
Abstract
Random copolymers of P(MMA-co-styrene) were synthesized via single electron transfer-living radical polymerization (SET LRP) at 25 °C in N,N-dimethylformamide (DMF) and benzene using CCl4 as initiator and Fe(0) wire/N,N,N′,N′-tetramethyl-1,2-ethanediamine (TMEDA)/hydrazine (NH2NH2) complexes as catalyst in the presence of air. Fe(0) wire-mediated single electron transfer-living radical copolymerization of MMA and styrene represented a robust and versatile technique to synthesize the well-defined copolymers. The copolymerization rate was faster in DMF than in benzene, as determined by the apparent rate constants. The results showed that the copolymerization followed first-order kinetics model in the presence of polar DMF and non-polar benzene. The molecular weights increased linearly with the increase of monomer conversion with a narrow polydispersity index when the conversion was beyond 25 %. The polarity and the quantity of solvent had significant effects on the polymerization, and the apparent rate constants were 1.28 × 10−4, 1.21 × 10−4, and 9.23 × 10−5 s−1 in the order of DMF amount, 5, 10, and 15 mL. The conversion increased from 29.3 to 48.5 % and the polydispersity index (PDI) changed from 1.24 to 1.21 with [CCl4]0/[TMEDA]0 molar ratio changing from 1:0.5 to 1:5. The chain extension experiment demonstrated that the copolymerization exhibited a living characteristic.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the past decades, a number of controlled/living radical polymerization methods have been developed and the three most promising types are: atom transfer radical polymerization (ATRP), stable free radical polymerization (SFRP), and reversible addition-fragmentation chain transfer (RAFT) polymerization. Among them, ATRP has rapidly attracted growing interest because of its versatility in the synthesis of polymers with predictable molecular weights, low polydispersities, and specific functionalities [1, 2]. In ATRP, an equilibrium is established between the dormant species and the active propagating radicals mediated by metal complexes. However, normal ATRP has some limits, such as water- and oxygen-free systems. Therefore, some improved ATRP techniques have been developed, for example, reverse ATRP (RATRP) [3, 4], initiators for continuous activator regenerated by electron transfer ATRP (ICAR ATRP) [5, 6], simultaneous reverse and normal initiation (SRNI ATRP) [7], activators generated by electron transfer ATRP (AGET ATRP) [8, 9], activators regenerated by electron transfer ATRP (ARGET ATRP) [10, 11] and single electron transfer-living radical polymerization (SET LRP) [12–14]. SET LRP was first investigated by Percec et al. [15]. SET LRP technique has numerous advantages in synthesizing well-defined polymers with narrow molecular weight distribution at ambient temperature and in friendly solvent. In a short time, since its appearance, SET LRP has provided the end-group control and the ability to synthesize the polymers with complex architectures, such as branched polymers [16], star polymers [17, 18], graft polymers [19, 20], etc.
The most commonly used transition metal for SET LRP catalyst is copper. In a typical SET LRP system, Cu(0) and N-ligands complexes were used as catalyst and an appropriate equilibrium between free radical and dormant species is mediated via outer-sphere single electron transfer process with low activation energy, which is different from the mechanism of ATRP via a homogeneous inner-sphere single electron transfer. The activator Cu(0) reacted with an initiator containing halide to generate Cu(I) through redox process, Cu(I) was disproportioned in situ to generate Cu(0) and Cu(II) in appropriate dipolar aprotic and polar solvents in the presence of different N-ligands. Thus, solvents and ligands play an important role in SET LRP. Some ligands, such as tris(2-(dimethylamino)ethyl)amine (Me6-TREN), tris(2-aminoethyl)amine (TREN), N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA), 2,2′-bipyridine (Bpy), and poly(ethylene imine) (PEI), and some solvents such as dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF), water, and other alcohols, are commonly used. For example, Cu(0)-mediated living radical copolymerization of styrene and methyl methacrylate with PMDETA as ligand and N,N-dimethylformamide as solvent at ambient temperature was reported [23], and the molecular weight of the prepared styrene and methyl methacrylate copolymers were up to 3,800 g/mol with a low polydispersity of 1.16. The reactivity ratios of methyl methacrylate and styrene in the system were 0.545 and 0.507, respectively. However, the copolymerization rate was very slow. Our group [24] has reported Cu(0)-mediated copolymerization of methyl methylacrylate (MMA) and styrene (St) in the presence of phenol. The copolymerization rate increased in the presence of phenol. The most commonly used transition metal for SET LRP catalyst is copper, other transition metal complexes, such as Fe, have also been used successfully. In comparison to Cu catalysts, Fe catalysts have both the least toxicity of the suitable transition metals and the lowest price. Recently, Liu et al. [21] reported the Fe(0)-mediated single electron transfer-living radical polymerization of acrylonitrile in N,N-dimethylformamide at 65 °C with well-controlled behavior. Fe(0)/EDTA catalyst complex has already been used in the SET LRP of MMA [22]. However, Fe-mediated copolymerization of styrene and methyl methacrylate has not been reported. The reducing agent is an important component of ATRP because it determines the active catalyst and maintains the controlled/living process. Hydrazine is a convenient reducing agent because the by-products are typically nitrogen gas and water compared with other reducing agents. Thus, it is used as an antioxidant and an oxygen scavenger. To the best of the authors’ knowledge, Fe-mediated single electron transfer-living radical copolymerization of styrene and methyl methacrylate has not been reported with NH2NH2 as reducing agent in the presence of air with iron wire. Herein, we aim to develop a novel copolymerization of styrene and methyl methacrylate mediated by iron wire in the presence of air. One of the advantages of iron wire used as catalyst was easy to handle when the polymerization was over. The polymerization was carried out using CCl4 as initiator, Fe(0) wire as catalyst, N,N,N′,N′-tetramethyl-1,2-ethanediamine (TMEDA) as ligand, and NH2NH2 as reducing agent.
Experimental
Materials
Styrene and methyl methacrylate were purchased from Tianjin Fuchen Chemical Reagents Factory, China, which were distilled under reduced pressure prior to use. Carbon tetrachloride (CCl4, 99 %), obtained from Hunan Huihong Reagent Co., was used without further purification. Fe wire (10#, 99 %) was obtained from Shanghai Shenle Iron Wire Co., China, which was cut to 50 mm length before use. N,N-Dimethylformamide (DMF), purchased from Tianjin Tianda Chemical Reagents Factory, China, was distilled under reduced pressure prior to use. Hydrazine (AR) was obtained from Sinopharm Chemical Reagent Co., Shanghai, China. TMEDA was purchased from Jinjinle Industry Co., Shanghai, China. Other reagents were used without further purification.
Polymerization
The single electron transfer-living radical copolymerization of MMA and styrene in the presence of air was carried out in the ratio of [MMA]0/[styrene]0/[CCl4]0/[TMEDA]0/[NH2NH2] = 100:100:1:2:5. In a typical experiment, 1.04 g styrene (0.01 mol), 1 g MMA (0.01 mol), Fe wire, 0.0232 g TMEDA (0.0002 mol), 10 mL DMF, 0.0154 g CCl4 (0.0001 mol), and 0.016 g NH2NH2 (0.0005 mol) were in turn placed into a 100 mL three-neck round-bottom flask equipped with magnetic stirring bar. The reaction mixture was placed in oil bath at 25 °C. After a desired time, the polymer was precipitated in a large excess of methanol. The resultant polymer was filtered and dried at 60 °C in vacuo. The monomer conversions were determined gravimetrically.
Characterization
1H NMR spectra of the copolymer samples were recorded with Bruker 400 MHz spectrometer and using deuterated chloroform (CDCl3) solvent and tetramethyl silane as standard. The GPC analysis of the copolymers was carried out at 30 °C by Waters 1515 gel permeation chromatography (GPC) system which was equipped with refractive index detector, using HR1, HR3, and HR4 column. Molecular weight calibration curve was polystyrene (PSt) narrow standards in the molecular weight range 100–500,000. THF was used as a mobile phase at a flow rate of 1.0 mL/min and with column temperature of 30 °C.
Results and discussion
Preparation of copolymer of MMA and styrene by SET LRP in the presence of air. Styrene and methyl methacrylate copolymer was prepared by SET LRP process using Fe wire/TMEDA/CCl4/NH2NH2 catalyst system in the presence of air as described in Scheme 1.

Mechanism of SET LRP of styrene and MMA
To obtain information on structural characteristics of MMA and styrene copolymer prepared in the present study, the 1H NMR spectra of the copolymer by single electron transfer-living radical copolymerization methods were carried out. Figure 1 shows the chemical shifts due to methyl protons in the region of 0–1 ppm, methylene protons in the region of 1–1.9 ppm, and methine protons in the region of 2.1–2.5 ppm. The resonance peaks of methoxy protons appear in the region of 2.5–3.6 ppm and the phenyl protons appear in the region of 6.4–7.5 ppm. It demonstrated that MMA and styrene were copolymerized. The signal at 2.85 ppm indicated that the obtained copolymer was a random copolymer and was consistent with what Mays et al. reported [25]. The molar compositions were determined by comparing the relative intensities of the resonance signals at 6.4–7.5 ppm and 2.5–3.8 ppm.

1H NMR spectrum of copolymer of P(MMA-co-styrene) prepared in DMF at 25 °C. Experimental conditions: [MMA]0/[styrene]0/[CCl4]0/[TMEDA]0/[NH2NH2] = 100:100:1:2:5. Solvent DMF 10 mL. Polymerization temperature 25 °C, Time 150 min, Conversion 62.3 %, M n,GPC = 12,700 g/mol; PDI = 1.19
Fe wire-mediated single electron transfer-living radical polymerization of MMA and styrene at ambient temperature in the presence of air.
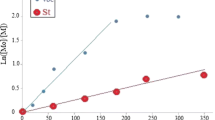
The feasibility of Fe wire-mediated single electron transfer-living radical copolymerizations of MMA and styrene was investigated at 25 °C in the presence of air in polar solvent (DMF) and apolar solvent (benzene) with TMEDA as ligand, CCl4 as initiator, and NH2NH2 as a reducing agent. The results are illustrated in Fig. 2.

Single electron transfer-living radical copolymerization of MMA and styrene in different solvents (DMF and benzene) in the presence of air: conversion versus time and ln([M]0/[M]) versus time. Experimental conditions: [MMA]0/[styrene]0/[CCl4]0/[TMEDA]0/[NH2NH2] = 100:100:1:2:5. Solvent DMF 10 mL, benzene 10 mL. Polymerization temperature 25 °C
As shown in Fig. 2, the first-order reactions were observed in both the experiments under the same experimental conditions except for solvent, indicating that the propagating radical concentrations were kept constant and the copolymerization was well controlled. In addition, an induction period was observed, which corresponded to the decomposition of CCl4 and to the establishment of the equilibrium between Fe(II) and Fe(III). However, there was no induction period in DMF but there was an induction period in benzene. It indicated that the solvent played an important role in SET LRP process. The conversion reached about 75.6 % in DMF and 50.8 % in benzene within 210 min, respectively. According to Fig. 2, the apparent rate constants (\( k_{\text{p}}^{\text{app}} \)), derived from \( \ln ([{\text{M}}]_{0} /[{\text{M}}]) = k_{{\text{p}}}^{{{\text{app}}}} t \), were 1.21 × 10−4 s−1 in DMF and 5.77 × 10−5 s−1 in benzene, respectively. The copolymerization rate was higher in DMF than in benzene, indicating that the polarity of solvent could affect the copolymerization rate. Therefore, it can be inferred that the copolymerization of MMA and styrene in DMF is favored by disproportionation of Fe(II). A similar result was observed by Lligadas et al. [26], who performed a comparison of copper(0) mediated polymerization in DMSO and toluene. The polymerization rate was faster in DMSO than in toluene because the use of DMSO favored the disproportionation. Meanwhile, in the previous reports [24], the apparent rate constant of Cu-mediated copolymerization of St and MMA at 25 °C in DMF was 2.54 × 10−5 s−1 in the presence of phenol. However, in our study, the copolymerization rate of St and MMA was higher because NH2NH2 showed a higher rate of copolymerization. As discussed above, Fe-mediated SET LRP with NH2NH2 as reducing agent in the presence of air was a robust and fast method for the synthesis of well-defined poly(St-co-MMA).
The proposed mechanism for Fe-mediated SET LRP in DMF and benzene is illustrated in Schemes 2 and 3.

The proposed mechanism for Fe-mediated SET LRP in DMF with NH2NH2 as reducing agent in the presence of air

The proposed mechanism for Fe-mediated SET LRP in benzene with NH2NH2 as reducing agent in the presence of air
As a possible mechanism, the initiator reacted with Fe(0) to generate Fe(II) and radical, and at the same time, Fe(0) was oxidized to produce FeO in the presence of air. Then FeO was reduced to form Fe(0) by NH2NH2. Fe(II) complex was disproportionated in the presence of DMF to produce Fe(III) complex and Fe(0), and then Fe(III) complex was reduced by reducing agent (NH2NH2) to generate Fe(II) complex.
However, there were different mechanisms in benzene (Scheme 3) and there was no disproportionation process in it. Fe(0) reacted with alkyl halide (R–X) to generate Fe(II)X2/L and radical R· in the presence of appropriate ligands. Radical R· was added to monomer to form a propagating species. Meanwhile, Fe(II)X2/L reacted with R–X to generate Fe(III)X3/L and radical R·. Fe(III)X3/L was reduced to Fe(II)X2/L using NH2NH2.
Figure 3 shows the values of molecular weights and PDI measured by GPC. The molecular weight increases linearly with respect to monomer conversion, and the obtained values are in good agreement with theoretical values. The polydispersity values are relatively low when the conversions were beyond 25 %. However, the PDI values are a little broader at the beginning of copolymerization process, which at the early stage was due to low concentration of Fe(II) generated by disproportionation resulting in too low radical concentration to polymerize. From the discussion above, the Fe wire-mediated single electron transfer-living radical polymerization was a robust and versatile technique for copolymerization of MMA and styrene.

Single electron transfer-living radical copolymerization of MMA and styrene in different solvents (DMF and benzene) in the presence of air: dependence of molecular weight, M n, and polydipersities on monomer conversion, same reaction conditions as Fig. 2
Quantity of solvent and its effect on copolymerization
To investigate the effect of amount of solvent on the copolymerization, a series of experiments were carried out in the presence of varied quantities of solvent. The results are shown in Figs. 4 and 5.

Single electron transfer-living radical copolymerization of MMA and styrene in varied amount of DMF in the presence of air: conversion versus time and ln([M]0/[M]) versus time. Experimental conditions: [MMA]0/[styrene]0/[CCl4]0/[TMEDA]0/[NH2NH2] = 100:100:1:2:5. Solvent DMF 5, 10, 15 mL. Polymerization temperature 25 °C

Single electron transfer-living radical copolymerization of MMA and styrene in varied amount of DMF in the presence of air: dependence of molecular weight, M n, and polydipersities on monomer conversion, same reaction conditions as Fig. 4
It can be seen from Fig. 4 that the linearity of the semi-logarithmic plot of ln([M]0/[M]) versus time is first order by various amounts of DMF, as expected for a copolymerization involving constant concentration of living species. The conversions increase with the decreasing amount of DMF. When 5 mL DMF was used, the conversion was about 78.6 %. However, when 15 mL DMF was used, the conversion was about 67 %. The apparent rate constants were 1.28 × 10−4, 1.21 × 10−4, and 9.23 × 10−5 s−1 in the order of DMF quantities, 5, 10, and 15 mL, respectively. The induction periods in 10 and 15 mL DMF were found to be 11 and 17 min, respectively, and there was no induction period in 5 mL DMF. A fast dynamic equilibrium reached between the dormant and the active species. It indicated that the amount of solvent played an important role in SET LRP. This was because the decrease in the amount of solvent was equal to the increase in the concentration of monomer.
Figure 5 shows the molecular weight versus conversion plot for three copolymerizations of MMA and styrene in 5, 10, and 15 mL DMF along with polydispersities. In three instances, the evolution of M n is linear, and it is in extremely close agreement with the theoretical values. Such linearity is entirely consistent with a controlled copolymerization. However, at low conversion, the polydispersities are broad, which are attributed to variation in number of activation/deactivation cycles that the polymer chains undergo at low conversion. In the case of 5 mL DMF, the polydispersities are higher than that in 10 and 15 mL. However, the copolymerization was controllable when the conversion was beyond 35 % (PDI < 1.25).
Quantity of ligand and its effect on copolymerization
Ligands are one of the most important components of catalyst system used in ATRP. It helps to solubilize the transition metal salts in organic media and alter the redox potential of the metal center for the appropriate dynamics of exchange between the dormant and the active species with atom transfer reaction. To investigate the effect of ligand’s quantity on the copolymerization, many experiments were performed with different amounts of ligand at 25 °C in DMF in the presence of air. The results are listed in Table 1 and the corresponding GPC traces of the copolymer are illustrated in Fig. 6. As shown in Table 1, the conversions increase with increase in the amount of TMEDA, indicating that the amount of TMEDA had a profound effect on the copolymerization rate. That was to say, under these conditions, increasing the concentration of ligand would facilitate the disproportionation of Fe(II). For [MMA]0/[styrene]0/[CCl4]0/[TMEDA]0/[NH2NH2] = 100:100:1:0.5:5, a lower conversion of 29.3 % was obtained than 62.3 % conversion with [MMA]0/[styrene]0/[CCl4]0/[TMEDA]0/[NH2NH2] = 100:100:1:5:5. It could be shown that the molar ratio of [MMA]0/[styrene]0/[CCl4]0/[TMEDA]0/[NH2NH2] = 100:100:1:0.5:5 was enough to offer a sufficient control over the Fe(0)-mediated living radical copolymerization of St and MMA (PDI < 1.25).

Evolution of GPC traces of the prepared copolymers performed with different amounts of ligand at 25 °C in DMF in the presence of air
The copolymerization rate decreased when the ratio was [MMA]0/[styrene]0/[CCl4]0/[TMEDA]0/[NH2NH2] = 100:100:1:5:5, probably due to poor solubility of ligand in DMF and the potential for degradative chain transfer [27]. The experimental molecular weights followed the corresponding theoretical values. The PDIs are between 1.19 and 1.24. Figure 6 shows GPC curves of poly(MMA-co-St) which were unimodal, symmetric, and narrow (PDI < 1.25) with no visible evidence of either high or low molecular mass impurities. The experimental results indicated that the attempt to obtain the poly(MMA-co-St) with high monomer conversion was successful by iron wire-mediated LRP technique at room temperature. This method showed some superiority in comparison with the reported single electron transfer-living radical copolymerization of AN [23, 24]. All the evidences indicated that the copolymerization proceeded in a living/controlled process.
Chain extension of poly(MMA and styrene)
A predetermined poly(MMA-co-styrene), obtained by single electron transfer-living radical polymer, was used as initiator. The molar ratio of [MMA]0/[styrene]0/[MMA-co-St]0/[TMEDA]0/[NH2NH2] was kept at 100:100:1:2:5. The chain extension experiment was carried out under the same condition as described above. The experimental result is shown in Fig. 7. As shown, the GPC curve is monomodal and symmetrical in nature, the curve is clean and clear shift is toward higher molar mass region with copolymerization, indicating the chain extension process was successful.

GPC curves before and after chain extension with poly(MMA-co-styrene) as macroinitiator. Experimental conditions: [MMA]0/[styrene]0/[macroinitiator]0/[TMEDA]0/[NH2NH2] = 100:100:1:2:5. Solvent DMF 10 mL; polymerization temperature 25 °C, time 120 min, conversion 50.3 %, M n,GPC = 14,600 g/mol, PDI = 1.42
Conclusion
Single electron transfer-living radical copolymerization of MMA and styrene was conducted using CCl4 as initiator and Fe(0) wire/TMEDA/[NH2NH2] combination as ligand in the presence of air in DMF at 25 °C. The copolymerization was found to be a random copolymerization. The copolymerization process was well controlled with predominant formation of relatively uniform primary chains. The polarity and amount of solvent had a profound effect on the copolymerization. With an increasing amount of ligand, the copolymerization rate increased. The chain-end functionality was well preserved during the copolymerization, as shown by successful chain extension experiments.
References
Matyjaszewski K, Xia J (2001) Atom transfer radical polymerization. Chem Rev 101:2921–2990
Kamigaito M, Ando T, Sawamoto M (2001) Metal-catalyzed living radical polymerization. Chem Rev 101:3689–3746
Limer A, Haddleton DM (2006) Reverse atom transfer radical polymerisation (RATRP) of methacrylates using copper(I)/pyridinimine catalysts in conjunction with AIBN. Eur Polym J 42:61–68
Ozturk T, Cakmak I (2008) Synthesis of poly(ethylene glycol-b-styrene) block copolymers by reverse atom transfer radical polymerization. J Polym Res 15:241–247
Plichta A, Li WW, Matyjaszewski K (2009) ICAR ATRP of styrene and methyl methacrylate with Ru(Cp*)Cl(PPh3)2. Macromolecules 42:2330–2332
Zhu G, Zhang L, Zhang Z, Zhu J, Tu Y, Cheng Z (2011) Iron-mediated ICAR ATRP of methyl methacrylate. Macromolecules 44:3233–3239
Gromada J, Matyjaszewski K (2001) Simultaneous reverse and normal initiation in atom transfer radical polymerization. Macromolecules 34:7664–7671
Mueller L, Jakubowski W, Matyjaszewski K, Pietrasik J, Kwiatkowski P, Chaladaj W, Jurczak J (2011) Synthesis of high molecular weight polystyrene using AGET ATRP under high pressure. Eur Polym J 47:730–734
Li WW, Matyjaszewski K (2011) Cationic surface-active monomers as reactive surfactants for AGET emulsion ATRP of n-butyl methacrylate. Macromolecules 44:5578–5585
Jonsson M, Nystrom D, Nordin O, Malmstrom E (2009) Surface modification of thermally expandable microspheres by grafting poly(glycidyl methacrylate) using ARGET ATRP. Eur Polym J 45:2374–2382
Dong H, Matyjaszewski K (2008) ARGET ATRP of 2-(dimethylamino)ethyl methacrylate as an intrinsic reducing agent. Macromolecules 41:6868–6870
Turan E, Caykara T (2011) SET–LRP of N-isopropylacrylamide in the presence of chain transfer agent. J Polym Sci, Part A: Polym Chem 49:2818–2822
Vlček P, Raus V, Janata M, Kříž J, Sikora A (2011) Controlled grafting of cellulose esters using SET-LRP process. J Polym Sci, Part A: Polym Chem 49:164–173
Nguyen NH, Percec V (2011) Acid dissolution of copper oxides as a method for the activation of Cu(0) wire catalyst for SET-LRP. J Polym Sci, Part A: Polym Chem 49:4241–4252
Percec V, Guliashvili T, Ladislaw JS, Wistrand A, Stjerndahl A, Sienkowska MJ, Monteiro MJ, Sahoo S (2006) Ultrafast synthesis of ultrahigh molar mass polymers by metal-catalyzed living radical polymerization of acrylates, methacrylates, and vinyl chloride mediated by SET at 25°C. J Am Chem Soc 128:14156–14165
Tang X, Liang X, Yang Q, Fan X, Shen Z, Zhou Q (2009) AB2-type amphiphilic block copolymers composed of poly(ethylene glycol) and poly(N-isopropylacrylamide) via single-electron transfer living radical polymerization: synthesis and characterization. J Polym Sci, Part A: Polym Chem 47:4420–4427
Whittaker MR, Urbani CN, Monteiro MJ (2008) Synthesis of linear and 4-arm star block copolymers of poly(methyl acrylate-b-solketal acrylate) by SET-LRP at 25°C. J Polym Sci, Part A: Polym Chem 46:6346–6357
Zhang W, Zhang W, Zhang Z, Cheng Z, Tu Y, Qiu Y, Zhu X (2010) Thermo-responsive fluorescent micelles from amphiphilic A3B miktoarm star copolymers prepared via a combination of SET-LRP and RAFT polymerization. J Polym Sci, Part A: Polym Chem 48:4268–4278
Zhai S, Song X, Yang D, Chen W, Hu J, Lu G, Huang X (2011) Synthesis of well-defined pH-responsive PPEGMEA-g-P2VP double hydrophilic graft copolymer via sequential SET-LRP and ATRP. J Polym Sci, Part A: Polym Chem 49:4055–4064
Zhai S, Wang B, Feng C, Li Y, Yang D, Hu J, Lu G, Huang X (2010) Thermoresponsive PPEGMEA-g-PPEGEEMA well-defined double hydrophilic graft copolymer synthesized by successive SET-LRP and ATRP. J Polym Sci, Part A: Polym Chem 48:647–655
Liu DL, Chen H, Yin P, Ji NY, Zong GX, Qu RJ (2011) Synthesis of polyacrylonitrile by single-electron transfer-living radical polymerization using Fe(0) as catalyst and its adsorption properties after modification. J Polym Sci, Part A: Polym Chem 49:2916–2923
Wang GX, Lu M, Wu H (2012) SET LRP of MMA mediated by Fe(0)/EDTA in the presence of air. Polym Bull 69:417–427
Gao J, Zhang Z, Zhou N, Cheng Z, Zhu J, Zhu X (2011) Copper(0)-mediated living radical copolymerization of styrene and methyl methacrylate at ambient temperature. Macromolecules 44:3227–3232
Wang GX (2012) Copolymerization of styrene and methyl methacrylate mediated by copper (0)/2,2′-bipyridine in the presence and absence of phenol. J Macromol Sci Part A Pure Appl Chem 49:55–59
Zhang HW, Hong KL, Mays JW (2002) Synthesis of block copolymers of styrene and methyl methacrylate by conventional free radical polymerization in room temperature ionic liquids. Macromolecules 35:5738–5741
Lligadas G, Percec V (2008) A comparative analysis of SET-LRP of MA in solvents mediating different degrees of disproportionation of Cu(I)Br. J Polym Sci, Part A: Polym Chem 46:6880–6895
Bednarek M, Biedroń T, Kubisa P (2000) Studies of atom transfer radical polymerization (ATRP) of acrylates by MALDI TOF mass spectrometry. Macromol Chem Phys 201:58–66
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Wang, GX., Lu, M., Li, J. et al. Copolymerization of styrene and methyl methacrylate mediated by iron wire/N,N,N′,N′-tetramethyl-1,2-ethanediamine as catalyst in the presence of air. Iran Polym J 22, 109–116 (2013). https://doi.org/10.1007/s13726-012-0109-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13726-012-0109-z