
Abstract
Hypochloraemia is a common electrolyte abnormality in patients with heart failure (HF). It has a strong association with adverse outcome regardless of HF phenotype and independent of other prognostic markers. How hypochloraemia develops in a patient with HF and how it might influence outcome are not clear, and in this review we explore the possible mechanisms. Patients with HF and hypochloraemia almost invariably take higher doses of loop diuretic than patients with normal chloride levels. However, renal chloride and bicarbonate homeostasis are closely linked, and the latter may be influenced by neurohormonal activation: it is likely that the etiology of hypochloraemia in patients with HF is multifactorial and due to more than just diuretic-induced urinary losses. There are multiple proposed mechanisms by which low chloride concentrations may lead to an adverse outcome in patients with HF: by increasing renin release; by a stimulatory effect on the with-no-lysine kinases which might increase renal sodium-chloride co-transporter activity; and by an adverse effect on myocardial conduction and contractility. None of these proposed mechanisms are proven in humans with HF. However, if true, it might suggest that hypochloraemia is a therapeutic target that might be amenable to treatment with acetazolamide or chloride supplementation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Low serum chloride levels are associated with adverse prognosis in patients with acute or chronic heart failure (HF) regardless of left ventricular ejection fraction and independently of other prognostic markers such as N-terminal pro-B-type natriuretic peptide levels. |
It is not clear how hypochloraemia develops in patients with HF but it may be linked to neurohormonal activation, high-dose loop diuretic usage, and metabolic alkalosis. |
It is not known whether hypochloraemia is a marker or mediator of adverse outcome in patients with heart failure, although there are several putative mechanisms that might suggest the latter. For example, hypochloraemia might be linked to increased neurohormonal activation, diuretic resistance, and increased risk of sudden cardiac death. |
Acetazolamide may increase natriuresis and diuresis while also increasing chloride reabsorption and bicarbonate excretion and thus might be a useful treatment for patients with HF, hypochloraemia, metabolic alkalosis, and diuretic resistance. |
Introduction
A link between low serum chloride concentrations, loop diuretics, and risk of death in patients following a heart attack was first reported in 1979 [1], and the first reported association between low serum chloride concentrations and increased risk of death amongst patients with heart failure (HF) was in 2007 [2]. The authors of neither paper made even a passing reference to the chloride findings in the discussion [1, 2], perhaps owing to a lack of understanding regarding the importance of serum chloride: the potential prognostic significance of low chloride has, until recently, not been appreciated.
Hypochloraemia is a common electrolyte disturbance and marker of adverse outcome amongst patients with HF independent of other prognostic markers, including hyponatremia (Table 1) [3,4,5,6,7,8,9,10,11,12]. The mechanisms are poorly understood. In this review, we will discuss the etiology of hypochloraemia in patients with HF, explore the possible mechanisms behind its association with adverse outcome, and consider what, if anything, might be done about it. The present article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Hypochloraemia and Heart Failure
Chloride is the main anion in the plasma and extracellular fluid [13], and is freely filtered in the glomerulus of the kidney into the urinary space (tubular lumen). Renal tubular cells are asymmetric with an apical surface facing the urinary space and a basolateral membrane facing the renal interstitium (peritubular capillaries). The majority of chloride reabsorption occurs in the proximal convoluted tubule (PCT), paracellularly in the intercellular space passively along an electrochemical gradient as the permeability to chloride anions exceeds that of other anions such as bicarbonate [14, 15]. Active, trans-cellular, reabsorption occurs via Cl−/anion counter transports (antiporters or exchangers) in particular formate amongst others (sulphate, iodide, oxalate, hydroxyl, and bicarbonate) on the apical membrane, and by a sodium-driven Cl−/HCO3− antiporter and K+/Cl− symporter on the basolateral membrane [16]. In the loop of Henle (LoH), further chloride reabsorption takes place via Na+/K+/2Cl− co-transporters (NKCC2) on the apical membrane (the site of action of loop diuretics) and voltage-gated chloride channels on the basolateral membrane [17]. In the distal convoluted tubule (DCT) and collecting duct (CD) (responsible for ~ 5% of chloride reabsorption) chloride is reabsorbed by thiazide-sensitive Na+/Cl− co-transporter and Cl−/HCO3− antiporter and returns to the bloodstream via voltage-gated chloride channels on the basolateral membrane (Fig. 1) [18].

Chloride reabsorption along the nephron. The majority of renal chloride reabsorption occurs in the proximal convoluted tubule, paracellularly along an electrochemical gradient although transcellular Cl−/anion transport also plays a role
At first sight, the origin of hypochloraemia seems likely to be similar to the putative etiology of hyponatremia in patients with HF: low chloride results from either haemodilution or depletion due to loop diuretics [19]. However, patients with hypochloraemia appear to fall into two phenotypes; those with concurrent hyponatremia and those with normal sodium concentrations [3]. The group with normal sodium has higher bicarbonate, and lower potassium concentrations (and a higher rate of clinically significant hypokalaemia (defined as a serum K+ < 3.5 mmol/l) [3].
An Association with Metabolic Alkalosis?
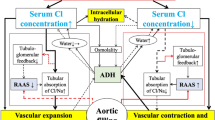
In other disease states, such as severe vomiting or mineralocorticoid excess, hypochloraemia is associated with metabolic alkalosis (HCO3− > 30 mmol/l) [20, 21]. Metabolic alkalosis is the most common acid-base abnormality in patients with HF, affecting up to half of patients admitted to hospital [22]. While activation of the renin–angiotensin–aldosterone system (RAAS) is usually linked to sodium homeostasis [23], data from in vitro and in vivo animal studies suggest that neurohormonal activation might play a significant role in the development and maintenance of a metabolic alkalosis in patients with HF (Fig. 2).

Possible association between hypochloraemia, metabolic alkalosis, and neurohormonal activation in patients with heart failure. Our proposed theoretical link between hypochloraemia, metabolic alkalosis, and neurohormonal activation in patients with heart failure is based on various in vivo and in vitro animal experiments. The dotted greyed lines denote that loop diuretics are only a contributing factor in this proposed model, rather than the driving force
In vitro and in vivo studies suggest that when noradrenaline [24], and angiotensin II [25] levels increase, bicarbonate reabsorption in the first segment of the PCT increases. Additionally, in vivo studies in rats show that aldosterone increases the activity of the H+-ATPase pump in the CD which increases H+ secretion into the urine [26]. The increased acidification of the urine might result in a net gain of bicarbonate by the body.
Loop diuretics might also contribute to a metabolic alkalosis: a so-called “contraction-alkalosis” due to decreased extracellular fluid volume resulting in increased bicarbonate concentration [27] is well recognized in the literature, but may be an over-simplification. In vivo, increased sodium delivery to the CD (due to apical NKCC2 co-transporter inhibition) increases the activity of the H+-ATPase pump, increasing H+ secretion into the urine [28]. In vitro studies in rats have found that hypokalaemia (a potential complication of loop diuretic use) promotes bicarbonate reabsorption in the PCT [29, 30], and hypokalaemia increases RAAS activation in humans with HF [31, 32], which might further drive bicarbonate reabsorption (Fig. 2).
In vitro and in vivo studies in both rabbits and rats suggest that increased bicarbonate reabsorption is accompanied by increased chloride excretion [33,34,35]. The same process may occur in humans [36]. One small study (N = 51) found that patients with HF and hypochloraemia had higher serum bicarbonate, and greater fractional chloride excretion than those with normal chloride levels while having similar fractional sodium excretion (Table 1) [8]. “Chloride wasting nephropathy”—persistent urinary chloride excretion—is seen in patients with hyperaldosteronism [37] and/or severe potassium depletion [38], and similar metabolic states have been reported in patients with HF [39].
An additional factor contributing to the maintenance of an alkalosis is that as serum concentrations of chloride fall (either due to increased excretion in response to increased bicarbonate reabsorption, or diuretic use, or both), there is less and less chloride filtered into in the urinary space. A threshold of low serum chloride may be reached beyond which bicarbonate excretion is inhibited as there is less chloride in the urine to exchange with bicarbonate [36, 37].
Patients can thus be trapped in a cycle of hypochloraemia and alkalosis, which is only partly due to loop diuretic usage (Fig. 2): for example, among patients admitted with HF, those with serum bicarbonate concentrations above the median (≥ 28 mmol/l) had more severe disease (lower left ventricular ejection fraction, worse renal function, and higher natriuretic peptide levels) but were on lower doses of loop diuretic than patients with serum bicarbonate below the median [25].
Chloride and Outcome
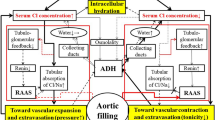
Whether a low chloride concentration is a marker or a mediator of adverse outcome is unknown although there are possible pathophysiological mechanisms, which might suggest the latter (Fig. 3).

Confirmed and possible associations between hypochloraemia and adverse outcome in patients with heart failure. The dotted lines denote possible links demonstrated in animal studies and the thick lines denote confirmed links in patients with heart failure
Diuretic Resistance
The with-no-lysine (WNK) kinases (WNK1, WNK3, and WNK4) are the first step in an enzymatic cascade which increases activity of the Na+/K+/2Cl− and Na+/Cl− co-transporters [40,41,42,43]. Chloride binds to the catalytic site of the kinases, thus inactivating them [44, 45]. In vitro and in vivo studies suggest that the activity of WNK1 and WNK4 is reduced at high chloride concentrations [44, 46], but increased at lower concentrations [47, 48]. Thus, hypochloraemia may increase the activity of both Na+/K+/2Cl− and Na+/Cl− co-transporters, meaning greater doses of loop diuretic are required to induce a diuresis. In addition, chronic use of loop diuretic leads to an increase in sodium delivery to the distal tubule with consequent hypertrophy of cells in the distal nephron. The hypertrophied cells reabsorb sodium more avidly, an effect that can be mitigated by increasing the dose of loop diuretic and/or the additional use of a thiazide diuretic [49].
Consistent with this idea, patients with hypochloraemia take higher doses of loop diuretics than those with normal chloride levels [3,4,5,6,7,8,9,10,11,12], but whether a high-dose diuretic is the cause of hypochloraemia or becomes necessary because of hypochloraemia-induced diuretic resistance is uncertain.
Effect on the RAAS
Renin secretion is controlled by the macula densa. These specialized cells are sensitive to sodium chloride, low concentrations of which in the urinary space leads to renin secretion from the juxtaglomerular cells of the afferent and efferent arterioles. Increased chloride (but not increased sodium) delivery to the macula densa suppresses renin release from the granular cells in the afferent arteriole and a subsequent fall in angiotensin II levels [50, 51]. Chloride and renin are inversely related in patients with HF [8]. This is the rate-limiting event in the RAAS.
Sudden Death
Chloride channels play a role in ventricular repolarization [52, 53], and in regulating the positive chronotropic effect of cardiac pacemaker activity [54]. Myocyte volume and pH are regulated, in part, by chloride-dependent co-transporters [55, 56]. Abnormalities of the chloride channels and co-transporters may be arrhythmogenic [57, 58] and can impair contractility [59]. Consistent with these observations, a large study of outpatients with HF found that patients with hypochloraemia had an increased risk of sudden death (Table 1) [3].
A Therapeutic Target?
Hypertonic saline (HS) increases diuresis and may improve outcome when given alongside intravenous furosemide in patients admitted with HF [60, 61]. However, data on changes in chloride levels are absent from almost all reports of HS and whether any observed benefit is due a change in chloride levels is pure speculation. A proof-of-concept study of oral chloride supplementation in patients with HF (N = 10) found that lysine chloride increased chloride levels but required enormous doses to affect only small changes in serum chloride (Table 1) [8]. Further work is ongoing (NCT03446651) [62].
Acetazolamide
Acetazolamide (ACZ) is a carbonic anhydrase (CA) inhibitor. CA catalyses the interconversion between carbon dioxide and water on the one hand, and hydrogen (H+) and bicarbonate ions on the other (Fig. 4). CA on the apical membrane of the PCT cell converts free H+ and bicarbonate to water and carbon dioxide in the urinary space; the water then diffuses back into the cell via aquaporin 1 channels, carbon dioxide freely diffuses across the apical membrane [63, 64]. There, the water and carbon dioxide are converted back to H+ and bicarbonate ions by intracellular CA.

Renal carbonic anhydrase and acetazolamide. Inhibition of renal carbonic anhydrase with acetazolamide might increase luminal bicarbonate concentrations, reduce intracellular hydrogen ion concentrations thus reducing sodium reabsorption via the Na+/H+ antiporter, and reduce movement of chloride out of the peritubular capillaries. ACZ acetazolamide
The newly formed H+ ions in the cell are excreted in exchange for urinary sodium via Na+/H+ co-transporters on the apical membrane [65, 66], and bicarbonate returns to the circulation via Na+/HCO3− and Cl−/HCO3− antiporters on the basolateral membrane (Fig. 4) [67]. Inhibition of intracellular CA reduces production of intracellular H+, thus reducing sodium reabsorption via the Na+/H+ antiporters on the apical membrane, and inhibition of luminal CA reduces production of water and carbon dioxide, thus increasing urinary bicarbonate levels (Fig. 4).
ACZ increases bicarbonate excretion and chloride reabsorption in vivo [32, 68], and increases serum chloride levels in humans [69, 70]. The reasons behind this are not clear but may result from two potential mechanisms: firstly, increased HCO3− in the urinary space increases the negative charge thus increasing the electrochemical gradient along which chloride is reabsorbed in the PCT. Secondly, in vivo studies suggest that ACZ, separately from CA inhibition, also inhibits the basolateral Cl-/HCO3− antiporter in the PCT thus reducing movement of chloride out of the blood and into the cell (Fig. 4).
There are thus three ways in which ACZ might be beneficial for patients with HF: (1) increasing sodium excretion and increasing diuresis [71, 72]; (2) increasing bicarbonate excretion, which may reduce metabolic alkalosis [73, 74]; and (3) increasing renal chloride reabsorption, which may reverse hypochloraemia [69, 70].
The ADVOR study of ACZ in patients admitted with HF is aiming to recruit ~ 500 patients, the largest study of ACZ in patients with HF to date. The primary endpoint is treatment success (i.e., clinical decongestion defined as the absence of pleural effusion, ascites, and significant peripheral edema) after 3 days of treatment. Secondary endpoints include mortality and morbidity alongside changes in natriuresis, body weight, and natriuretic peptide levels [75]. There is no planned analysis of either chloride or bicarbonate changes but the data will give an insight into the usefulness of ACZ as a treatment for patients with HF.
Future Perspective: Is Prevention Better Than Cure?
Amongst patients admitted with HF, those with hypochloraemia that resolves by the time of discharge have a similar post-discharge prognosis to those with normal chloride concentrations throughout admission [5]. Conversely, incident hypochloraemia during admission is associated with an increased risk of adverse outcome post-discharge [5]. If hypochloraemia results from the inevitable combination of severe HF and high-dose loop diuretics, it may be that prevention of hypochloraemia, rather than the correction of an existing abnormality, may have the greater effect on outcome. Whether acetazolamide might be best employed as a preventative measure is unknown, but should be the focus of future research.
Conclusions
Hypochloraemia is a common electrolyte abnormality in patients with HF and is an important marker of poor prognosis. There are many unknowns as to how hypochloraemia develops and whether it has a pathophysiological effect in patients with HF. If the latter is true, it may be a therapeutic target. As ever, more work is needed.
References
Flear CT, Hilton P. Hyponatraemia and severity and outcome of myocardial infarction. Br Med J. 1979;1(6173):1242–6.
Felker GM, Allen LA, Pocock SJ, Shaw LK, McMurray JJ, Pfeffer MA, Swedberg K, Wang D, Yusuf S, Michelson EL, Granger CB, CHARM Investigators. Red cell distribution width as a novel prognostic marker in heart failure: data from the CHARM Program and the Duke Databank. J Am Coll Cardiol. 2007;50(1):40–7.
Cuthbert JJ, Pellicori P, Rigby A, Pan D, Kazmi S, Shah P, Clark AL. Low serum chloride in patients with chronic heart failure: clinical associations and prognostic significance. Eur J Heart Fail. 2018;20(10):1426–35.
Grodin JL, Simon J, Hachamovitch R, Wu Y, Jackson G, Halkar M, Starling RC, Testani JM, Tang WH. Prognostic role of serum chloride levels in acute decompensated heart failure. J Am Coll Cardiol. 2015;66(6):659–66.
Ter Maaten JM, Damman K, Hanberg JS, Givertz MM, Metra M, O’Connor CM, Teerlink JR, Ponikowski P, Cotter G, Davison B, Cleland JG, Bloomfield DM, Hillege HL, van Veldhuisen DJ, Voors AA, Testani JM. Hypochloremia, diuretic resistance, and outcome in patients with acute heart failure. Circ Heart Fail. 2016;9(8):e003109.
Grodin JL, Verbrugge FH, Ellis SG, Mullens W, Testani JM, Tang WH. Importance of abnormal chloride homeostasis in stable chronic heart failure. Circ Heart Fail. 2016;9(1):e002453.
Testani JM, Hanberg JS, Arroyo JP, Brisco MA, Ter Maaten JM, Wilson FP, Bellumkonda L, Jacoby D, Tang WH, Parikh CR. Hypochloraemia is strongly and independently associated with mortality in patients with chronic heart failure. Eur J Heart Fail. 2016;18(6):660–8.
Hanberg JS, Rao V, Ter Maaten JM, Laur O, Brisco MA, Perry Wilson F, Grodin JL, Assefa M, Samuel Broughton J, Planavsky NJ, Ahmad T, Bellumkonda L, Tang WH, Parikh CR, Testani JM. Hypochloremia and diuretic resistance in heart failure: mechanistic insights. Circ Heart Fail. 2016;9(8):e003180.
Grodin JL, Sun JL, Anstrom KJ, Chen HH, Starling RC, Testani JM, Tang WH. Implications of serum chloride homeostasis in acute heart failure (from ROSE-AHF). Am J Cardiol. 2017;119(1):78–83.
Ferreira JP, Girerd N, Duarte K, Coiro S, McMurray JJ, Dargie HJ, Pitt B, Dickstein K, Testani JM, Zannad F, Rossignol P, High-Risk Myocardial Infarction Database Initiative Investigators. Serum chloride and sodium interplay in patients with acute myocardial infarction and heart failure with reduced ejection fraction: an analysis from the high-risk myocardial infarction database initiative. Circ Heart Fail. 2017;10(2):e003500.
Grodin JL, Testani JM, Pandey A, Sambandam K, Drazner MH, Fang JC, Tang WHW. Perturbations in serum chloride homeostasis in heart failure with preserved ejection fraction: insights from TOPCAT. Eur J Heart Fail. 2018;20(10):1436–43.
Marchenko R, Sigal A, Wasser TE, Reyer J, Green J, Mercogliano C, Khan MS, Donato AA. Hypochloraemia and 30 day readmission rate in patients with acute decompensated heart failure. ESC Heart Fail. 2020. https://doi.org/10.1002/ehf2.12587.
Yunos NM, Bellomo R, Story D, Kellum J. Bench-to-bedside review: chloride in critical illness. Crit Care. 2010;14(4):226.
Berend K, van Hulsteijn LH, Gans RO. Chloride: the queen of electrolytes? Eur J Intern Med. 2012;23(3):203–11.
Planelles G. Chloride transport in the renal proximal tubule. Pflugers Arch. 2004;448(6):561–70.
Sindić A, Chang MH, Mount DB, Romero MF. Renal physiology of SLC26 anion exchangers. Curr Opin Nephrol Hypertens. 2007;16(5):484–90.
Jeck N, Seyberth HW. Loop disorders: insights derived from defined genotypes. Nephron Physiol. 2011;118:7–14.
Romero MF. Molecular pathophysiology of SLC4 bicarbonate transporters. Curr Opin Nephrol Hypertens. 2005;14(5):495–501.
Verbrugge FH, Steels P, Grieten L, Nijst P, Tang WH, Mullens W. Hyponatremia in acute decompensated heart failure: depletion versus dilution. J Am Coll Cardiol. 2015;65(5):480–92.
Maciel AT. Severe metabolic alkalosis due to the combination of unmeasured cations and hypochloraemia in a patient with gastroparesia and frequent emesis. BMJ Case Rep. 2009;2009:bcr09.2008.1011.
Khanna A, Kurtzman NA. Metabolic alkalosis. J Nephrol. 2006;19(Suppl 9):S86–96.
Cooper LB, Mentz RJ, Gallup D, Lala A, DeVore AD, Vader JM, AbouEzzeddine OF, Bart BA, Anstrom KJ, Hernandez AF, Felker GM. Serum bicarbonate in acute heart failure: relationship to treatment strategies and clinical outcomes. J Card Fail. 2016;22(9):738–42.
Zannad F. Aldosterone and heart failure. Eur Heart J. 1995;16(Suppl N):98–102.
Chan YL. Adrenergic control of bicarbonate absorption in the proximal convoluted tubule of the rat kidney. Pflugers Arch. 1980;388(2):159–64.
Liu F, Cogan M. Angiotensin II. A potent regulator of acidification in the rat early proximal convoluted tubule. J Clin Investig. 1988;82:601–7.
Eiam-Ong S, Kurtzman NA, Sabatini S. Effect of furosemide-induced hypokalemic metabolic alkalosis on renal transport enzymes. Kidney Int. 1993;43(5):1015–20.
Palmer BF. Metabolic complications associated with use of diuretics. Semin Nephrol. 2011;31(6):542–52.
Bartoli E, Satta A, Faedda R, Olmeo NA, Soggia G, Branca G. A furosemide test in the functional evaluation of the human nephron in vivo. J Clin Pharmacol. 1983;23:53–6.
Chan YL, Biagi B, Giebisch G. Control mechanisms of bicarbonate transport across the rat proximal convoluted tubule. Am J Physiol. 1982;242:F532–43.
Soleimani M, Bergman JA, Hosford MA, McKinney TD. Potassium depletion increases luminal Na+/H+ exchange and basolateral Na+ :CO3=:HCO3− cotransport in rat renal cortex. J Clin Investig. 1990;86(4):1076–83.
Williams GH. Aldosterone and heart failure: the rest of the story. Heart Fail Rev. 2005;10:5–6.
Cleland JG, Dargie HJ, Robertson I, Robertson JI, East BW. Total body electrolyte composition in patients with heart failure: a comparison with normal subjects and patients with untreated hypertension. Br Heart J. 1987;58:230–8.
Bomsztyk K. Chloride transport by rat renal proximal tubule: effects of bicarbonate absorption. Am J Physiol. 1986;250(6 Pt 2):F1046–54.
Star RA, Burg MB, Knepper MA. Bicarbonate secretion and chloride absorption by rabbit cortical collecting ducts. Role of chloride/bicarbonate exchange. J Clin Investig. 1985;76(3):1123–30.
Levine DZ, Vandorpe D, Iacovitti M. Luminal chloride modulates rat distal tubule bidirectional bicarbonate flux in vivo. J Clin Investig. 1990;85(6):1793–8.
Luke RG, Galla JH. It is chloride depletion alkalosis, not contraction alkalosis. J Am Soc Nephrol. 2012;23(2):204–7.
Wakabayashi Y, Mishina T, Marumo F, Kikawada R. Two cases of saline-responsive metabolic alkalosis associated with high urinary chloride concentrations. Tohoku J Exp Med. 1986;150(4):427–33.
Garella S, Chazan JA, Cohen JJ. Saline-resistant metabolic alkalosis or “chloride-wasting nephropathy”. Report of four patients with severe potassium depletion. Ann Intern Med. 1970;73(1):31–8.
Peixoto AJ, Alpern RJ. Treatment of severe metabolic alkalosis in a patient with congestive heart failure. Am J Kidney Dis. 2013;61(5):822–7.
Kahle KT, Rinehart J, Lifton RP. Phosphoregulation of the Na–K–2Cl and K–Cl cotransporters by the WNK kinases. Biochim Biophys Acta. 2010;1802(12):1150–8.
Yang CL, Zhu X, Ellison DH. The thiazide-sensitive Na–Cl cotransporter is regulated by a WNK kinase signaling complex. J Clin Investig. 2007;117(11):3403–11.
Ponce-Coria J, San-Cristobal P, Kahle KT, Vazquez N, Pacheco-Alvarez D, de Los Heros P, Juarez P, Munoz E, Michel G, Bobadilla NA, Gimenez I, Lifton RP, Hebert SC, Gamba G. Regulation of NKCC2 by a chloride-sensing mechanism involving the WNK3 and SPAK kinases. Proc Natl Acad Sci USA. 2008;105:8458–63.
Terker AS, Castañeda-Bueno M, Ferdaus MZ, Cornelius RJ, Erspamer KJ, Su XT, Miller LN, McCormick JA, Wang WH, Gamba G, Yang CL, Ellison DH. With no lysine kinase 4 modulates sodium potassium 2 chloride cotransporter activity in vivo. Am J Physiol Ren Physiol. 2018;315(4):F781–90.
Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 2016;89(1):127–34.
Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal. 2014;7(324):ra41.
Chen JC, Lo YF, Lin YW, Lin SH, Huang CL, Cheng CJ. WNK4 kinase is a physiological intracellular chloride sensor. Proc Natl Acad Sci USA. 2019;116(10):4502–7.
Richardson C, Alessi DR. The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J Cell Sci. 2008;121(Pt 20):3293–304.
Bazúa-Valenti S, Chávez-Canales M, Rojas-Vega L, González-Rodríguez X, Vázquez N, Rodríguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, García-Valdés J, Hadchouel J, Gamba G. The effect of WNK4 on the Na+–Cl−cotransporter is modulated by intracellular chloride. J Am Soc Nephrol. 2015;26(8):1781–6.
Gupta R, Testani J, Collins S. Diuretic resistance in heart failure. Curr Heart Fail Rep. 2019;16(2):57–66.
Schnermann J, Ploth DW, Hermle M. Activation of tubulo-glomerular feedback by chloride transport. Pflugers Arch. 1976;362(3):229–40.
Lorenz JN, Weihprecht H, Schnermann J, Skøtt O, Briggs JP. Renin release from isolated juxtaglomerular apparatus depends on macula densa chloride transport. Am J Physiol. 1991;260(4 Pt 2):F486–93.
Carmeliet EE. Chloride ions and the membrane potential of Purkinje fibres. J Physiol. 1961;156:375–88.
Hutter O, Noble D. Anion conductance of cardiac muscle. J Physiol. 1961;157:335–50.
Huang ZM, Prasad C, Britton FC, Ye LL, Hatton WJ, Duan D. Functional role of CLC-2 chloride inward rectifier channels in cardiac sinoatrial nodal pacemaker cells. J Mol Cell Cardiol. 2009;47(1):121–32.
Clemo HF, Feher JJ, Baumgarten CM. Modulation of rabbit ventricular cell volume and Na+/K+/2Cl− cotransport by cGMP and atrial natriuretic factor. J Gen Physiol. 1992;100(1):89–114.
Vaughan-Jones RD, Spitzer KW, Swietach P. Intracellular pH regulation in heart. J Mol Cell Cardiol. 2009;46(3):318–31.
Duan DY, Liu LL, Bozeat N, Huang ZM, Xiang SY, Wang GL, Ye L, Hume JR. Functional role of anion channels in cardiac diseases. Acta Pharmacol Sin. 2005;26(3):265–78.
Orchard CH, Cingolani HE. Acidosis and arrhythmias in cardiac muscle. Cardiovasc Res. 1994;28(9):1312–9.
Orchard CH, Kentish JC. Effects of changes of pH on the contractile function of cardiac muscle. Am J Physiol. 1990;258(6 Pt 1):C967–81.
Griffin M, Soufer A, Goljo E, Colna M, Rao VS, Jeon S, Raghavendra P, D’Ambrosi J, Riello R, Coca SG, Mahoney D, Jacoby D, Ahmad T, Chen M, Tang WHW, Turner J, Mullens W, Wilson FP, Testani JM. Real world use of hypertonic saline in refractory acute decompensated heart failure: a U.S. center’s experience. JACC Heart Fail. 2020;8(3):199–208.
Gandhi S, Mosleh W, Myers RB. Hypertonic saline with furosemide for the treatment of acute congestive heart failure: a systematic review and meta-analysis. Int J Cardiol. 2014;173(2):139–45.
US National Institutes of Health. ClinicalTrials.gov Mechanism and Effects of Manipulating Chloride Homeostasis in Acute Heart Failure. https://clinicaltrials.gov/ct2/show/NCT03446651?term=chloride&cond=Heart+Failure&draw=2&rank=2. Accessed 09 June 2020.
Skelton LA, Boron WF, Zhou Y. Acid–base transport by the renal proximal tubule. J Nephrol. 2010;23(Suppl 16):S4–18.
Agre P, Sasaki S, Chrispeels MJ. Aquaporins: a family of water channel proteins. Am J Physiol. 1993;265:F461.
Pichake J, Kharkar PS, Ceruso M, Supuran CT, Toraskar MP. Carbonic anhydrase inhibitors: design, synthesis, and biological evaluation of novel sulfonyl semicarbazide derivatives. ACS Med Chem Lett. 2014;5:793–6.
Dobyan DC, Bulger RE. Renal carbonic anhydrase. Am J Physiol. 1982;243(4):F311–24.
Breton S. The cellular physiology of carbonic anhydrases. JOP. 2001;2(4 Suppl):159–64.
Seki G, Frömter E. Acetazolamide inhibition of basolateral base exit in rabbit renal proximal tubule S2 segment. Pflugers Arch. 1992;422:60–5.
Kataoka H. Acetazolamide as a potent chloride-regaining diuretic: short- and long-term effects, and its pharmacologic role under the ‘chloride theory’ for heart failure pathophysiology. Heart Vessels. 2019;34(12):1952–60.
Khan MI. Treatment of refractory congestive heart failure and normokalemic hypochloremic alkalosis with acetazolamide and spironolactone. Can Med Assoc J. 1980;123(9):883–7.
Greenston JP. Carbonic anhydrase inhibitors for hypercapnic ventilatory failure in chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2001;1:CD002881.
Faisy C, Meziani F, Planquette B, et al. Effect of acetazolamide vs placebo on duration of mechanical ventilation among patients with chronic obstructive pulmonary disease: a randomized clinical trial. JAMA. 2016;315:480–8.
Verbrugge FH, Martens P, Ameloot K, Haemels V, Penders J, Dupont M, Tang WHW, Droogné W, Mullens W. Acetazolamide to increase natriuresis in congestive heart failure at high risk for diuretic resistance. Eur J Heart Fail. 2019;21(11):1415–22.
Wongboonsin J, Thongprayoon C, Bathini T, Ungprasert P, Aeddula NR, Mao MA, Cheungpasitporn W. Acetazolamide therapy in patients with heart failure: a meta-analysis. J Clin Med. 2019;8(3):E349.
Mullens W, Verbrugge FH, Nijst P, Martens P, Tartaglia K, Theunissen E, Bruckers L, Droogne W, Troisfontaines P, Damman K, Lassus J, Mebazaa A, Filippatos G, Ruschitzka F, Dupont M. Rationale and design of the ADVOR (Acetazolamide in Decompensated Heart Failure with Volume Overload) trial. Eur J Heart Fail. 2018;20(11):1591–600.
Acknowledgements
Funding
No funding was received for this study. No Rapid Service Fee was received by the journal for the publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Joseph Cuthbert, Sunil Bhandari, and Andrew L. Clark declare that they have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12674555.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Cuthbert, J.J., Bhandari, S. & Clark, A.L. Hypochloraemia in Patients with Heart Failure: Causes and Consequences. Cardiol Ther 9, 333–347 (2020). https://doi.org/10.1007/s40119-020-00194-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-020-00194-3