
Abstract
The structural and electronic properties of non-metal atoms (X = C, N, Si, P) doped (6,0) boron nanotube (BNT) have been considered in a systematic study by performing periodic spin polarized density functional theory (DFT) calculations. The studies showed that cylindrical shape of the nanotube is changed by doping, except for C substitution. Notably, all the substitution processes are endothermic and the C-substituted becomes energetically more stable than the other dopants. It is revealed that the C-doped BNT is semi-metal, whereas the nanotube in the presence of the other dopants remains semiconductor. Hence, the substitution is an effective way in narrowing band gap of the nanotube. Doping by N atom changes the band gap from indirect to direct, which can be suitable for optical applications. Thus, electronic structure of the tube has been controlled by type of the dopant atoms. Our study predicted that the BNTs in the presence of the dopants are promising candidate as interconnects for nano-devices as well as field emission devices.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Boron atom is one of the appealing and challenging elements to create novel structures [1]. Boron nanostructures (BNS) have been received special attention as a result of their innovative uses in medicine [2], superconductivity [3], hydrogen storage [4,5,6,7,8], and electrical interconnects [9, 10]. Up to date, many experimental efforts have been performed aiming to synthesize the BNSs [11,12,13,14,15]. Boron nanotube (BNT) was originally predicted for the first time by Boustani et al. [16] and then the BNTs have been investigated in many experimental and theoretical studies [3,4,5, 10,11,12,13, 17,18,19,20,21].
The previous studies confirmed that, hexagonal and triangular patterns were more stable than a buckled triangular or hexagonal boron sheet, which two and three center bonds of these sheets (nominated as α-sheet) are main reasons for their stability [17, 18]. Recently, tetragonal, hexagonal and dodecagonal cycles for carbon, boron nitride (BNNT) and aluminum nitride nanotubes have been proposed [22]. Theoretical studies also showed that a series of the BNTs may be obtained by rolling up the stable α-sheet [17, 18]. In all these studies, researchers have found out that the surface buckling is a common phenomenon for the BNTs. Unlike carbon nanotube (CNT) [23], silicon carbide nanotube (SiCNT) [24], C3N nanotube [25, 26], and g-C3N4 nanotube [27], the BNTs based on the α-sheet are predicted to be metallic [20, 21, 28,29,30] and semiconductor for small diameters (diameter <17 Å) [1, 17,18,19,20].
The structural and electronic properties of structures can be modified by chemical doping. Many experimental and theoretical investigations on substitution of non-metal impurity in CNT [31], boron nitride nanotube [32], SiCNT [33], phosphorene [34], TiO2 [35], and BNT [36,37,38], encouraged us to explore the properties of the BNT in the presence of non-metal impurity atoms. In the BNT case, it is noted that the previous studies [36,37,38] have been carried out only on hexagonal form (as an unstable form). Our main concerns on the stable form are listed as follows. (1) What is the effect of C, Si, N, and P atoms on the electronic properties of the BNT? (2) Do these atoms change the structural properties of the BNT? (3) What is the best site for doping and which substitution has the minimum formation energy (E Form)?
Computational methods
The spin-polarized DFT calculations were performed by means of the Quantum ESPRESSO package [39] on (6,0) armchair BNT. A (p,0) BNT corresponds to a (n,n) (n = p) CNT and a (p,p) BNT corresponds to a (n,o) (n = 3p) CNT [17]. The system is represented by a tetragonal supercell of 25 Å × 25 Å × 17.79 Å, containing 192 atoms for the perfect BNT, which the distance of the impurity adjacent images in z-axis is suitable. The distance between the outer walls of neighbor periodic images of the tube is at least 25 Å to negligible the interaction between the tubes. The Monkhorst–Pack meshes [40] of 1 × 1 × 7 were used for sampling the Brillouin zone and the tetrahedron method [41] was also used for the calculation of density of state (DOS) to achieve high accuracy. A plane-wave basis set with a cutoff value of 600 eV was used in combination with ultrasoft pseudopotentials. All structures were fully relaxed until the forces on each atom were less than 0.01 eV Å−1. To further analyze the electronic charge population analysis, we have performed the analysis with the Löwdin method [42]. Losing and gaining charge density are represented by negative and positive values for charge transfer (CT), respectively. Further, work function (Φ) is defined as the minimum energy required to remove an electron from the surface to a point in the vacuum [43].
Results and discussion
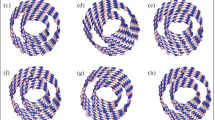
Full geometry optimization without any constraints on the (6,0) BNT was performed. The optimized structures are depicted in Fig. 1. The average calculated B–B bond length and diameter of the nanotube are about 1.71 and 9.5 Å, respectively. These values are in good agreement with the previous reports [17, 18]. As shown in Fig. 1a, buckling pattern is observed for the nanotube: the two atoms in the triangular regions become inequivalent with one moving radially outward (B atom labeled B1) and the other moving radially inward (B atom labeled B3). Based on Löwdin charge analysis [42], we found that the BNT is an amphoteric molecule, containing acidic sites [B atoms which lose charge like B1 (+0.19 |e|), B3 (+0.20 |e|), and B4 (+0.03 |e|)] and basic sites [B atoms which gain charge like B2 (−0.03 |e|)]. As a result of dissimilarity in the B atoms, four various positions for further analysis were considered.

Optimized structures of a perfect BNT, b C-doped BNT, c Si-doped BNT, d N-doped BNT, and e P-doped BNT. The tubes have been shown in different views. Pink, gray, cyan, blue, and orange colors represent B, C, Si, N, and P atoms, respectively
Let us consider first the structural properties of the doped BNT. For the doped BNT, one B atom is substituted by another atom in the supercell. As shown in Fig. 1, C atom retains on the surface of the tube, while the other dopants will leave the surface. As shown in Table 1, the minimum distance (d min) between C, Si, N, and P with the nearest B atom is found to be 1.60, 2.19, 1.37, and 2.09 Å, respectively. Due to large atomic radius of Si and P atoms, the long Si–B and P–B bond length are expected. Furthermore, Si, N, and P in the doped BNT are moving radially outward, outward, and inward, respectively. The protrusions of the dopant atom with respect to the surface of the tube are 2.0, 1.30, and −1.20 Å, for N, Si, and P atoms, respectively (the minus value is represented for inward moving). The drastic changes in the geometric structure of BNT can be seen in Fig. 1d where the N atom, as smaller study atoms, can locate among two B atoms and bonds with pyramidal like configurations, with bond angles close to 99.4°. The N-B bond length is quite smaller than the B–B bond.
To probe the relative stability of the X-doped BNT, the computational methods of the doped systems were estimated. The enhancement of energetic stability is supported by the E form. The E form of the most stable configurations is presented in Table 1. Through the total energy calculation, the E Form are computed using:
where E [impurity tube] and E [pure tube] are the total energy of the doped tube with the impurity atoms and the undoped case, respectively. Further, the µ B and µ X are the chemical potentials for B and X (C, Si, N, and P) atoms, respectively. The µ B, µ C, µ Si, µ N, and µ P are calculated as the total energy per atom in the rhombohedral structure of α-boron, pure graphene, Si bulk, gaseous N2 molecule, and P4 molecule, respectively. The positive value indicates that the process is endothermic. Due to unique structure of the BNT (hexagonal and triangular pattern), endothermic process can be expected. Endothermic processes for doping with non-metal atoms in different structures were obtained [44,45,46,47]. The most favorite site for C, Si, N, and P atoms is 2, 1, 1, and 3 sites, respectively. As a result, the E form depends on the occupation sites. It is noted that the doping with C atom is energetically more favorable the other dopants. The results showed that the doping with P atom is energetically more expensive than the doping with Si and N atoms which need to a high external energy. Our investigations showed that four parameters may be affected on stability of the tubes. These parameters are the number of electrons in the dopants, structural distortion of the tubes, the magnetic moment of the structures, and the amount of charge variation between the guest and host atoms. C and Si atoms have an extra valence electron with respect to B atom, while the other dopants have two more electros. It seems that the BNT does not have a good ability to accept two more electrons in the system. A small distortion, as another criterion for stability, has been observed in C- and Si-doped BNT systems. An important aspect of non-metal-doped BNT is the magnetic behavior of the system. The results of magnetic moments are also presented in Table 1. From the results, we can see that no magnetism is observed when N and P atoms are doped in the tube, while magnetic moment is observed after doping with C and Si atoms. This evidence can be explained by the number of electrons in the system (odd numbers of electrons in the N- and P-doped BNT). Unlike Si atom, C atom located at the surface of the nanotube; thus, C atom can hybridize with their neighboring atoms, showing small diamagnetic ordering. Interestingly, the tubes with large magnetic moment are favorable than the other systems. As an important result, the numbers of valence electrons, atomic radius of the dopants, the amounts of structural distortion, and total magnetic moment have synergic effects on the E form.
The electronic properties of small gap semiconducting (6,0) pure and impurity BNTs were also studied, as summarized in Table 1. Based on Löwdin analysis, the elements of the second (third) row of the periodic table gain (lose) charges. This charge variation can be explained by Pauling electronegativity. Although Si atom has higher electronegativity with respect to B atom, the B atoms in the BNT have more tendencies toward electrons respect than B in atomic form.
In addition, the electronic band structures and DOSs of the structures were also studied, as shown in Figs. 2 and 3. After doping with the atoms, several impurity states below the Fermi level (FL) will appear which can easily donate electron to conduction band minimum (CBM). The BNT with a donor impurity is known as n-type semiconductor. In the case of Si-, N- and, P-doped systems, the FL moves to valence band maximum (VBM) and some impurity states are introduced between the VBM and the CBM. These impurity states are filled (in the case of N and P) or present unpaired electrons (in the case of C and Si). As a result of these new states, charge carrier would become easier to transport from the VBM to the CBM. These transfers make a hole in the states under the FL. In other words, the conductivity of the BNT is enhanced by the impurity atoms.

Band structures of a perfect BNT, b C-doped BNT, c Si-doped BNT, d N-doped BNT, and e P-doped BNT. The band gaps have been assigned by green color

Density of states of a perfect BNT, b C-doped BNT, c Si-doped BNT, d N-doped BNT, and e P-doped BNT. The Fermi level is shown by dashed red line. The surfaces of the valence and conduction regions are shown by green color
The results of Table 1 revealed that the C, Si, and P can increase the field emission current of BNT by reducing the work function (Φ). In semiconductors, the field emission strongly depends on the Φ. By definition, the Φ (computed here as the difference between the energies of FL and the vacuum region) is the minimum energy needed by one electron to escape the surface. The inverse relation between Φ and electron emission from the semiconductor surface in the presence of an electric field can be observed [43]. We believe that the electronic and chemical properties of semiconductor BNT can alter in the presence of the doped atoms; therefore, it could be potentially used in several applications.
Conclusion
In summary, a first-principles method has been applied to investigate the influence of non-metal (C, Si, N, and P atoms) impurities on the stability and electronic properties of the BNT. The obtained results showed that the number of electrons in the dopants, structural distortion, the amount of charge variation between the guest and the host atoms, and the magnetic moment of the structures have impact role in the E form. Doping with C atom is energetically more favorable than the other dopants. The results showed that the doping with Si and N atoms needs to a high external energy and doping with P atom is more expensive energetically. The calculations indicated that the performance of BNT in field emission devices should be affected by non-metal impurities, and the magnetic moment is very sensitive to the number of electrons of guest atom. The calculated results showed that the substitution of B atom by the non-metal may induce change in the electronic structure, where the band gaps are narrowed for all the doped systems. Our study proposes that the BNT in the presence of the doped atom is potentially useful for spintronic applications and the development of magnetic nanostructures.
References
Lau, K.C., Pandey, R.: Ab initio study of single-walled boron nanotubes. In: Saxena, S. (ed.) Handbook of Boron Nanostructures, p. 49. Pan Stanford Publishing, Danvers (2016)
Sethia, K., Roy, I.: Applications of boron nanostructures in medicine. In: Saxena, S. (ed.) Handbook of Boron Nanostructures, p. 101. Pan Stanford Publishing, Danvers (2016)
Zhao, Y., Zeng, S., Ni, J.: Superconductivity in two-dimensional boron allotropes. Phys. Rev. B 93(1), 014502 (2016)
Li, M., Li, Y., Zhou, Z., Shen, P., Chen, Z.: Ca-coated boron fullerenes and nanotubes as superior hydrogen storage materials. Nano Lett. 9(5), 1944–1948 (2009)
An, H., Liu, C.-S., Zeng, Z.: Radial deformation-induced high-capacity hydrogen storage in Li-coated zigzag boron nanotubes. Phys. Rev. B 83(11), 115456 (2011)
Li, F., Wei, W., Sun, Q., Yu, L., Huang, B., Dai, Y.: Prediction of single-wall boron nanotube structures and the effects of hydrogenation. J. Phys. Chem. C 121(10), 5841–5847 (2017)
Wang, J., Du, Y., Sun, L.: Ca-decorated novel boron sheet: a potential hydrogen storage medium. Int. J. Hydrog. Energy 41(10), 5276–5283 (2016)
Molani, F., Jalili, S., Schofield, J.: Al-doped B80 fullerene as a suitable candidate for H2, CH4, and CO2 adsorption for clean energy applications. J. Saudi Chem. Soc. (2017). doi:10.1016/j.jscs.2017.07.001
Lin, B., Dong, H., Du, C., Hou, T., Lin, H., Li, Y.: B40 fullerene as a highly sensitive molecular device for NH3 detection at low bias: a first-principles study. Nanotechnology 27(7), 075501 (2016)
Baňacký, P., Noga, J., Szöcs, V.: Electronic structure of boron nanotubes: perspective material for nanotechnologies. Quantum Matter 4(4), 367–372 (2015)
Liu, F., Shen, C., Su, Z., Ding, X., Deng, S., Chen, J., Xu, N., Gao, H.: Metal-like single crystalline boron nanotubes: synthesis and in situ study on electric transport and field emission properties. J. Mater. Chem. 20(11), 2197–2205 (2010)
Ciuparu, D., Klie, R.F., Zhu, Y., Pfefferle, L.: Synthesis of pure boron single-wall nanotubes. J. Phys. Chem. B 108(13), 3967–3969 (2004)
Patel, R.B., Chou, T., Iqbal, Z.: Synthesis of boron nanowires, nanotubes, and nanosheets. J. Nanomater. 2015, 14 (2015)
Mannix, A.J., Zhou, X.-F., Kiraly, B., Wood, J.D., Alducin, D., Myers, B.D., Liu, X., Fisher, B.L., Santiago, U., Guest, J.R.: Synthesis of borophenes: anisotropic, two-dimensional boron polymorphs. Science 350(6267), 1513–1516 (2015)
Zhai, H.-J., Zhao, Y.-F., Li, W.-L., Chen, Q., Bai, H., Hu, H.-S., Piazza, Z.A., Tian, W.-J., Lu, H.-G., Wu, Y.-B.: Observation of an all-boron fullerene. Nat. Chem. 6(8), 727–731 (2014)
Boustani, I., Quandt, A.: Nanotubules of bare boron clusters: Ab initio and density functional study. EPL 39(5), 527 (1997)
Yang, X., Ding, Y., Ni, J.: Ab initio prediction of stable boron sheets and boron nanotubes: structure, stability, and electronic properties. Phys. Rev. B 77(4), 041402 (2008)
Tang, H., Ismail-Beigi, S.: First-principles study of boron sheets and nanotubes. Phys. Rev. B 82(11), 115412 (2010)
Singh, A.K., Sadrzadeh, A., Yakobson, B.I.: Probing properties of boron α-tubes by ab initio calculations. Nano Lett. 8(5), 1314–1317 (2008)
Bezugly, V., Kunstmann, J., Grundkötter-Stock, B., Frauenheim, T., Niehaus, T., Cuniberti, G.: Highly conductive boron nanotubes: transport properties, work functions, and structural stabilities. ACS Nano 5(6), 4997–5005 (2011)
Gonzalez Szwacki, N., Tymczak, C.: The symmetry of the boron buckyball and a related boron nanotube. Chem. Phys. Lett. 494(1), 80–83 (2010)
BabaeiPour, M., Safari, E.K., Shokri, A.: First-principles study of nanotubes within the tetragonal, hexagonal and dodecagonal cycle structures. Phys. E 86, 129–135 (2017)
Odom, T.W., Huang, J.-L., Kim, P., Lieber, C.M.: Structure and electronic properties of carbon nanotubes. J. Phys. Chem. B 104(13), 2794–2809 (2000)
Molani, F., Jalili, S., Schofield, J.: A computational study of platinum adsorption on defective and non-defective silicon carbide nanotubes. Monatsh. Chem. 146(6), 883–890 (2015)
Jalili, S., Molani, F., Akhavan, M., Schofield, J.: Role of defects on structural and electronic properties of zigzag C3N nanotubes: a first-principle study. Phys. E 56, 48–54 (2014)
Molani, F., Jalili, S., Schofield, J.: A novel candidate for hydrogen storage: Ca-decorated zigzag C3N nanotube. Int. J. Hydrog. Energy 41(18), 7431–7437 (2016)
Jalili, S., Molani, F., Schofield, J.: First principles study on energetic, structural, and electronic properties of defective g-C3N4-zz3 nanotubes. J. Theor. Comput. Chem. 13(04), 1450021 (2014)
Lau, K.C., Pati, R., Pandey, R., Pineda, A.C.: First-principles study of the stability and electronic properties of sheets and nanotubes of elemental boron. Chem. Phys. Lett. 418(4), 549–554 (2006)
Kunstmann, J., Quandt, A.: Broad boron sheets and boron nanotubes: an ab initio study of structural, electronic, and mechanical properties. Phys. Rev. B 74(3), 035413 (2006)
Boustani, I., Quandt, A., Hernández, E., Rubio, A.: New boron based nanostructured materials. J. Chem. Phys. 110(6), 3176–3185 (1999)
Ye, J., Shao, Q., Wang, X., Wang, T.: Effects of B, N, P and B/N, B/P pair into zigzag single-walled carbon nanotubes: a first-principle study. Chem. Phys. Lett. 646, 95–101 (2016)
Li, K., Ye, J., Zhang, J., Wang, X., Shao, Q.: The stability and electronic structures of Si/O/Al/P atom doped (5, 0) boron nitrogen nanotubes with Stone-Wales defects: density functional theory studies. Phys. E 87, 112–117 (2017)
Gali, A.: Ab initio study of nitrogen and boron substitutional impurities in single-wall SiC nanotubes. Phys. Rev. B 73(24), 245415 (2006)
Boukhvalov, D.W.: Atomic and electronic structure of nitrogen-and boron-doped phosphorene. Phys. Chem. Chem. Phys. 17(40), 27210–27216 (2015)
Di Valentin, C., Pacchioni, G.: Trends in non-metal doping of anatase TiO2: B, C, N and F. Catal. Today 206, 12–18 (2013)
Jain, S.K., Srivastava, P.: Structural stability of nitrogen-doped ultrathin single-walled boron nanotubes: an Ab initio study. Appl. Nanosci. 2(3), 345–349 (2012)
Jain, S.K., Srivastava, P.: Electronic properties of C-substituted boron nanotubes: a first principles study. EPJ B 86(7), 1–7 (2013)
Jain, S.K., Srivastava, P.: Effect of nitrogen impurity on electronic properties of boron nanotubes. Adv. Cond. Matter Phys. 2014, 706218 (2014)
Giannozzi, P., Baroni, S., Bonini, N., Calandra, M., Car, R., Cavazzoni, C., Ceresoli, D., Chiarotti, G.L., Cococcioni, M., Dabo, I.: QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21(39), 395502 (2009)
Monkhorst, H.J., Pack, J.D.: Special points for Brillouin-zone integrations. Phys. Rev. B 13(12), 5188–5192 (1976)
Blöchl, P.E., Jepsen, O., Andersen, O.K.: Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 49(23), 16223 (1994)
Löwdin, P.O.: On the non-orthogonality problem connected with the use of atomic wave functions in the theory of molecules and crystals. J. Chem. Phys. 18(3), 365–375 (1950)
Kittel, C.: Introduction to Solid State Physics, 8th edn. Wiley, Hoboken (2005)
Zhou, Z., Gao, X., Yan, J., Song, D., Morinaga, M.: A first-principles study of lithium absorption in boron-or nitrogen-doped single-walled carbon nanotubes. Carbon 42(12), 2677–2682 (2004)
Cruz-Silva, E., Lopez-Urias, F., Munoz-Sandoval, E., Sumpter, B.G., Terrones, H., Charlier, J.-C., Meunier, V., Terrones, M.: Electronic transport and mechanical properties of phosphorus- and phosphorus-nitrogen-doped carbon nanotubes. ACS Nano 3(7), 1913–1921 (2009)
Audiffred, M., Elías, A.L., Gutiérrez, H.R., López-Urías, F., Terrones, H., Merino, G., Terrones, M.: Nitrogen–silicon heterodoping of carbon nanotubes. J. Phys. Chem. C 117(16), 8481–8490 (2013)
Yuan, N., Bai, H., Ma, Y., Ji, Y.: First-principle simulations on silicon-doped armchair single-walled carbon nanotubes of various diameters. Phys. E 64, 195–203 (2014)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Molani, F. The effect of C, Si, N, and P impurities on structural and electronic properties of armchair boron nanotube. J Nanostruct Chem 7, 243–248 (2017). https://doi.org/10.1007/s40097-017-0234-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40097-017-0234-5