
Abstract
Objective
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic challenges national health systems and the global economy. Monitoring of infection rates and seroprevalence can guide public health measures to combat the pandemic. This depends on reliable tests on active and former infections. Here, we set out to develop and validate a specific and sensitive enzyme linked immunosorbent assay (ELISA) for detection of anti-SARS-CoV-2 antibody levels.
Methods
In our ELISA, we used SARS-CoV-2 receptor-binding domain (RBD) and a stabilized version of the spike (S) ectodomain as antigens. We assessed sera from patients infected with seasonal coronaviruses, SARS-CoV-2 and controls. We determined and monitored IgM-, IgA- and IgG-antibody responses towards these antigens. In addition, for a panel of 22 sera, virus neutralization and ELISA parameters were measured and correlated.
Results
The RBD-based ELISA detected SARS-CoV-2-directed antibodies, did not cross-react with seasonal coronavirus antibodies and correlated with virus neutralization (R2 = 0.89). Seroconversion started at 5 days after symptom onset and led to robust antibody levels at 10 days after symptom onset. We demonstrate high specificity (99.3%; N = 1000) and sensitivity (92% for IgA, 96% for IgG and 98% for IgM; > 10 days after PCR-proven infection; N = 53) in serum.
Conclusions
With the described RBD-based ELISA protocol, we provide a reliable test for seroepidemiological surveys. Due to high specificity and strong correlation with virus neutralization, the RBD ELISA holds great potential to become a preferred tool to assess thresholds of protective immunity after infection and vaccination.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coronaviruses (CoV) are known to cause respiratory diseases in humans. The alphacoronaviruses HCoV-229E and HCoV-NL63 as well as the betacoronaviruses HCoV-OC43 and HCoV-HKU1 circulate seasonally in humans and cause common colds [1]. In contrast, the severe acute respiratory syndrome-related coronavirus 1 (SARS-CoV-1), the Middle East respiratory syndrome-related coronavirus (MERS-CoV) and SARS-CoV-2 are zoonotic betacoronaviruses and can cause life-threatening severe respiratory distress syndromes and pandemics [2].
The immune response to the seasonal CoVs and SARS-CoV and MERS-CoV has been studied intensively [3, 4]. They are able to trigger a humoral immune response that correlates with disease severity [3]. Of note, mild infections resulted in short-lived and very low antibody titers near the detection limit. This loss of humoral immunity has been linked to the occurrence of (re-)infections with seasonal CoVs [3].
The current SARS-CoV-2-triggered COVID-19 pandemic started in December 2019 in Wuhan, Hubei province of China and has led with August 2020 to > 18 million confirmed infections [5]. Diagnosis of COVID-19 relies on PCR testing of respiratory specimen [6]. However, to assess whether a patient had recovered from a previous infection serological analyses are required. Serological surveys for SARS-CoV-2 antibody responses are not only important to monitor how much of a given population has been infected, but also serology will be required to assess vaccine responses. Finally, serology will be key to identify the quality of convalescent plasma that can be applied within clinical (compassionate) trials [7].
To allow for reliable serology, specific and sensitive assays are urgently needed. Here, we provide and validate a robust and simple ELISA protocol which is based on the SARS-CoV-2 receptor-binding domain (RBD). With this protocol, we performed a cross-sectional analysis of sera from PCR-proven COVID-19 patients. We demonstrate correlation of the ELISA test results with neutralizing antibody levels in a neutralization assay using a clinical isolate.
Materials and methods
Patient samples
The SARS-CoV-2 ELISA was validated using pseudonymized samples from patients aged older than 18 years from the diagnostic repository of the Institute of Clinical Microbiology and Hygiene, University Hospital Regensburg, originating from summers of 2016–2019. Potential cross-reactivity of the ELISA protocol was analyzed using sera from patients with PCR-proven seasonal coronavirus infections and detectable antibody reactivity against seasonal coronavirus antigens. Sensitivity of the protocol was quantified with sera from patients with PCR-proven SARS-CoV-2 infection at different time points. This procedure was approved by the ethical commission of the Faculty for Medicine, University of Regensburg, Regensburg, Germany (ref. no. 20-1854-101).
Design of recombinant proteins
S-protein sequences used for sequence analysis and gene synthesis are given in Table S1. Phylogenetic analysis, synthesis and cloning of the S protein variants used in this study (Fig. S1) are described in the supplementary materials and methods.
Protein structure analysis and visualization
Protein structures were visualized and analyzed by Pymol (LLC Schrodinger) using the structural data from the Protein Data Bank (https://www.rcsb.org) repository using entry codes 6vsb and 6m17.
Protein production and purification
Proteins were expressed in Expi293 cells (Thermo Fisher Scientific) in different scales using the commercial ExpiFectamine™ system. Affinity purification of the proteins and quality controls are described in the supplementary materials and methods.
Line blot assay
CoV line blot assay (beta-version of recomLine Coronavirus IgG assay) was performed as described in the manufacturer’s protocol. Line blots were evaluated by visual inspection and using recomScan software (Mikrogen Diagnostik, Neuried, Germany).
Enzyme-linked immunosorbent assay
A detailed ELISA protocol is provided in the supplementary materials and methods. Commercial Anti-SARS-CoV-2-ELISA IgG (EUROIMMUN, Lübeck, Germany) was performed according to the manufacturer’s recommendations.
Virus isolation and virus load quantification
SARS-CoV-2 isolation from respiratory specimen, determination of the viral loads using quantitative SARS-CoV-2 real-time RT-PCR (RT-qPCR) and 50% tissue culture infective dose (TCID50) are described in the supplementary materials and methods.
Virus neutralization assay
Virus neutralization assay using SARS-CoV-2 from respiratory specimen is described in the supplementary materials and methods.
Data evaluation and curve fitting
Experimental data were evaluated and plotted using GraphPad Prism (GraphPad Prism version 8.4.2 for Windows, GraphPad Software, San Diego, California USA).
Results
S-protein’s RBD has been shown to be SARS-CoV-1’s Achilles’ heel [8]. Due to the homology between the S-proteins as well as the RBDs of SARS-CoV-1 and SARS-CoV-2 (Fig. 1a), this may also hold true for the virus driving the current pandemic. This is why we decided to establish an ELISA protocol, which uses the SARS-CoV-2 RBD and stabilized ectodomain (StabS [9]) as antigens (Fig. 1b). To promote robust production of the two recombinant proteins, coding sequences were codon-optimized and the autologous S-protein signal peptide was replaced by a minimal version of the tissue plasminogen activator (tPA) signal peptide developed in our lab. RBD yielded 70 mg/l supernatant for the RBD and 14 mg/l supernatant for the StabS protein. Purity and homogeneity were verified by reducing SDS-PAGE and size exclusion chromatography (Fig. 1c, d).

Antigens used in SARS-CoV-2 ELISA. a Phylogenetic trees calculated for the RBD and complete S-protein of clinically relevant members of the coronavirus family. Trees are calculated on the basis of percent identity between each pair of sequences in the respective alignment. b Structure of the S-protein ectodomain in complex with its receptor, angiotensin-converting enzyme 2 (ACE2). c Reducing SDS-PAGE (linear gradient of 8–16% polyacrylamide) of the purified RBD and StabS protein. d Size exclusion chromatography of the purified RBD and StabS protein. e ELISA titrations of SARS-CoV-1 and SARS-CoV-2-binding antibody CR3022 against immobilized StabS and RBD [OD450–630: optical density (OD) at 450 nm after background subtraction at 630 nm]. Resulting dissociation constants (KD) are given in the diagram
A number of SARS-CoV-2 serology ELISA protocols have been published recently [10,11,12,13,14,15]. In our protocol, the antigens were directly absorbed to the plate’s plastic surface. To control protein integrity, we used our ELISA protocol in combination with the structure-dependent monoclonal anti-RBD antibody CR3022 [16,17,18]. High affinity binding as reflected by a KD of 0.27 nM for the RBD protein and 0.48 nM for StabS as well as a strong absorption (OD450–630 > 1 at saturation after 4 min of development time) in both cases demonstrated sufficient amounts of well-folded protein (Fig. 1e). We used CR3022 at saturation concentration to test the stability of RBD-coated plates, which had been coated up to 8 days earlier. No performance loss was detected during this period when plates were stored at room temperature in PBS-T (Fig. S2).
We used our ELISA to screen for anti-S and anti-RBD antibodies in sera from COVID-19 patients and controls. In a first step, we quantified anti-RBD responses and anti-S responses in 22 sera, which displayed differential ELISA signals (Fig. S3).
We found that antibody responses against StabS correlated very well with antibody response to RBD as determined by correlation of OD450–630 at 1:100 dilution, area under the curve (AUC), effective concentration at 50% signal (EC50) and titer (Fig. S4). Moreover, comparable signal strengths in these analyses suggest that RBD is a key immunogenic determinant of anti-S responses (Fig. S3). Therefore, anti-RBD antibody levels represent a valuable surrogate for testing anti-S-directed antibody responses.
High-throughput titration of sera for the purpose of determining end point titers or calculating EC50 values—in particular in seroepidemiological surveillance studies on broader population scale—is a time- and material-consuming procedure. Thus, we tested whether OD450–630 at 1:100 dilution of sera correlate with standard serum characteristics such as area under the curve (AUC), effective concentration at 50% signal (EC50) and titer. This analysis revealed that 1:100 dilution of sera reflects total serum antibody responses (Fig. S5). Therefore, we used single-point measurements for all further assays.
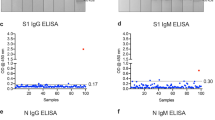
Next, we wanted to establish cutoff values for anti-RBD-directed IgG, IgA and IgM antibody responses following the requirements for validation of diagnostic assays in clinical virology [19]. For that purpose, we measured 190 SARS-CoV-2 naïve sera that had been collected before the current SARS-CoV-2 pandemic (Fig. S6). As recently proposed by Okba et al. [11], we used the mean of the background signals plus six standard deviations (SD) to define the cutoff value. Using these parameters, we determined the specificity of our assay by measuring 1000 independent SARS-CoV-2 infection naïve sera. We obtained a false positive rate of 7 out of 1000 sera, corresponding to a specificity of 99.3% (Fig. 2).

Assay specificity of RBD ELISA. To define the specificity of the assay, 1000 sera isolated in the summers of 2016 and 2018 (SARS-CoV-2 naïve control group) and 34 sera of patients with seasonal corona virus infection (seasonal CoV) were measured (median is shown). The cutoff is set at six standard deviations above the mean of the reference panel (borderline ± 10% cutoff)
To further verify the specificity of our ELISA, we tested whether sera from patients, that suffered from infection with seasonal CoVs, cross-react with our assay. For that purpose, we retrieved in a total of 43 sera from patients that had PCR-proven seasonal corona virus infection from our diagnostic repository. In a commercial line blot assay, 34 sera out of 43 scored positive for seasonal CoV-specific antibodies (Table S2). We used these 34 sera to further validate our RBD ELISA. Of utmost importance, none of these seasonal coronavirus sera showed any cross-reactivity in our ELISA (Fig. 2).
To verify precision and reproducibility of our ELISA [19], we used a minimal panel of seasonal (N = 5), naïve (N = 15), weakly IgG reactive (N = 10) and strongly IgG reactive (N = 10) sera. For weakly IgG reactive sera, the relative standard deviation (σrel) was 3.98% for IgG, 1.72% for IgM and 7.15% for IgA; for strongly reactive IgG positive sera σrel was determined to be 0.16% for IgG, 0.22% for IgM, and 2.73% for IgA. Combined inter-assay and inter-operator variabilities for weakly IgG reactive sera was determined as a σrel of 10.26% and for strong IgG reactive sera σrel was calculated to be 4.12%.
To determine the sensitivity of our assay, we analyzed sera from patients suffering from COVID-19 (N = 144), assuming that after symptomatic SARS-CoV-2 infection eventually all subjects develop antibodies. We quantified the anti-RBD IgG responses, which correlated well (R2 = 0.8812, Spearmen’s ρ = 0.917, p value < 0.0001) with anti-SARS-CoV-2 responses measured using a commercial IgG ELISA (EUROIMMUN, Fig. S7) that has been validated recently [20]. Next, we determined the IgM, IgG and IgA levels at different time frames after the first detection of SARS-CoV-2 RNA by RT-qPCR in our cohort (proven infection; Fig. 3a–c). At > 10 days after proven infection, these assays displayed sensitivities of 92% for IgA, 96% for IgG and 98% for IgM. However, we found that the antibody responses were already remarkably elevated at early time points after proven infection and, in concordance with previous findings, did not display a steady increase over time [21]. We speculated that infection preceded virus testing by several days. For 41% of the subjects (N = 59), the time point of symptom onset was available and we could calculate the average period between symptom onset and detection of SARS-CoV-2 RNA by RT-qPCR (5.7 days; Fig. S8). This suggests that the majority of patients who were subjected to PCR testing had already suffered from COVID-19 for 5.7 days. Therefore, an analysis of anti-RBD responses in relation to days after symptom onset should result in lower anti-RBD responses at early time points and a steady increase of antibody levels over time. Indeed, in the subgroup of patients where the time point of symptom onset was available, we detected very low antibody responses early after symptom onset and a steady increase in all anti-RBD antibody isotypes (Fig. 3d–f).

Serological testing of COVID-19 patients using RBD ELISA. a–c Serum reactivities of different immunoglobulin isotypes at 1:100 dilution of 144 sera of different time points after positive SARS-CoV-2 RT-qPCR, 1 per patient and time point (S/CO = signal/cutoff). Sampling of sera > 10 days post-PCR positive ranges from 11 to 29 days. Sera from patients with < 300 RNA copies/ml in respiratory specimen are shown as open circles, all others as closed circles. d–f Serum reactivities at 1:100 dilution (S/CO values) of 59 sera of different time points after onset of symptoms (subset of values from a–c with known case history), one per patient and time point
Neutralizing antibodies correlate with protection against several pathogens. Since we used the RBD of SARS-CoV-2, we expected the serum reactivity measured in our ELISA to correlate with virus neutralization due to sterical hindrance of RBD’s binding to its receptor ACE2 (Fig. 1b). In a first step, we isolated ten SARS-CoV-2 strains from respiratory specimen of COVID-19 patients. Of three different cell lines, kidney epithelial cells (Vero) supported virus propagation more efficiently than hepatocarcinoma-derived Huh-7 cells and lung carcinoma-derived A549 cells (Fig. 4a). For this reason, Vero cells were selected for neutralization experiments, using a highly replicative and cytopathic SARS-CoV-2 isolate (strain CA).

Virus isolation and neutralization assay. a Isolation of ten SARS-CoV-2 strains from respiratory specimen using three different cell lines (A549, derived from lung carcinoma; Huh7, hepatocyte-derived carcinoma; Vero, kidney epithelial cells from African green monkey; nd = not done). Viral loads in cell culture supernatants were determined using RT-qPCR. b After titration of the cytopathic SARS-CoV-2 strain CA, Vero cells were infected at a multiplicity of infection (MOI) of 0.05. Prior to infection, the virus was incubated with dilutions of 22 serum samples (fourfold serial dilutions starting at 1:20). Of these, six (S1–S6, black), eight (S7–S14, orange), and eight (S15–S22, red) samples displayed negative, medium, and high SARS-CoV-2 IgG reactivities, respectively, in the RBD-based ELISA. Two days post-infection, viral loads were determined in cell culture supernatants using RT-qPCR
Prior to infection of Vero cells, the virus was incubated with serial dilutions of the 22 sera, which displayed differential ELISA titrations (Fig. S3). After removal of the inoculum at 12–24 h post-infection, viral loads were determined in cell culture supernatants at 48 h post-infection using SARS-CoV-2 RT-qPCR. All sera which were negative in the ELISA (N = 6) did not inhibit virus entry and replication (Fig. 4b). In contrast, sera with borderline or positive ELISA values reduced SARS-CoV-2 viral loads by two (N = 2) to six log10 (N = 14). Notably, the reduction of viral loads showed a varying pattern, from a sudden to a more gradual inhibition of viral replication. Neutralization capacity was more pronounced in sera with high ELISA values. Most importantly, neutralization titers (given as IC50 values) correlated strongly with anti-RBD (R2 = 0.8943, Spearmen’s ρ = 0.965, p value < 0.0001) and anti-StabS (R2 = 0.9057, Spearmen’s ρ = 0.964, p value < 0.0001) antibody levels (Fig. 5a, b and Fig. S9a–f).

Correlation of antibody levels with SARS-CoV-2 neutralization. Correlation of OD from IgG ELISA measurements at 1:100 serum dilution with log IC50 values obtained from the neutralization experiments for a panel of 22 reference sera using RBD (a) and StabS (b) as an antigen. Coefficients of determination (R2), Spearmen’s ρ, p value, linear regression line (solid line) and 95% confidence intervals (dashed line) are given in the diagrams
Discussion
We established and validated a protocol for an easy-to-perform, robust and sensitive ELISA for detection of S-directed SARS-CoV-2 antibody responses in sera that correlate with SARS-CoV-2 virus neutralization.
As reported for a commercial assay our ELISA is highly specific (99.3%) as well, and sensitivity is dependent on the time point after infection when the serum sample is taken [20]. In concordance with findings of others, we detected robust antibody levels at approximately 2 weeks after onset of symptoms [22, 23]. Moreover, our data suggest that seroconversion for all Ig isotypes requires at least 6–10 days after onset of symptoms. In our cohort, IgM levels tend to appear first, followed by IgG conforming to earlier reports on SARS-CoV-2 Ig responses [21, 24,25,26,27,28]. Of note, we detected a late rise of the median IgA levels in our cohort. It is possible that this is an etiopathologic predictor of COVID-19 [29]. Taken into account, that the cohort of our cross-sectional study contained only symptomatic COVID-19 cases, seroconversion rates in oligo- or asymptomatic patients, the persistence of antibody levels and the role of antibodies in mucosal fluids warrant further investigation. Longitudinal epidemiological surveillance studies to address these questions are urgently needed. We provide an assay that can be easily implemented to quantify antibody responses in COVID-19.
The presence of RBD-directed antibodies, as determined by the herein described approach, correlate with SARS-CoV-2 neutralization. These findings strongly suggest that anti-RBD antibodies may confer protection and that the RBD as well as StabS proteins used in our study can be applied for sorting B cells and generating neutralizing monoclonal antibodies. Surveillance of anti-RBD and anti-StabS antibody responses therefore represent, as far as we analyzed, a reliable, simple, quick and high-throughput compatible alternative for any type of state of the art neutralization assay such as standard plaque reduction or lentiviral- and VSV-derived pseudotype assays. Such robust correlates will gain further importance once we start determining thresholds for protective immunity as a result of natural infection and, more importantly, following vaccination.
References
van der Hoek L. Human coronaviruses: what do they cause? Antivir Ther. 2007;12:651–8.
Fung TS, Liu DX. Human coronavirus: host–pathogen interaction. Annu Rev Microbiol. 2019;73:529–57. https://doi.org/10.1146/annurev-micro-020518-115759.
Kellam P, Barclay W. The dynamics of humoral immune responses following SARS-CoV-2 infection and the potential for reinfection. J Gen Virol. 2020. https://doi.org/10.1099/jgv.0.001439.
Dijkman R, Jebbink MF, El Idrissi NB, Pyrc K, Muller MA, Kuijpers TW, et al. Human coronavirus NL63 and 229E seroconversion in children. J Clin Microbiol. 2008;46:2368–73. https://doi.org/10.1128/JCM.00533-08.
Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020;20:533–4. https://doi.org/10.1016/S1473-3099(20)30120-1.
WHO. Laboratory testing for 2019 novel coronavirus (2019-nCoV) in suspected human cases: interim guidance. 2020. www.who.int.
Li L, Zhang W, Hu Y, Tong X, Zheng S, Yang J, et al. Effect of Convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19: a randomized clinical trial. J Am Med Assoc. 2020. https://doi.org/10.1001/jama.2020.10044.
Berry JD, Hay K, Rini JM, Yu M, Wang L, Plummer FA, et al. Neutralizing epitopes of the SARS-CoV S-protein cluster independent of repertoire, antigen structure or mAb technology. MAbs. 2010;2:53–66. https://doi.org/10.4161/mabs.2.1.10788.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–3. https://doi.org/10.1126/science.abb2507.
Stadlbauer D, Amanat F, Chromikova V, Jiang K, Strohmeier S, Arunkumar GA, et al. SARS-CoV-2 seroconversion in humans: a detailed protocol for a serological assay, antigen production, and test setup. Curr Protoc Microbiol. 2020;57:e100. https://doi.org/10.1002/cpmc.100.
Okba NMA, Muller MA, Li W, Wang C, Geurtsvankessel CH, Corman VM, et al. Severe acute respiratory syndrome coronavirus 2 - specific antibody responses in coronavirus disease patients. Emerg Infect Dis. 2019. https://doi.org/10.3201/eid2607.200841.
Sun B, Feng Y, Mo X, Zheng P, Wang Q, Li P, et al. Kinetics of SARS-CoV-2 specific IgM and IgG responses in COVID-19 patients. Emerg Microbes Infect. 2020;9:940–8. https://doi.org/10.1080/22221751.2020.1762515.
Amanat F, Stadlbauer D, Strohmeier S, Nguyen THO, Chromikova V, McMahon M, et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat Med. 2020. https://doi.org/10.1038/s41591-020-0913-5.
Freeman B, Lester S, Mills L, Rasheed MAU, Moye S, Abiona O, et al. Validation of a SARS-CoV-2 spike protein ELISA for use in contact investigations and serosurveillance. bioRxiv. 2020. https://doi.org/10.1101/2020.04.24.057323.
Guo L, Ren L, Yang S, Xiao M, Chang YF, et al. Profiling early humoral response to diagnose novel coronavirus disease (COVID-19). Clin Infect Dis. 2020. https://doi.org/10.1093/cid/ciaa310.
ter Meulen J, van den Brink EN, Poon LL, Marissen WE, Leung CS, Cox F, et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. 2006;3:e237. https://doi.org/10.1371/journal.pmed.0030237.
Tian X, Li C, Huang A, Xia S, Lu S, Shi Z, et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microbes Infect. 2020;9:382–5. https://doi.org/10.1080/22221751.2020.1729069.
Yuan M, Wu NC, Zhu X, Lee CD, So RTY, Lv H, et al. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science. 2020;368:630–3. https://doi.org/10.1126/science.abb7269.
Rabenau HF, Kessler HH, Kortenbusch M, Steinhorst A, Raggam RB, Berger A. Verification and validation of diagnostic laboratory tests in clinical virology. J Clin Virol. 2007;40:93–8. https://doi.org/10.1016/j.jcv.2007.07.009.
Meyer B, Torriani G, Yerly S, Mazza L, Calame A, Arm-Vernez I, et al. Validation of a commercially available SARS-CoV-2 serological immunoassay. Clin Microbiol Infect. 2020. https://doi.org/10.1016/j.cmi.2020.06.024.
Wolfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Muller MA, et al. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581:465–9. https://doi.org/10.1038/s41586-020-2196-x.
Long QX, Liu BZ, Deng HJ, Wu GC, Deng K, Chen YK, et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med. 2020;26:845–8. https://doi.org/10.1038/s41591-020-0897-1.
Caruana G, Croxatto A, Coste AT, Opota O, Lamoth F, Jaton K, et al. Diagnostic strategies for SARS-CoV-2 infection and interpretation of microbiological results. Clin Microbiol Infect. 2020. https://doi.org/10.1016/j.cmi.2020.06.019.
Zhao J, Yuan Q, Wang H, Liu W, Liao X, Su Y, et al. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clin Infect Dis. 2020. https://doi.org/10.1093/cid/ciaa344.
Whitman JD, Hiatt J, Mowery CT, Shy BR, Yu R, Yamamoto TN, et al. Test performance evaluation of SARS-CoV-2 serological assays. medRxiv. 2020. https://doi.org/10.1101/2020.04.25.20074856.
To KK, Tsang OT, Leung WS, Tam AR, Wu TC, Lung DC, et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect Dis. 2020;20:565–74. https://doi.org/10.1016/S1473-3099(20)30196-1.
Tan W, Lu Y, Zhang J, Wang J, Dan Y, Tan Z, et al. Viral kinetics and antibody responses in patients with COVID-19. medRxiv. 2020. https://doi.org/10.1101/2020.03.24.20042382.
Lassaunière R, Frische A, Harboe ZB, Nielsen AC, Fomsgaard A, Krogfelt KA, et al. Evaluation of nine commercial SARS-CoV-2 immunoassays. medRxiv. 2020. https://doi.org/10.1101/2020.04.09.20056325.
Cervia C, Nilsson J, Zurbuchen Y, Valaperti A, Schreiner J, Wolfensberger A, et al. Systemic and mucosal antibody secretion specific to SARS-CoV-2 during mild versus severe COVID-19. bioRxiv. 2020. https://doi.org/10.1101/2020.05.21.108308.
Acknowledgements
Open Access funding provided by Projekt DEAL. We acknowledge sponsoring of reagents by Mikrogen GmbH and financial support through the pandemic responsiveness fund of The Bavarian Ministry of Science and Art.
Author information
Authors and Affiliations
Contributions
Conception and design of the study was accounted by DP, BSc, RW and AG. Samples were provided by FHa, ML, TM, DL, FA, VH, RO, MM, SS, TG, and BG. Data were collected by DP, BSc, AS, VG, MV, PN, VA, SF, MKi, PP, MW, NR, JJW, LB, and MD. DP, BSc, MV, and PS analysed and interpreted the data with valuable intellectual input by RW, HHN, JJ, JJW, MKo, RB, FHi, and BSa. Drafting the manuscript was done by DP and BSc. Manuscript was revised by RW and JJ. All authors approved the submitted version.
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peterhoff, D., Glück, V., Vogel, M. et al. A highly specific and sensitive serological assay detects SARS-CoV-2 antibody levels in COVID-19 patients that correlate with neutralization. Infection 49, 75–82 (2021). https://doi.org/10.1007/s15010-020-01503-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s15010-020-01503-7