P, Rizzo A, Radaelli M, et al. Efficacy and tolerability of natalizumab in relapsing-remitting multiple sclerosis patients: a post-marketing observational study. Neurol Sci. 2011;31(Suppl 3):299–302.
Saida T, Kira J-I, Kishida S, Yamamura T, Sudo Y, Ogiwara K, et al. Efficacy, safety, and pharmacokinetics of natalizumab in Japanese multiple sclerosis patients: a double-blind, randomized controlled trial and open-label pharmacokinetic study. Mult Scler Relat Disord. 2016 (Epub ahead of print).
Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 Revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302.
Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444–52.
Sorensen PS, Jensen PE, Haghikia A, Lundkvist M, Vedeler C, Sellebjerg F, et al. Occurrence of antibodies against natalizumab in relapsing multiple sclerosis patients treated with natalizumab. Mult Scler. 2011;17(9):1074–8.
Gorelik L, Lerner M, Bixler S, Crossman M, Schlain B, Simon K, et al. Anti-JC virus antibodies: implications for PML risk stratification. Ann Neurol. 2010;68(3):295–303.
Lee P, Plavina T, Castro A, Berman M, Jaiswal D, Rivas S, et al. A second-generation ELISA (STRATIFY JCV™ DxSelect™) for detection of JC virus antibodies in human serum and plasma to support progressive multifocal leukoencephalopathy risk stratification. J Clin Virol Off Publ Pan Am Soc Clin Virol. 2013;57(2):141–6.
Tysabri (natalizumab). Summary of product characteristics. Cambridge: Biogen; 2016.
Calabresi PA, Giovannoni G, Confavreux C, Galetta SL, Havrdova E, Hutchinson M, et al. The incidence and significance of anti-natalizumab antibodies: results from AFFIRM and SENTINEL. Neurology. 2007;69(14):1391–403.
Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366(20):1870–80.
Miller DH, Khan OA, Sheremata WA, Blumhardt LD, Rice GP, Libonati MA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348(1):15–23.
Acknowledgements
The authors would like to acknowledge Daniel Mikol, MD, PhD, formerly of Biogen, for his significant contribution to the interpretation of data, and Tetsuhiro Shiota, Biogen Japan, for his contribution to data validation. Sponsorship and article processing charges for this study were funded by Biogen. All named authors met the International Committee of Medical Journal Editors criteria for authorship for this manuscript. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data, the accuracy of the data analysis, and the work as a whole. Biogen provided funding for medical writing support in the development of this manuscript; Linda A. Goldstein, PhD, and Alison Adams, PhD, of Ashfield Healthcare Communications (Middletown, CT), based on input from the authors, wrote the first draft and revised subsequent drafts of the manuscript, and Joshua Safran of Ashfield Healthcare Communications copyedited and styled the manuscript per journal requirements. Biogen reviewed and provided feedback on the manuscript to the authors. The authors had full editorial control of the manuscript, and provided their final approval of all content.
Disclosures
Dr. Saida (coordinating investigator) has received funding from, held board membership for, spoken at scientific meetings for, prepared manuscripts for, and/or had consulting agreements with Astellas, Biogen, Bayer-Schering, Daiichi-Sankyo, Eisai, Kaketsuken, Merck Serono, Mitsubishi-Tanabe, Nihon, Novartis, Ono, Sanofi, TDS Japan, and Teijin. Dr. Kira (coordinating investigator) has been a consultant for Biogen and Novartis and has received honoraria from Bayer HealthCare, Biogen, Boehringer Ingelheim, Eisai, Kyowa Kirin, Mitsubishi Tanabe, Otsuka, Pfizer Japan, and Teijin; funding for travel from Bayer HealthCare and Biogen; and grants from the Japanese Ministry of Health, Labour and Welfare, the Japanese Science and Technology Agency, and the Japanese Ministry of Education, Culture, Sports, Science and Technology. Dr. Kishida (PML consultant) has been a consultant for Ajinomoto. Dr. Yamamura (scientific advisor) has been a member of scientific advisory boards for Biogen and Chugai and has received research support from Asahi Kasei Medical, Chugai, Mitsubishi Tanabe, Ono, and Teva; speaker honoraria from Abbot Japan, Astellas, Bayer, Biogen, Dainippon Sumitomo, Eisai, Mitsubishi Tanabe, Nihon, Novartis, and Santen; and funding from the Japanese Ministry of Health, Labour and Welfare and the Japan Society for the Promotion of Science. Dr. Ling is an employee of and holds stock and/or stock options in Biogen. Dr. Torii is an employee of and holds stock and/or stock options in Biogen. Dr. Lucas is an employee of and holds stock and/or stock options in Biogen. Dr. Kuesters was an employee of Biogen at the time this analysis was performed. Dr Steiner is an employee of and holds stock and/or stock options in Biogen. Dr. Tibung is an employee of and holds stock and/or stock options in Biogen. Ms. Lucas is an employee of and holds stock and/or stock options in Biogen. Dr. Ohtsuka was an employee of Biogen at the time this analysis was performed.
Compliance with Ethics Guidelines
The study protocol was approved by the ethics committees at each participating site, and all patients provided written informed consent. The study was conducted in accordance with the International Conference on Harmonisation Guideline for Good Clinical Practice and the ethical principles outlined in the Declaration of Helsinki of 1964, as revised in 2013.
Data Availability
The datasets generated and/or analyzed during the current study are not publicly available and are fully owned by Biogen, but are available from Nisha Lucas at nisha.lucas@biogen.com on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/C527F0604BCD82AC.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Saida, T., Kira, JI., Kishida, S. et al. Safety and Efficacy of Natalizumab in Japanese Patients with Relapsing-Remitting Multiple Sclerosis: Open-Label Extension Study of a Phase 2 Trial. Neurol Ther 6, 39–55 (2017). https://doi.org/10.1007/s40120-016-0059-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-016-0059-z