
Abstract
Cardiac senescence and age-related disease development have gained general attention and recognition in the past decades due to increased accessibility and quality of health care. The advancement in global civilization is complementary to concerns regarding population aging and development of chronic degenerative diseases. Cardiac degeneration has been rigorously studied. The molecular mechanisms of cardiac senescence are on multiple cellular levels and hold a multilayer complexity level, thereby hampering development of unambiguous treatment protocols. In particular, the synergistic exchange of the senescence phenotype through a senescence secretome between myocytes and stem cells appears complicated and is of great future therapeutic value. The current review article will highlight hallmarks of senescence, cardiac myocyte and stem cell senescence, and the mutual exchange of senescent secretome. Future cardiac cell therapy approaches require a comprehensive understanding of myocardial senescence to improve therapeutic efficiency as well as efficacy.
Similar content being viewed by others

Introduction
The word senescence derives from the Latin senescere, which translates literally as “to grow old.” Aging and inter-species differences in longevity have fascinated scientists and philosophers since the beginning of recorded history in ancient Egypt. Aristotle’s Parva Naturalia (350 B.C.) called attention to aging and degenerative conditions in organisms. In more recent times, 40 years ago, initial insight into the cellular biology of senescence was achieved by Hayflick in his description of senescence as “a process that limits proliferation” [1]. Subsequent decades witnessed increasing cumulative knowledge in the causes, nature, and organ-specific effects of senescence.
Aging and senescence are often used interchangeably and synonymously, but there are deviations in the evolutionary and biological bases of these processes and underlying mechanisms. Aging refers to biological, behavioral, and social variability occurring over the course of life that does not necessarily increase the risk of death. Senescence, however, is a biological process of inexorable dysfunctional alterations leading to reduced probability of reproduction and an increased susceptibility to death [2].
Intermingling of aging and senescence as related phenomena stems from the fact that aging is an indisputable major risk factor for promotion of senescence. A 2012 report by the United Nations Fund for Population Activities (UNFPA) states that the number of “old” people has increased to 810 million, is projected to reach 1 billion in less than 10 years, and will reach 2 billion by 2050. Longevity goes hand in hand with prolonged exposure to additional risk factors such as high cholesterol, hypertension, obesity, diabetes, and high levels of stress. Collectively, age-related alterations coincident with exposure to risk factors that adversely affect vitality represent a substantially increasing risk of senescence and progression of degenerative diseases among the population. Furthermore, medical advances have replaced early mortality with increased chronic morbidity. The many examples include: 1) developments in chemotherapeutics have turned selected fetal cancers from postnatal lethality into chronic disease, 2) implementation of PTCA in clinics have transformed lethal myocardial infarctions into chronic conditions, 3) diabetes management has improved, and 4) many veterans survive injuries that would have been fatal in past wars, leaving veterans with polytrauma rather than causing loss of life [3]. A military journal report estimates that veterans (and other war survivors worldwide) are at risk for a variety of chronic and degenerative diseases and will require decades of health care [3]. Thus, the perspective on aging and senescence will inevitably shift from a “luxury problem” for a select few lucky individuals to a legitimate global concern affecting both bourgeois and proletariat alike and requiring an effective remedy. The fundamental basis for combating senescence originates in a comprehensive understanding of the underlying mechanisms in order to design interventional strategies to alter mechanisms and complement longevity with improved quality of life.
Cellular senescence refers to a permanent arrest of cell division functionally linked with deterrence of potential maladaptive threats stemming from oncogenic stress or DNA damage. In other words, stringent cell cycle arrest in senescent cells enforces a safety mechanism to defeat potential development of cancer. Cellular senescence can be induced by genetic damage and/or epigenetic disruption. The major initiator and facilitator of senescence is the DNA damage response (DDR) that causes permanent cell cycle arrest. Persistent DDR is initiated by telomere erosion, nontelomeric DNA damage, DNA double-strand breakage, the effect of histone deacetylase inhibitors on chromatin modification, and strong mitogenic stimuli that cause misfired replication origins, which culminates in a senescent phenotype by inducing and maintaining cell cycle arrest. Continual DDR activity in a senescent cell is reflected by distinct nuclear foci containing DDR proteins and phospho-ATM/ATR substrates designated as DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS) [4–6, 7••, 8].
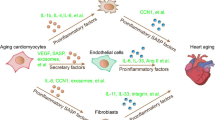
The major difference between a senescent and quiescent cell is that, under physiological conditions, quiescent cells can re-enter the cell cycle, while senescent cells require rigorous non-physiologic interventions that reinforce cell cycle entry (e.g., inactivation of p53). The majority of senescent cells express cell cycle inhibition markers, such as p16INK4a and p53, and have increased lysosomal content, which is indicated by senescence-associated beta-galactosidase activity [9–12]. Unlike quiescent cells, permanent cell cycle arrest increases cell size and metabolic activity more than twofold compared to healthy cells. Perhaps the most important hallmark of a senescent cell is inability to contribute to tissue repair and regeneration and maintenance of normal homeostasis. In addition, senescent cells possess the peculiar ability to propagate their senescent phenotype through proactive secretion of proteases, growth factors, and cytokines that ultimately lead to the senescence-associated secretory phenotype (SASP) [13•, 14–19]. Senescent cells are involved in multiple degenerative diseases. This review is focused predominantly on the process of senescence in the heart, the hegemonic perpetuation of this phenotype throughout the myocardium, and approaches for reversing a senescent phenotype and rejuvenation of the myocardium.
Cardiomyocyte Senescence
Aging and cardiovascular disease are positively correlated with cardiac diseases reaching epidemic proportions in the elderly. With the progression of aging, the myocardium undergoes degenerative alterations and processes that ultimately lead to myocyte death. Pathologic changes in expression of contractile protein isoforms and altered levels of calcium-handling proteins lead to gradual but increasing deterioration of contractile function [20, 21]. Longitudinal echocardiographic assessments reveal prominent age-related hypertrophy and decrease in diastolic function [22, 23]. In a compensatory effort, remaining myocardium undergoes structural and functional alterations, culminating in reduced myocardial performance as measured by MPI (myocardial performance index) [23]. Age-related cardiac histopathologic alterations present as interstitial and subendocardial fibrosis, hyaline cytoplasmic changes, vacuolization, collapsed sarcomeres, etc. [24]. Morphometeric measurements of aging hearts reveal myocyte hypertrophy, increased myocyte apoptosis, and increased fibrosis and amyloid deposition. The mechanistic basis for the accrual of myocyte age-related changes and senescence involves multiple pathways influenced at multiple levels in the cells [25].
Myocyte Senescence and Mitochondria
Cardiac aging and susceptibility to senescence are associated with mitochondrial function. During oxidative phosphorylation, mitochondria generate high levels of reactive oxygen species (ROS) as a byproduct of electron transfer. Because signaling is altered by protein oxidation, cellular nucleic acids and proteins are influenced by ROS. In addition, oxidized proteins accumulate in insoluble protease-resistant aggregates and interfere with cellular function. Ironically, ROS also attack mitochondrial nucleic acids, lipids, and proteins, which culminate in mitochondrial DNA mutations and dysfunction by damaging respiratory enzymes. Defects in respiratory enzymes further increase ROS generation, which causes decreased mitochondrial function that promotes cell and organ dysfunction. High metabolic demands of the heart and ROS production as a byproduct of oxidative phosphorylation renders the myocardium prone to oxidative damage and gradual organ malfunction [26, 27]. Initial evidence for the role of mitochondrial ROS in cardiac aging was gathered from overexpression of mitochondria-specific catalase in mice [28]. Unlike overexpression of catalase in the nucleus, mitochondrial catalase increased animal lifespan and resistance to phenotypic changes indicative of cardiac aging.
The rescue of premature aging and senescent cardiac phenotype in hearts of mice by mitochondrial-specific overexpression of catalase supports the premise that ROS production, DNA damage, and biogenesis are contributors of a vicious circle of ROS-induced ROS-release, leading inevitably to functional deterioration. Damaged mitochondria undoubtedly increase metabolic stress upon cells, which in turn stimulates mitochondrial biogenesis through PGC-1 alpha-mediated signaling. Indeed, PCG-1 knockout mice exhibit decreased mitochondrial biogenesis and reduced cardiac energy production [29, 30]. Although cells naturally posses the ability to produce additional mitochondria to avoid metabolic stress, the protection of mitochondrial biogenesis can be disrupted by the damaging effects of ROS. Mitochondrial biogenesis requires well-orchestrated expression of mitochondrial DNA and transcription factors. ROS-mediated mitochondrial DNA damage and mutations are thought to be a major cause of reduced mitochondrial biogenesis in senescent hearts. Mutation of mitochondrial polymerase in middle-aged mice prompts increased oxidative stress, with cardiomyopathic changes [31]. Aged heart cells have a reduced threshold for ROS-induced ROS-release and thus are more sensitive to mitochondrial permeability transition pore (MPTP) induction. MPTP opening leads to mitochondrial swelling and collapse, leading to ATP deprivation, oxidative stress, and apoptosis.
Further validation of the role of mitochondria in cardiac aging was derived from the study of a mutant mouse with alteration of p66shc, a mitochondrial redox enzyme localized in the intermembranous space that forms ROS using electrons, resulting in H2O2 production [32]. Additionally, p66shc accumulates within mitochondria and activates mitochondrial calcium responses to subsequently induce apoptosis. The p66shc genetic mutation increases lifespan and decreases cardiac functional deterioration through decreased oxidative damage in transgenic mice [32]. Along with the natural process of aging, “authentic” cardiac risk factors such as smoking, diabetes, and high levels of homocysteine are additional inducers of oxidative stress, and contribute to furthering the vicious cycle of mitochondrial deterioration. In addition, paracrine effectors such as insulin-like growth factor 1 (IGF-1) induce ROS production and modulate cellular signaling via regulation of redox reactions [33].
Collectively, these findings illustrate the prominent role that increased mitochondrial ROS production and decreased mitochondrial biogenesis have in aging and organ deterioration. As a coincident process developing with advanced age, suboptimal mitochondrial function alone may not necessarily appear problematic. However, imposition of additional risk factors, stress, or pathologic challenges such as myocardial infarction, where substrate availability decreases in a highly energy-demanding environment, places demands upon mitochondrial energy production and biogenesis, which ultimately fail, leading to senescence and severe functional deterioration in time.
Myocyte Senescence and Adrenergic Signaling
Chronic activation of beta-adrenergic signaling is deleterious to cardiac function [34]. Increases in heart rate, contractility, peripheral vasoconstriction, and wall stress are tied to heightened catecholamine-driven increases in cardiac metabolic demand in conditions when oxygen availability is limited, such as coronary atherosclerosis. Stimulation of beta-adrenergic receptors decreases anti-apoptotic and anti-oxidative stress signaling, contributing to senescence and functional deterioration in a cascading effect, starting with adenylate cyclase 5 activation, with downstream induction of cyclic-AMP and PKA that act as inhibitors of the cardioprotective Raf/MEK/Erk pathway [35]. In knockout mice, genetic deletion of adenylate cyclase 5 increases lifespan through upregulation of the Raf/Erk-pathway and increased anti-apoptotic signaling and stress resilience [36, 37].
Myocyte Senescence and Renin-Angiotensin-Aldosterone System (RAAS)
RAAS is another critical player for regulation of cardiac function through angiotensin II (Ang II). RAAS normally acts as a protective mechanism to increase blood pressure following a disturbance of systemic pressure sensed by renal artery flow. Ang II is a vasoconstrictor and plays a crucial role in fluid balance. Persistent RAAS activation with chronic Ang II exposure of myocytes promotes development of cardiac failure [38]. Even more than the systemic production of Ang II, increases in local Ang II are particularly detrimental to cardiomyocytes [39], which prompted worldwide usage of Ang II blockers for the prevention and treatment of cardiac remodeling. The detrimental effects of Ang II on cardiac myocytes have been studied in transgenic mice that exhibit cardiac-specific elevation of Ang II production. Mice with cardiac overexpression of Ang II reveal a dilated cardiomyopathic and aged phenotype in addition to increased mortality. The major cause of dilated cardiomyopathy was the induced down-regulation of the sarcoplasmic reticulum pump (SERCA2) and diminution of Ca2+ transients, indicative of disruption on calcium homeostasis [40]. Along with the effects on vasoconstriction and fluid balance, Ang II plays an important role in cardiac contractility by increasing cardiac pump function independent of systemic blood pressure, which with persistent stimulation will exacerbate cardiac dysfunction. Cardiac tissue Ang II is significantly elevated in aged rodents, presumably in conjunction with angiotensin-converting enzyme elevation. Although further studies are needed to fully decipher the mechanisms involved in the elevation of cardiac Ang II, cumulative evidence clearly shows that Ang receptor inhibition and disruption decreases age-related cardiac pathology and prolongs lifespan in rodents [41, 42].
Myocyte Senescence and Mammalian Target of Rapamycin Complex1(mTORC1)
The serine/threonine protein kinase mTORC 1 acts as the cellular sensor for nutrients, stress, cellular growth, and metabolism, and is a major player in cellular aging, metabolic disorders, and cancer. The relationship of mTORC1 to longevity has been established in a variety or organisms [43]. Pharmacologic inhibition or genetic inactivation of mTORC1 increase longevity in rodents [44]. Additionally, suppression of mTORC1 through restriction of caloric intake antagonizes aging in monkeys, suggestive of a potential role for mTORC1 in human longevity. Cardiac aging correlates with maladaptation to stress, hypertrophy, and cardiac failure, so a causal link with mTORC1 is plausible. Cardiac pathologies involving mTORC1 link synergistic increases of stress-induced protein synthesis with decreased protein degradation due to impaired autophagy. Overstimulation of mTORC1 by excessive growth hormone (Ang II, IGF-1, or catecholamines) promotes cardiac senescence, with IGF-1 hyperactivity, in particular, leading to insulin resistance through phosphorylation of insulin receptor-1 (IRS-1) [45] that increases myocardial risk to pathologic changes. Consolidation of all mTORC1 activities in cardiac aging has provided insight and understanding in a new cluster of potential targets for cardiac rejuvenation.
Senescence of Cardiac Stem Cells
Stem cells contributors to repair and regeneration following tissue damage. As primitive and naïve cells, stem cells are highly susceptible to signals and stimuli from their habitat and systemic environment. Thus, stem cells from an unexploited niche are presumably exposed to a “younger” and healthier environment than stem cells from an overly actively proliferating niche. Essentially, the natural process of aging is detrimental to cardiac stem cell quality. Age-related changes such as DNA damage, impaired catabolism, altered epigenetics, and exposure to environmental stress factors contribute to deterioration of stem cell function and negative signaling from the niche towards stem cells, which culminates in a vicious cycle of unfavorable effects on tissue repair and regeneration. Unlike myocytes, stem cells possess a relatively high capacity for proliferation, resulting in an increased susceptibility to single- and double-DNA breaks, chromosomal translocation, telomere erosion, and other types of mutations, ultimately leading to replicative senescence. In addition, cardiac stem cells in a pathologic heart are exposed to chronic elevated levels of Ang II and sympathetic hyperactivity that contribute to initiation and maintenance of senescence. Cardiac stem cells, and particularly c-kit + human cardiac stem cells (hCSCs), undergo senescence in aged and diseased hearts as measured by p16 expression. Stem cells derived from the myocardial tissue of chronic heart failure patients show increased numbers of dual-positive c-kit+/p16+ hCSCs, with concurrent evidence of telomeric shortening [46•, 47].
Further elucidation of this mechanism in hCSCs has revealed that hCSCs derived from pathologically injured aged hearts have reduced telomerase activity, increased dysfunction foci, and elevated expression of p16 and p21 [48]. The molecular pathways studied in cardiac stem cell senescence appear to those previously identified described in cardiomyocytes. hCSCs senescence is influenced by the IGF-2/IGF2R, HGF/c-Met, and RAAS-activated angiotensin II pathways. Protective IGF-1 increases telomerase activity, maintains telomere length, blocks the occurrence of replicative senescence, and preserves a functional population of cardiac stem cells capable of tissue repair and regeneration, whereas IGF-2 appears to promote adverse effects. HGF production decreases upon age, inhibiting the migratory capability of CSCs in response to tissue damage and external stimuli [49•]. The addition of HGF to rescue migratory potential of CPCs, indeed, increases activity, tissue repair, and regeneration [48]. Additionally, hCSCs are also sensitive to local Ang II and increasingly express AT1R with age. Ang II induces oxidative stress through increased ROS generation. The lifespan of aged hCSCs has been improved through the use of ACE-inhibitors, demonstrating the detrimental effect of Ang II exposure on stem cell functional capacity [50].
Myocyte and CSC-senescence Synergy; the Role of SASP
The environment within tissue is characterized by the collective phenotypes of constituent cells in the contiguous area and their chemical properties as revealed by chemokines, cytokines, and growth factors. In addition, cells communicate and interact with each other in a dynamic network that forms and alters their microenvironment. Simultaneously, extracellular signals that impinge upon cell behavior promote additional phenotypic changes such that the collective population of cells as a whole are intimately related and exhibit solidarity in maintaining tissue homeostasis. The presumptive role of senescent cells to function primarily in suppression of tumor formation through irreversible cell cycle arrest renders them guardians who must notify their local environment when potential replicative errors appear within the population. Indeed, through the development of SASP, senescent cells profoundly alter their transcriptome to provide protection from uncontrolled cell growth through the propagation of permanent cell cycle arrest in a hegemonic fashion throughout the environment . Existence of a senescent secretome was initially demonstrated in fibroblasts undergoing replicative senescence, as revealed by array analysis of the classic wound healing response in surrounding senescent fibroblasts [51, 52].
SASP factors generally can be subdivided into three major categories: 1) soluble signaling factors, 2) secreted proteases, and 3) insoluble proteins/extracellular matrix components. IL-6, IL-1, chemokines (e.g., IL-8), as well as IGF or colony stimulating factors, are examples of soluble SASP factors that affect neighboring cells through cell-surface receptors. Additionally, select extracellular proteases are known SASP agents, including matrix metalloproteinases and urokinases, that upregulate in multiple cell types upon progression to senescence19. Although a wide range of SASP factors are upregulated during senescence, the severity of phenotype and mechanistic basis of senescence appear to be influential in the determination of patterns and levels of secreted factors. Replicative senescence, telomere erosion, DNA damage, and chromatin disruption display a closely related pattern of SASP factors. However, p16-mediated senescent cells do not exhibit any potential for secretion of SASP factors [53]. Another attribute of the SASP is a temporal development of intensity, with progression and severity of SASP driving higher levels of SASP factor production.
Although SASP displays a markedly beneficial role by empowering one particular cell to prevent progression of tumor growth in oncogenesis, the dark side of SASP becomes apparent when it is provoked in the myocardium, where exposure of myocytes to risk factors and/or pathologic challenge ultimately culminate in myocyte senescence. In a proliferative organ that exhibits a high regenerative potential, restricted induction of senescence in troublesome cells and their neighboring brethren may not necessarily lead to dramatic consequences. However, the limited regenerative potential of the heart means that myocyte losses from death or the twilight functional consequences of senescence cannot be adequately compensated by generation of new cardiomyocytes. In addition to a limited baseline regenerative capacity within the myocytes themselves, the cohabitating hCSC are similarly susceptible to risk factors and pathologic injury that lead to conversion into senescence over time. Notably, as post-mitotic myocytes undergo senescence as a result of combined effects of metabolic disruptions, RAAS, catecholamines, and DNA damage, bystander hCPCs must cope not only with their chronological aging and premature senescence, but also undergo replicative senescence. Considering the prominent role of SASP in maintaining tissue similitude, SASP factors secreted by cardiomyocytes influence other cardiac cell types, including hCSC. Thus, already-impaired hCSC are further vanquished by SASP factors from myocytes that accelerate deterioration of cardiac regenerative and reparative potential. As the dominos fall, the hCSCs, in turn, are stimulated to secrete stem-cell-specific SASP factors that influence other cell types in the heart, propagating a vicious circle and culminating in the spread of senescence throughout the entire myocardium. Cardiac senescence, therefore, is not solely a “one-cell type concern”. Once the process of senescence is initiated, it develops into hegemony of cellular deterioration that ultimately dominates the cardiac function and leaves the heart impaired. Considering the multilayer extent of senescence in the heart, efforts are underway to identify interventional approaches in this senescence-driven downward spiral to inhibit progression of global cardiac deterioration.
Senescence Reversal
If spreading senescence is ultimately detrimental to myocardial performance, then reasonable molecular interventional strategies could be valuable in slowing or reversing the acquisition of a senescent phenotype. Two areas that have been explored in cardiomyocytes are mTORC1 or telomerase manipulation [44, 54–56]. Correlations that link metabolic activity to aging and senescence have prompted approaches to restrict caloric intake to 30 % of typical daily intake levels in order to study the effects on the cardiovascular systems of rodents and humans. Caloric restriction findings reveal a very powerful ability to prevent structural detrimental changes in the cardiovascular system, partially through reduction of ROS. Inhibition of mTORC1 as a target for longevity and reversal of senescence has also been studied using rapamycin, resulting in reduced cardiac inflammation, increased cardiac ejection fraction, decreased ANP levels, and an overall reduction in the hallmarks associated with myocardial hypertrophy [54, 55]. Alternatively, exploring the influence of telomere length on senescence, hTERT expression in human somatic cells maintains telomere length and antagonizes them reaching their replicative limit. In addition to elongated telomeres, these cells displayed a more “youthful” phenotype defined by their gene expression pattern. Although telomerase expression does not possess the ability to create new myocytes through initiation of myocyte proliferation from a quiescent state, evidence from mouse studies supports a role for telomerase overexpression as an inducer of hypertrophy and protector from cardiac failure through inhibition of apoptosis. Telomerase expression and activity in the heart is an intriguing field to explore, but it is important to be mindful that telomerase can perpetuate in vitro cell immortalization with evidence of associated karyotypic abnormalities. Cautious and rigorous studies are required to assess the feasibility and safety of the further application of telomerase manipulation in the heart.
In the context of stem cells, molecular pathways associated with senescence include IGF-2 signalling, mTOR/autophagy, NO/telomerase/sirtuins, and p38MAPK [50]. Despite this roster of potential signaling players, interference with senescence-promoting pathways in hCPCs remains essentially unexplored. Recently, our group implemented a novel strategy for rejuvenation of hCPCs by overexpression of the proto-oncogene Pim-1. Pim-1 is a serine/threonine kinase that autophosphorylates and plays an important anti-apoptotic and pro-proliferative role in hCPCs. Working with hCPCs isolated from terminal heart failure patients undergoing left ventricular assist device (LVAD) placement implantation, adoptive transfer studies proved hCPCs engineered for Pim-1 overexpression displayed a remarkable increase in cell survival and engraftment, as well as contributing to durable improvement of cardiac function for up to 6 months [57••]. As might be expected, hCPCs derived from failing hearts are exposed to an environmental milieu that would place the heart under severe stress and would likely accelerate development of senescence characteristics. Indeed, subsequent phenotypic characterization of hCPCs isolated from LVAD patients revealed a senescent phenotype characterized by shortened telomeres, high levels of p16 and p53, and limited proliferation capacity. Importantly, modification of hCPCs using Pim-1 ameliorated senescent characteristics, with restoration of youthful telomeric length, enhanced replicative capacity, and decreased levels of p16 and p53 [58••]. To date, Pim-1 engineering of hCPCs has been performed using a lentiviral delivery approach, and further safety testing is warranted. However, of note in the recent study is the fact that even Pim-1-engineered hCPCs underwent in vitro replicative senescence at comparable cell passaging to youthful hCPC controls isolated from fetal hearts [58••]. The efficacy for enhancing myocardial regeneration, together with molecular antagonism of senescence, makes Pim-1 a promising candidate for future interventional approaches in combating loss of functional and reparative potential in the failing heart.
Conclusions
Human beings possess the unique characteristic of remembering the past and worrying about the future, which sets them apart from other organisms. With the knowledge of past statistics and predictions of the future, today’s scientific community is engaging in an earnest discussion of aging. It is worth emphasizing that, under normal health conditions, chronologic aging does not necessarily lead to cellular senescence. However, advanced age does provide a protracted platform for exposure to harmful factors that lead to older-age-mediated pathologic alterations and occurrence of senescence. As a mechanistic basis for the development of degenerative diseases, senescence and age-related pathologies has been a fascinating topic for decades in the cardiac field. Molecular studies at multiple levels of cellular biology on myocyte aging have revealed the complexity of senescence in the context of the myocardium. In addition to intracellular biology, the paracrine aspect of senescent cells in a multi-cellular organ with limited regenerative potential contributes to the complexity of senescence-mediated cardiac degeneration. There is more skill and experienced in other fields regarding the interpretation of senescence and primitive cells, and the concept of stem-cell-mediated repair and regeneration of the heart is relatively new. On one hand, the recognition of the concept of myocardial stem-cell-mediated regeneration has gained momentum. On the other hand, the understanding of myocardial senescence had added an additional layer of complexity by situating a new cell type to the equation of age-mediated functional deterioration. Stem-cell-mediated SASP holds particular potential for inducing permanent cell cycle arrest in the remaining myocardial stem cell population, leaving the heart with a further diminished regenerative potential. Not only is this phenomenon concerning in the context of endogenous myocardial repair and regeneration, but it also reveals the reality that isolated CSCs from a senescent heart for future adoptive transfer may not represent a “primitive” and highly proliferative cell type that is capable of combating a senescent environment in addition to regenerating new healthy myocytes. Collectively, the treatment of heart failure requires a stringent remedy against myocardial senescence. Future therapeutic interventions require rejuvenation not only of myocytes and the myocardium, but also of isolated CSCs with interventional strategies currently under investigation, such as Pim-1. The cardiac field has a tremendous base of expertise in protective signaling cascades. Perhaps it is time to utilize the accumulated knowledge to proactively encounter senescence using gene therapy, modified-cell therapy, and other timely approaches.
References
Papers of particular interest, Published recently, have been highlighted as: • Of importance •• Of major importance
Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–36.
Crews DE. Human senescence: Evolutionary and biocultural perspectives. 2003:4–5.
Geiling J, Rosen JM, Edwards RD. Medical costs of war in 2035: Long-term care challenges for veterans of iraq and afghanistan. Milit Med. 2012;177:1235–44.
Campisi J. Aging, cellular senescence, and cancer. Annu Rev Plant Physiol Plant Mol Biol. 2013;75:685–705.
Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: A link between cancer and age-related degenerative disease? Sem Cancer Biol. 2011;21:354–9.
Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–56.
•• Campisi J. Cellular senescence: Putting the paradoxes in perspective. Curr Opin Genet Dev. 2011;21:107–12. This paper points out the paradoxes in the field of senescence and provides clarification for the confusions.
Rodier F, Munoz DP, Teachenor R, Chu V, Le O, Bhaumik D, et al. DNA-scars: Distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2011;124:68–81.
Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 2012;130:1715–25.
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16ink4a. Cell. 1997;88:593–602.
Dimri GP, Campisi J. Molecular and cell biology of replicative senescence. Cold Spring Harb Symp Quant Biol. 1994;59:67–73.
Severino J, Allen RG, Balin S, Balin A, Cristofalo VJ. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp Cell Res. 2000;257:162–71.
• Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–90. This manuscript explains and elaborates on specifics of SASP.
Aoshiba K, Tsuji T, Kameyama S, Itoh M, Semba S, Yamaguchi K, Nakamura H. Senescence-associated secretory phenotype in a mouse model of bleomycin-induced lung injury. Experimental and toxicologic pathology : official journal of the Gesellschaft fur Toxikologische Pathologie. 2013.
Liu Y, Drozdov I, Shroff R, Beltran LE, Shanahan CM. Prelamin a accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res. 2013;112:e99–e109.
Cahu J, Bustany S, Sola B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012;3:e446.
Salminen A, Kauppinen A, Kaarniranta K. Emerging role of nf-kappab signaling in the induction of senescence-associated secretory phenotype (sasp). Cell Signal. 2012;24:835–45.
Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence-associated secretory phenotype by nf-kappab promotes senescence and enhances chemosensitivity. Gene Dev. 2011;25:2125–36.
Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Ann Rev Pathol. 2010;5:99–118.
Lakatta EG. Cardiovascular regulatory mechanisms in advanced age. Physiol Rev. 1993;73:413–67.
Lakatta EG. Changes in cardiovascular function with aging. Eur Heart J. 1990;11(Suppl C):22–9.
Hees PS, Fleg JL, Lakatta EG, Shapiro EP. Left ventricular remodeling with age in normal men versus women: Novel insights using three-dimensional magnetic resonance imaging. Am J Cardiol. 2002;90:1231–6.
Czuriga D, Paulus WJ, Czuriga I, Edes I, Papp Z, Borbely A. Cellular mechanisms for diastolic dysfunction in the human heart. Curr Pharm Biotechnol. 2012;13:2532–8.
Waller BF. The old-age heart: Normal aging changes which can produce or mimic cardiac disease. Clin Cardiol. 1988;11:513–7.
Anversa P, Palackal T, Sonnenblick EH, Olivetti G, Meggs LG, Capasso JM. Myocyte cell loss and myocyte cellular hyperplasia in the hypertrophied aging rat heart. Circ Res. 1990;67:871–85.
Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: The role of mitochondrial oxidative stress. Trend Cardiovasc Med. 2009;19:213–20.
Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95.
Zhang X, Klein AL, Alberle NS, Norby FL, Ren BH, Duan J, et al. Cardiac-specific overexpression of catalase rescues ventricular myocytes from ethanol-induced cardiac contractile defect. J Mol Cell Cardiol. 2003;35:645–52.
Rowe GC, Jiang A, Arany Z. Pgc-1 coactivators in cardiac development and disease. Circ Res. 2010;107:825–38.
Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, et al. Transcriptional coactivator pgc-1 alpha controls the energy state and contractile function of cardiac muscle. Cell metabolism. 2005;1:259–71.
Hance N, Ekstrand MI, Trifunovic A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Human Mol Genet. 2005;14:1775–83.
Gertz M, Steegborn C. The lifespan-regulator p66shc in mitochondria: Redox enzyme or redox sensor? Antioxid Redox Signal. 2010;13:1417–28.
Handayaningsih AE, Takahashi M, Fukuoka H, Iguchi G, Nishizawa H, Yamamoto M, et al. Igf-i enhances cellular senescence via the reactive oxygen species-p53 pathway. Biochem Biophys Res Commun. 2012;425:478–84.
Zhou YY, Song LS, Lakatta EG, Xiao RP, Cheng H. Constitutive beta2-adrenergic signalling enhances sarcoplasmic reticulum ca2+ cycling to augment contraction in mouse heart. J Physiol. 1999;521(Pt 2):351–61.
Chakraborti S, Chakraborti T, Shaw G. Beta-adrenergic mechanisms in cardiac diseases: A perspective. Cell Signal. 2000;12:499–513.
Tang T, Lai NC, Hammond HK, Roth DM, Yang Y, Guo T, et al. Adenylyl cyclase 6 deletion reduces left ventricular hypertrophy, dilation, dysfunction, and fibrosis in pressure-overloaded female mice. J Am Coll Cardiol. 2010;55:1476–86.
Hu CL, Chandra R, Ge H, Pain J, Yan L, Babu G, et al. Adenylyl cyclase type 5 protein expression during cardiac development and stress. Am J Physiol Heart Circ Physiol. 2009;297:H1776–82.
Domenighetti AA, Wang Q, Egger M, Richards SM, Pedrazzini T, Delbridge LM. Angiotensin ii-mediated phenotypic cardiomyocyte remodeling leads to age-dependent cardiac dysfunction and failure. Hypertension. 2005;46:426–32.
Heymes C, Silvestre JS, Llorens-Cortes C, Chevalier B, Marotte F, Levy BI, et al. Cardiac senescence is associated with enhanced expression of angiotensin ii receptor subtypes. Endocrinology. 1998;139:2579–87.
Edwards A, Pallone TL. Mechanisms underlying angiotensin ii-induced calcium oscillations. Am J Physiol Renal Physiol. 2008;295:F568–84.
Basso N, Cini R, Pietrelli A, Ferder L, Terragno NA, Inserra F. Protective effect of long-term angiotensin ii inhibition. Am J Physiol Heart Circ Physiol. 2007;293:H1351–8.
de Cavanagh EM, Inserra F, Ferder L. Angiotensin ii blockade: A strategy to slow ageing by protecting mitochondria? Cardiovasc Res. 2011;89:31–40.
Ming XF, Montani JP, Yang Z. Perspectives of targeting mtorc1-s6k1 in cardiovascular aging. Front Physiol. 2012;3:5.
Flynn JM, O'Leary MN, Zambataro CA, Academia EC, Presley MP, Garrett BJ, et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging cell. 2013;12:851–62.
Volkers M, Toko H, Doroudgar S, Din S, Quijada P, Joyo AY, et al. Pathological hypertrophy amelioration by pras40-mediated inhibition of mtorc1. Proc Natl Acad Sci U S A. 2013;110:12661–6.
• Cesselli D, D'Aurizio F, Marcon P, Bergamin N, Beltrami CA, Beltrami AP. Cardiac stem cell senescence. Methods Mol Biol. 2013;976:81–97.
Anversa P, Kajstura J, Leri A, Bolli R. Life and death of cardiac stem cells: A paradigm shift in cardiac biology. Circulation. 2006;113:1451–63.
Sussman MA, Anversa P. Myocardial aging and senescence: Where have the stem cells gone? Ann Rev Physiol. 2004;66:29–48.
• Beltrami AP, Cesselli D, Beltrami CA. Stem cell senescence and regenerative paradigms. Clin Pharmacol Ther. 2012;91:21–9. This paper is an extensive description of senescence in stem cells and direction for future interventions.
Cesselli D, Beltrami AP, D'Aurizio F, Marcon P, Bergamin N, Toffoletto B, et al. Effects of age and heart failure on human cardiac stem cell function. Am J Pathol. 2011;179:349–66.
Calabro A, Orsini B, Fedi P, Falchini M, Surrenti C. Enzyme immunoassay of gastric luminal prostaglandin e2 in duodenal ulcer disease. Am J Gastroenterol. 1989;84:629–32.
Shelton DN, Chang E, Whittier PS, Choi D, Funk WD. Microarray analysis of replicative senescence. Curr Biol: CB. 1999;9:939–45.
Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(ink4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396–403.
Neff F, Flores-Dominguez D, Ryan DP, Horsch M, Schroder S, Adler T, et al. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest. 2013;123:3272–91.
Xie K, Jin B, Li Y, Luo X, Zhu J, Ma D, et al. Modulating autophagy improves cardiac function in a rat model of early-stage dilated cardiomyopathy. Cardiology. 2013;125:60–8.
Heist EK, Huq F, Hajjar R. Telomerase and the aging heart. Sc Aging Knowl Environ: SAGE KE. 2003;2003:PE11.
•• Mohsin S, Khan M, Toko H, Bailey B, Cottage CT, Wallach K, et al. Human cardiac progenitor cells engineered with pim-i kinase enhance myocardial repair. J Am Coll Cardiol. 2012;60:1278–87. This is the first report of human cardiac stem cell rejuventaion using Pim-1 and the application of these cells in vivo.
•• Mohsin S, Khan M, Nguyen J, Alkatib M, Siddiqi S, Hariharan N, Wallach K, Monsanto M, Gude NA, Dembitsky WP, Sussman MA. Rejuvenation of human cardiac progenitor cells with pim-1 kinase. Circulation research. 2013 This study describes rejuvenation of human cardiac stem cells using Pim-1 showing telomere elongation and other hallmarks of reversal of senescence.
Acknowledgements
This review article was supported by the National Institute of Health to Mark A. Sussman (R21HL102714, R01HL067245, R37HL091102, P01HL085577, RC1HL100891, R21HL102613, R21HL104544, and R01HL105759), American Heart Association (11POST7610164), the Netherlands Organization for Scientific Research (NWO Mozaiek Grant) to Sailay Siddiqi, Alexandre Suermann, MD/PhD, Stipendium UMC Utrecht to Sailay Siddiqi, and GRASP Research to Sailay Siddiqi.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Sailay Siddiqi and Mark A. Sussman declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Siddiqi, S., Sussman, M.A. Cardiac Hegemony of Senescence. Curr Transl Geriatr and Exp Gerontol Rep 2, 247–254 (2013). https://doi.org/10.1007/s13670-013-0064-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13670-013-0064-3