
Abstract
γ-Fe2O3@CuFAp nanocatalyst was synthesized and characterized by several methods including XRD, SEM, TEM, ICP, and FT-IR analyses. The γ-Fe2O3@CuFAp has been used as an efficient recyclable magnetic catalyst for synthesis of benzimidazoles. The γ-Fe2O3@CuFAp was conveniently separated from the reaction mixture by an external magnet, and could be reused at least 12 times without any considerable change in catalytic activity.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Benzimidazole is an important structural moiety which is present in many natural and synthetic compounds with a variety of interesting biological activities [1], such as anticancer [2], antiarrhythmic [3], antihistaminic [4], antiulcerative [5], antihypertensives, antiviral, antitumor [1], antibacterial, antifungals [6], anti-inflammatory [7], anticonvulsant, antidiabetic properties [8], and antidepressant [9]. The first benzimidazole was described by Hobrecker in 1872, who obtained 2,5 (or 2,6)-dimethylbenzimidazole by the reduction of 2-nitro-4-methylacetanilide [10]. Three years later, Ladenburg procured the identical compound by refluxing 3,4-diaminotoluene with acetic acid [11]. Various methodologies have been used for the synthesis of benzimidazole derivatives, which include conversion of esters using an aluminum reagent [12], the oxidative cyclodehydrogenation of Schiff bases, which is generated from o-phenylenediamine and aldehydes in the presence of several catalysts [13], such as cabalt(II) chloride hexahydrate [14], iodine [15], cetylpyridinium bromide [16], the condensation of o-aryldiamines and aldehyde in refluxing nitrobenzene [17], direct condensation of o-aryldiamines and carboxylic acids [18, 19] or their derivatives [20] in the presence of catalysts, such as polyphosphoric acid [21], ionic liquids [22], p-TsOH [23], SiO2 as solid support [24], microwave irradiation using polyphosphoric acid [25], and intramolecular N-arylations of amidines mediated by potassium hydroxide [26]. Although these reported approaches are significant, but some of them have drawbacks like strict reaction conditions, prolonged reaction period, low yield, expensive catalysts, non-recoverability of the catalysts, use of toxic solvents, and co-occurrence of several side reactions. In this report, we have synthesized a magnetic copper-incorporated fluorapatite (γ-Fe2O3@CuFAp) as higly efficient and green catalyst for the preparation of benzimidazoles derivatives.
Experimental part
Materials and reagents
FeCl2·4H2O, FeCl3·6H2O, ammonia solution (25%), Ca(NO3)2·4H2O, (NH4)2HPO4, o-phenyl diamine, Cu(OAc)2, TBAF, benzaldehydes, and other organic compounds were purchased from commercial sources.
Physical measurements
Melting points were recorded on a Büchi B-545 apparatus in open capillary tubes. 1H NMR spectra were recorded on a 400 MHz Bruker DRX-400, DMSO-d 6 as solvent, and TMS as an internal standard. FT-IR spectra were determined on an α-Bruker spectrometer. XRD was carried out on a Philips Analytical PC-APD. The Cu contents of samples were determined by ICP-OES on a Varian 730-ES instrument.
Synthesis of nano-Fe3O4 MNPs
Fe3O4 nanoparticles were synthesized by reported chemical co-precipitation technique of ferric and ferrous ions in alkali solution [27,28,29]. FeCl2·4H2O (0.368 g) and FeCl3·6H2O (1.0 g) were dissolved in deionized water (30 mL) under Ar atmosphere at room temperature. A magnetic stirring bar was added to the flask, and then, an ammonia solution (25%, 10 mL) was added into the mixed Fe(III)/Fe(II) solution with constant stirring at room temperature to increase pH until 11. After 1 h stirring, the black precipitate of Fe3O4 was collected by external magnet and washed with hot water three times.
Synthesis of γ-Fe2O3@FAp MNPs
γ-Fe2O3@FAp nanoparticles were synthesized using reported procedure with minor modifications [17]. The prepared Fe3O4 nanoparticles (0.429 g, 1.85 mmol) were dissolved in 15 mL deionized water and tetra-butyl ammonium fluoride (TBAF) (0.78 g, 3 mmol) was added. The solution was stirred vigorously for 10 min at room temperature. A solution of (NH4)2HPO4 (1.19 g, 9 mmol) and Ca(NO3)2.4H2O (3.54 g, 15 mmol) in 30 mL deionized water was added dropwise over 30 min to the mixture and heated at 80°C for 1 h. Subsequently, it cooled to room temperature and aged overnight. The solution was magnetic decanted and washed with hot deionized water (DW), three times. The product was air-dried and then kept in a furnace at 300°C for 3 h to give a dark-brown powder.
Synthesis of γ-Fe2O3@CuFAp MNPs
As shown in Scheme 1, 500 mg γ-Fe2O3@FAp was stirred with 8 mmol Cu(NO3)2·4H2O in 100 mL water at room temperature for a period of 2 h. The obtained slurry was magnetic decanted, washed with DW repeatedly, and dried at 100°C yielding γ-Fe2O3@CuFAp nanoparticles [30].

Synthesis of γ-Fe2O3@CuFAp MNPs
General procedure for the synthesis of benzimidazoles
To a mixture of an aldehyde (1 mmol) and 1,2-phenylenediamine (1.2 mmol) in acetonitrile (5 mL), γ-Fe2O3@CuFAp (0.04 g) was added. The mixture was stirred at 80°C, and the progress of reaction was monitored by TLC. After completion of the reaction, the catalyst was separated by an external magnet, washed with CH2Cl2, dried, and reused for a consecutive run under the same reaction conditions. To obtain the desired product, the solvent was evaporated and the mixture was extracted with EtOAc (3 × 10 mL). The extract was concentrated, and the crude product was purified by column chromatography using ethyl acetate-n-hexane (2:8) as eluent to afford the desired product 3.
2-Phenyl-1H-benzo[d]imidazole (Table 2, Entry 1)
m.p. 291–293 °C (lit.28, m.p. 292 °C); FT-IR (KBr): \(\bar{\nu };\) 3408, 3043, 1611, 1590, 1465, 755, 706 cm−1, 1H NMR (DMSO-d6, 400 MHz): δ 12.92 (s, 1H, NH), 8.55 (t, J = 7.2 Hz, 1H), 8.19 (d, J = 8.0 Hz, 2H), 7.68 (t, J = 7.2 Hz, 1H), 7.57–7.50 (m, 3H), 7.26–7.24 (m, 2H).
Results and discussion
Preparation and characterization of the catalyst
As described in the experimental section, Fe3O4 NPs were synthesized by a chemical co-precipitation technique of ferric and ferrous ions in alkali solution. Then, Fe3O4 NPs were coated with fluorapatite and was heated in the air at 100°C and then kept in the 300°C furnace for 3 h to give a reddish-brown powder γ-Fe2O3@FAp NPs. Structure of the catalyst was established by XRD, SEM, TEM, ICP, and FT-IR analyses.
Characterization of catalyst
XRD analysis
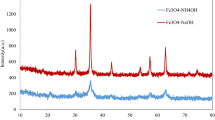
Figure 1 shows the XRD pattern of γ-Fe2O3@CuFAp NPs. The standard XRD data for γ-Fe2O3 and FAp are also depicted in the figure for comparison. Diffraction peaks at around 2θ = 17.4°, 21.4°, 27.7°, 30.4°, 35.3°, 37.5°, 39.6°, 41.6°, and 50.7 are readily distinguished from the XRD pattern. They agree with the cubic structure of maghemite (JCPDS file no. 39-1346).

XRD pattern of γ-Fe2O3@CuFAp NPs
Diffraction peaks at around 2θ = 12.7°, 19.6°, 25.5°, 26.7°, 29.7°, 30.2°, 32.8°, 33.9, 37.3°, 37.7°, 38.7°, 39.9°, 41.7°, 46.0°, 46.9°, 47.6°, 49.5°, 55.1°, 56.7°, 58.3°, 59.8°, 60.7°, 61.6°, 62.7°, 62.9°, and 66.3° are related to the FAp (JCPDS file no. 71-0880). The average crystallite size was calculated to be 12 nm for γ-Fe2O3 and 25 nm for FAp using the Scherrer equation.
Scanning electron microscopy (SEM)
The synthesized γ-Fe2O3@CuFAp nanoparticles were characterized by SEM (Fig. 2). The SEM image of the catalyst proved its spherical morphology. The average size of synthesized nanoparticles is about 50 nm according to the measurement software.

SEM image of γ-Fe2O3@CuFAp
Transmission electron microscopy (TEM)
TEM image of γ-Fe2O3@CuFAp is presented in Fig. 3. The average size of the nanoparticle was calculated to be 66 nm.

TEM image of γ-Fe2O3@CuFAp
Inductively coupled plasma-atomic emission spectroscopy (ICP-AES)
The ICP-AES was used to exact determination of the copper content of the material synthesized. Analysis confirmed 0.28 mmol Cu per gram of the γ-Fe2O3@CuFAp.
FT-IR
In the FT-IR spectrum of γ-Fe2O3@CuFAp, the bending mode of O–P–O which are overlapping with the stretching vibration of Fe–O appeared at 569 and 604 cm−1. The stretching vibrations of P–O bonds appeared at 1040 cm−1. The strong and broad band at 3024 cm−1 corresponds to the O–H groups and adsorbed water. The absence of a medium band at 632 cm−1 (relating to the stretching mode of OH in hydroxyapatite) refuses the presence of hydroxyapatite in the structure (Fig. 4).

FT-IR spectra of γ-Fe2O3@CuFAp
Catalytic activity of nano γ-Fe2O3@CuFAp for the synthesis of benzimidazoles
We have examined catalytic activity of nano-γ-Fe2O3@CuFAp for the synthesis of benzimidazoles (Scheme 2). At first, the reaction of o-phenylenediamine with benzaldehyde was chosen as a model reaction to optimize the reaction conditions (Table 1).

Synthesis of benzimidazoles
We found that in the presence of 0.04 g of nano γ-Fe2O3@CuFAp, the best yield of the product was obtained at 80°C in acetonitrile (Table 1, Entry 2). To show the role of the catalyst, similar reaction in the absence of the catalyst and in the presence of lower amount of nanomagnetic γ-Fe2O3@CuFAp was also examined. Under these conditions, the reactions led to the formation of the desired product in low yields and long reaction time (Entries 4 and 5).
To establish the generality of this method, the synthesis of various benzimidazoles was studied using different benzaldehydes under optimized reaction conditions (Table 2).
The reaction with aliphatic aldehydes such as acetaldehyde and phenylacetaldehyde resulted in a complex mixture due to aldol condensation.
A plausible mechanism for the formation of benzimidazoles catalyzed by γ-Fe2O3@CuFAp is shown in Scheme 3. The Lewis acid properties of γ-Fe2O3@CuFAp by coordination with the carbonyl group of aldehyde facilitate nucleophilic addition of 1,2-phenylenediamine to the activated aldehyde I to afford intermediate II. In the next step, the catalyst activates intermediate II followed by intramolecular Michael-type addition to afford the intermediate III. Finally, air oxidation of the intermediate III furnishes the desired products (3a–o).

Plausible mechanism for one-pot synthesis of benzimidazoles catalyzed by γ-Fe2O3@CuFAp
The reusability of a catalyst determines its lifetime. In this regard, the recyclability of γ-Fe2O3@CuFAp was investigated in a model reaction of o-phenylenediamine and benzaldehyde under optimized reaction conditions. The recovered catalyst washed with CH2Cl2, dried at room temperature, and reused for a similar reaction. The result showed that after 12 successive runs, catalytic activity of the catalyst was retained without any loss of activity (led to 83% average yield).
To examine the efficiency of the present method for the synthesis of benzimidazoles, preparation of compound 3a was compared with some of those reported in the literature (Table 3). It is evident from the results that the present protocol shows a very good comparability with previously reported data in terms of reaction times, yield, and reusability of the catalyst.
Conclusions
We have introduced the utilization of γ-Fe2O3@CuFAp, as a recyclable nanomagnetic catalyst, for green and efficient synthesis of aryl-substituted benzimidazoles through the condensation of 1,2-phenylenediamines with various aryl aldehydes. This method offers some advantages in terms of simplicity of performance, short reaction times, excellent yields, and mild condition. The current protocol could serve as a valuable alternative to known synthetic methods of benzimidazoles and is amenable for iterative combinatorial library generation.
References
Bansal, Y., Silakari, O.: The therapeutic journey of benzimidazoles: a review. Bioorg. Med. Chem. 20, 6208–6236 (2012)
Ibrahim, E.S.A., Omar, A.M.M.E., Mohsen, M.E., Khalil, M.A.: Novel potential anticancer agents derived from benzimidazole. J. Pharma. Sci. 69, 1348–1350 (1980)
Spasov, A.A., Smirnova, L.A., Iezhitsa, I.N., Sergeeva, S.A., Ozerov, A.A.: Pharmacokinetic of benzimidazole derivatives. Voprosy Meditsinskoj Khimii. 48(3), 252–258 (2002)
Janssens, F., Torremans, J., Janssen, M., Stokbroekx, R., Luyckx, M., Janssen, P.A.: New antihistaminic N-heterocyclic 4-piperidinamines. 1. Synthesis and antihistaminic activity of N-(4-piperidinyl)-1H-benzimidazol-2-amines. J. Med. Chem. 28(12), 1925–1933 (1985)
Santosh, P.C., Pandeya, S.N., Pathak, A.K.: Benzimidazole: a versatile chemical entity. Int. J. Res. Ayurveda Pharm. 2(6), 1126–1137 (2011)
Ozden, S., Atabey, D., Yildiz, S., Goker, H.: Synthesis and potent antimicrobial activity of some novel methyl or ethyl 1H-benzimidazole-5-carboxylates derivatives carrying amide or amidine groups. Bioorg. Med. Chem. 13, 1587–1597 (2005)
Chen, G., Liu, Z., Zhang, Y., Shan, X., Jiang, L., Zhao, Y., He, W., Feng, Z., Yang, S., Liang, G.: Synthesis and anti-inflammatory evaluation of novel benzimidazole and imidazopyridine derivatives. ACS Med. Chem. Lett. 4(1), 69–74 (2012)
Shingalapur, R.V., Hosamani, K.M., Keri, R.S., Hugar, M.H.: Derivatives of benzimidazole pharmacophore: synthesis, anticonvulsant, antidiabetic and DNA cleavage studies. Eur. J. Med. Chem. 45, 1753–1759 (2010)
Mathew, B., Suresh, J., Anbazhagan, S.: Development of novel (1-H) benzimidazole bearing pyrimidine-trione based MAO-A inhibitors: synthesis, docking studies and antidepressant activity. J. Saudi Chem. Soc. 20, S132–S139 (2016)
Wright, J.B.: The chemistry of the benzimidazoles. Chem. Rev. 48, 397–541 (1951)
Ladenburg, A.: Condensationsvorgänge in der Orthoreihe Ber. Eur. J. Org. Chem. 10(1), 1123–1131 (1877)
Neef, G., Eder, U., Sauer, G.: One-step conversions of esters to 2-imidazolines, benzimidazoles and benzothiazoles by aluminum organic reagents. J. Org. Chem. 46, 2824–2826 (1981)
Singh, M.P., Sasmal, S., Lu, W., Chatterjee, M.N.: Synthetic utility of catalytic Fe(III)/Fe(II) redox cycling towards fused heterocycles: a facile access to substituted benzimidazole, bisbenzimidazole and imidazopyridine derivatives. Synthesis 10, 1380–1390 (2000)
Khan, A.T., Parvin, T., Choudhury, L.H.: A simple and convenient one-pot synthesis of benzimidazole derivatives using cobalt(II) chloride hexahydrate as catalyst. Cheminform 40, 2339–2346 (2009)
Gogoi, P., Konwar, D.: An efficient and one-pot synthesis of imidazolines and benzimidazoles via anaerobic oxidation of carbon–nitrogen bonds in water. Tetrahedron Lett. 47(1), 79–82 (2006)
Chakrabarty, M., Karmakar, S., Mukherjee, R., Arima, S., Harigaya, Y.: A mild and expedient one-pot synthesis of substituted benzimidazoles in water using a phase-transfer catalyst. Monatsh. Chem. 140(4), 375–380 (2009)
Yadagiri, B., Lown, J.W.: Convenient routes to substituted benzimidazoles and imidazolo [4, 5-b] pyridines using nitrobenzene as oxidant. Synth. Commun. 20(7), 955–963 (1990)
Mohan, S.B., Behera, T.P., Kumar, B.R.: Microwave irradiation versus conventional method: synthesis of benzimidazolyl chalcone derivatives. Int. J. ChemTech. Res. 2(3), 1634–1637 (2010)
Tewari, A.K., Mishra, A.: Synthesis and antiviral activities of N-substituted-2-substituted-benzimidazole derivatives. Indian. J. Chem. B. 45B(2006), 489–493 (2006)
Perry, R.J., Wilson, B.D.: A novel palladium-catalyzed synthesis of 2-arylbenzimidazoles. J. Org. Chem. 58(25), 7016–7021 (1993)
Preston, P.N.: Benzimidazoles and congeneric tricyclic compounds. In: Weissberger, A., Taylor, E.C. (eds.) The chemistry of heterocyclic compounds, part 1, vol. 40, pp. 6–60. Wiley, New York (2008)
Ma, H., Wang, Y., Li, J., Wang, J.: Selective synthesis of 2-aryl-1-arylmethyl-1H-1,3-benzimidazoles promoted by Ionic liquid. Heterocycles 71, 135–140 (2007)
Xiangming, H., Huiqiang, M., Yulu, W.: p-TsOH catalyzed synthesis of 2-arylsubstituted benzimidazoles. Arkivoc 13, 150–154 (2007)
Ben-Alloum, A., Bakkas, S., Soufiaoui, M.: Benzimidazoles: oxydation heterocyclisante par le nitrobenzene ou le dimethylsulfoxyde sur silice et sous irradiation micro-ondes ou ultra-violet. Tetrahedron Lett. 39(25), 4481–4484 (1998)
Lu, J., Yang, B., Bai, Y.: Microwave irradiation synthesis of 2-substituted benzimidazoles using PPA as a catalyst under solvent-free conditions. Synth. Commun. 32(24), 3703–3709 (2002)
Baars, H., Beyer, A., Kohlhepp, S.V., Bolm, C.: Transition-metal-free synthesis of benzimidazoles mediated by KOH/DMSO. Org. Lett. 16, 536–539 (2014)
Massart, R., Dubois, E., Cabuil, V., Hasmonay, E.: Preparation and properties of monodisperse magnetic fluids. J. Magn. Magn. Mater. 149(1–2), 1–5 (1995)
Tang, B.Z., Geng, Y., Lam, J.W.Y., Li, B., Jing, X., Wang, X., Wang, F., Pakhomov, A.B., Zhang, X.X.: Processible nanostructured materials with electrical conductivity and magnetic susceptibility: preparation and properties of maghemite/polyaniline nanocomposite films. Chem. Mater. 11(6), 1581–1589 (1999)
Ho, K.M., Li, P.: Design and synthesis of novel magnetic core–shell polymeric particles. Langmuir 24(5), 1801–1807 (2008)
Mirfarjood, S.A., Mamahgani, M., Sheykhan, M.: Copper-exchanged magnetic-FAp: surface catalysis in decarboxylative coupling of a-oxocarboxylic acids with formamides. ChemistrySelect 2, 8650–8657 (2017)
Soleimani, E., Khodaei, M.M., Yazdani, H., Saei, P., Reza, J.Z.: Synthesis of 2-substituted benzimidazoles and benzothiazoles using Ag2CO3/Celite as an efficient solid catalyst. J. Iran. Chem. Soc. 12(7), 1281–1285 (2015)
Forouzani, M., Ghasemnejad-Bosra, H.: 1,3-Dibromo 5, 5-dimethylhydantoin (DBH)-catalyzed solvent-Free synthesis of 2-arylbenzimidazoles under microwave irradiation. E-J. Chem. 9(3), 1064–1069 (2012)
Shen, M., Driver, T.G.: Iron (II) bromide-catalyzed synthesis of benzimidazoles from aryl azides. Org. Lett. 10(15), 3367–3370 (2008)
Yu, J., Xia, Y., Lu, M.: Iron-catalyzed highly efficient aerobic oxidative synthesis of benzimidazoles, benzoxazoles, and benzothiazoles directly from aromatic primary amines under solvent-free conditions in the open air. Synth. Commun. 44(20), 3019–3026 (2014)
Brătulescu, G.: Mild, one-pot preparation of 2-substituted benzimidazoles from organic halides. Synth. Commun. 47(8), 811–817 (2017)
Shen, M.-G., Cai, C.H.: Ytterbium perfluorooctanesulfonates catalyzed synthesis of benzimidazole derivatives in fluorous solvents. J. Fluor. Chem. 128(3), 232–235 (2007)
Alvim, H.G.O., de Oliveira, H.C.B., Bataglion, G.A., Eberlin, M.N., Ramos, L.M., Silva, W.A.: Synthesis of 2-arylbenzimidazoles under mild conditions catalyzed by a heteropolyacid-containing task-specific ionic liquid and catalyst investigation by electrospray (tandem) mass spectrometry. RSC Adv. 5(88), 69418–69422 (2015)
Sharghi, H., Beyzavi, M.H., Doroodmand, M.M.: Reusable porphyrinatoiron(III) complex supported on activated silica as an efficient heterogeneous catalyst for a facile, one-pot, selective synthesis of 2-arylbenzimidazole derivatives in the presence of atmospheric air as a “green” oxidant at ambient temperature. Eur. J. Org. Chem. 2008(24), 4126–4138 (2008)
Behbahani, F.K., Rezaee, E., Fakhroueian, Z.: Synthesis of 2-substituted benzimidazoles using 25% Co/Ce-ZrO2 as a heterogeneous and nanocatalyst. Catal. Lett. 144(12), 2184–2190 (2014)
Acknowledgements
The authors gratefully acknowledge partial financial support from the Research Council of University of Guilan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mirfarjood, S.A., Mamaghani, M. & Sheykhan, M. Copper-incorporated fluorapatite encapsulated iron oxide nanocatalyst for synthesis of benzimidazoles. J Nanostruct Chem 7, 359–366 (2017). https://doi.org/10.1007/s40097-017-0245-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40097-017-0245-2