
Abstract
Three new C27-steroidal glycoalkaloids, spiralosides A–C (1–3), were obtained from the total alkaloids of Solanum spirale by chromatographic methods. On the basis of spectroscopic evidence, spiralosides A–C were elucidated as (22R,25S)-22,26-epiminocholest-5-ene-3β,16α-diol-N-acetyl-3-O-α-l-rhamnopyranosyl-(1→4)-β-d-glucopyranosyl (1), (22R,25S)-22,26-epiminocholest-5-ene-3β,16α-diol-N-acetyl-3-O-β-d-glucopyranosyl (2), (22R,25S)-22,26-epiminocholest-3β,16α-diol-N-acetyl-3-O-β-d-glucopyranosyl (3), respectively. The total alkaloids of S. spirale have been screened for their antitussive and expectorant effects in intact animal model.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Solanum spirale (Solanaceae), an erect shrub, is widely distributed in Yunnan, Guangxi and Hunan provinces of China. It usually grows forest-shrub edge and wasteland at the altitude between 500 and 1900 m [1]. S. spirale, popularly named “Li Fei San”, has been used in “Yi” ethnopharmacy as anti-tussive and anti-inflammatory agent historically. Besides, its tender leaves and fruits can be edible as a wild vegetable by “Dai” ethnopharmacy [2]. Previously, the reported phytochemical studies of S. spirale led to the isolation of a series of components, such as oil components, steroidal saponins, steroidal alkaloids and steroidal glycoalkaloids with antioxidant activity, and significant antibacterial activity and cytotoxicity [3–8]. The steroidal alkaloid glycosides from Solanum genus possess extensive activities including anticancer [9, 10], anti-acetylcholinesterase [11, 12], anticholesterol [13, 14], and antifungal [15] properties. In the course of searching for bioactive steroidal alkaloid glycosides from the fruits of S. spirale, three new steroidal glcoalkaloids named spiralosides A–C (1–3) were isolated. The total alkaloids were evaluated anti-tussive activity against ammonia liquor induced damage and the expectorant effects used by phenol red secretion test in mice. Reported herein are the isolation, structural elucidation, and the anti-tussive activities of the total alkaloids.
2 Result and Discussion
Compound 1 was obtained as a white, amorphous powder; \([\alpha ]_{\text{D}}^{23}\) −46.6 (c 0.03, MeOH). It displayed a positive reaction to Dragendorff’s reagent and gave the molecular formula of C41H67NO12 by HRESIMS at m/z 788.4553 [M+Na]+ (calcd for 788.4561), corresponding to nine degrees of unsaturation. The 1H-, 13C-NMR and DEPT spectra displayed two sugar units on basis of three proton signals at δ H 4.36 (1H, d, J = 7.8 Hz), 4.81 (1H, br s), and 1.23 (3H, d, J = 6.2 Hz), two anomeric carbons [δ C 102.3 (d), and 102.9 (d)], a methyl group (δ C 17.8), a methylene carbon (δ C 62.1), and other 8 methines signals between δ C 70.6 and δC 79.9. The coupling constant (J = 7.8 Hz) of the anomeric proton at δ H 4.36 (1H, d, H-1) indicated the β-configuration of the glucosyl residues [16]. Likewise the other anomeric configuration of the rhamnopyranosyl was confirmed as α-orientated on the basis of the chemical shift values of C-3″ (δ C 72.2), C-5″ (δ C 70.6) with those of the corresponding carbons of methyl α- and β-rhamnopyranoside [17].The identification of the sugar residues were continued by hydrolysis with 10 % HCl to afford d-glucose and l-rhamnose, which were confirmed by GC chromatographic analysis of their l-cysteine methyl ester-TMS derivates. Besides of two sugar units, 13C-NMR and DEPT spectra also showed 29 carbons, five methyl groups, ten methylenes, ten methines, and four quaternary carbons (Table 1). Comparison of above data with those of capsimine [18] and baikeine [19], pingbeinine [20] showed that the aglycone of 1 was similar to capsimine with exception of an additional acetyl group at δ C 22.2 (q) and 172.4 (s) in 1 (Fig. 1). This acetyl group (δ C 22.2, q and 172.4, s) was located at N in last ring by correlations of δ H 2.08 (3H, s, H-29) with δ C 172.4 (s, C-28) and 50.7 (t, C-26), of δ H 3.60 (1H, overlap, H-26a), 2.91 (1H, t, J = 12.4 Hz, H-26b), and 4.46 (1H, dt, J = 6.1, 6.0 Hz, H-22) with δC 172.4 (s, C-28). Signal at δ H 4.22 (1H, t, J = 7.4 Hz) corresponding to δ C 76.3 (t) in its HSQC spectrum showed cross peaks with δ C 61.9 (d, C-17), 55.2 (d, C-14), and 37.4 (t, C-15), which suggested that the hydroxyl substitute at C-16. The other oxygenic proton signal at δ H 3.53 (1H, m) placed at C-3, which was confirmed by HMBC correlations of δ H 1.84 (1H, m, H-1a), 1.03 (1H, d, J = 4.3 Hz, H-1b), 2.38 (1H, dd, J = 2.1, 13.1 Hz, H-4a), and 2.22 (1H, t, J = 12.4 Hz, H-4b) with δ C 79.9 (d, C-3) (Fig. 2). The glycositatic position was unambiguously ascribed to be at C-3 from the HMBC correlation of δ H 4.36 (1H, d, J = 7.8 Hz, H-1′) with C-3 (δ C 79.9). The 1H-1H COSY spectrum of 1 displayed five partial fragments a–e (Fig. 2). The signals of fragments a and b were from δH 4.36 (1H, d, J = 7.8 Hz, H-1′) to δH 4.76 and 3.62 (1H each, H-6′), from δH 4.81 (1H, br s, H-1″) to δH 1.23 (3H, q, J = 7.8 Hz, H-6″), respectively, were further suggested a rhamnose and a glucose appeared. On the basis of δH 4.81 (1H, br s, H-1″) correlating with C-4′ (δC 79.6), the presence of a rhamnopyranosyl (1→4)-glucopyranosyl moiety was deduced (Fig. 2).

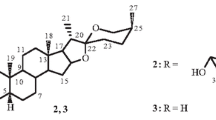
The chemical structures of spiralosides A–C (1–3)

Key 1H-1H COSY ( ) and HMBC (
) and HMBC ( ) correlations of 1
) correlations of 1
The ROESY correlations of Me-19/H-4b/H-2b/H-11b/H-8, H-3/H-2a, and H-11a/H-9/H-14 indicated the α-orientation of H-3, H-9 and H-14, β-orientation of H-8 (Fig. 3). ROESY corrections of Me-18 with H-20, of H-20 with H-16 positioned α-orientation for both Me-21 and OH-16, β-orientation for H-20. ROESY corrections of Me-18 with H-20 also indicated a 17β-side chain, as well as on the biogenetical derivation of C-27 steroidal alkaloids [21]. And a missing effect between H-17 and H-20 displayed, at least approximately, antiperiplanar positions of H-17 and H-20. The coupling constant 3 J 17,20 = 12.4 Hz was in agree with this assumption [7]. Furthermore, the coupling constant 3 J 20,22 = 6.0 Hz suggested that they should be on the same side and the C-22 of configuration is R [22]. Besides, ROESY corrections of H-22 with H-23b, of Me-27 with H-23a indicated H-22 and H-25 both are axial bonds and the C-25 of configuration is S. So compound 1 was determined as (22R,25S)-22,26-epiminocholest-5-ene-3β,16α-diol-N-acetyl-3-O-α-l-rhamnopyranosyl-(1→4)-β-d-glucopyranosyl, named spiraloside A. All of the signals of 1H- and 13C-NMR were assigned by HSQC, HMBC, 1H-1H COSY and ROESY spectra (Table 1).

Key ROESY ( ) correlations of 1
) correlations of 1
Compound 2 was isolated as a white, amorphous powder; \([\alpha ]_{\text{D}}^{23}\) −43.9 (c 0.03, MeOH). The molecular formula C35H57NO8 was established by the HRESIMS quasimolecular ion at m/z 642.3975 [M+Na]+ (calcd for m/z 642.3976), indicating eight degree of unsaturation. Its IR spectrum revealed the presence of hydroxyls (3440 cm−1) and an acylamino group (1630 cm−1). The 1D-NMR data (Table 1) were similar to those of 1 except the absence of the rhamnopyranosyl moiety, as supported by the HMBC correlations of δ H 4.34 (1H, d, J = 7.8 Hz, H-1′) with δ C 79.8 (d, C-3). Other parts of the structure were identical to those of 1 by detailed analysis of 2D-NMR and acid hydrolysis of 2. Consequently, spiraloside B (2) was determined as (22R,25S)-22,26-epiminocholest-5-ene-3β,16α-diol-N-acetyl-3-O-β-d-glucopyranosyl (Fig. 1).
Spiraloside C (3), obtained as an amorphous powder, had the molecular formula of C35H59NO8 as revealed by HRESIMS (calcd for C35H59NO8 at m/z 621.4241), corresponding to seven degrees of unsaturation. The IR data showed the presence of hydroxys (3440 cm−1) and an acylamino group (1619 cm−1). Comparison of 1D-NMR data of 3 with those of 2 (Table 1), showed that the two compounds were similar with exception of the absence of a double bond at C-5/6 in 3, which was suggested by the HMBC correlations of δ H 0.79 (3H, s, H-19) with δ C 46.0 (d, C-5), and 1.27 (1H, m, H-6b) with δ C 46.0 (d, C-5), as well as one less degree of unsaturation. The ROESY correlations of Me-19/H-2b, H-3/H-2a/H-5 indicated the α-orientation of H-3 and H-5. Other parts of the structure were identical to those of 2, by detailed analyses of 2D-NMR and acid hydrolysis of 3. Thus, spiraloside C (3) was determined as (22R,25S)-22,26-epiminocholest-3β,16α-diol-N-acetyl-3-O-β-d-glucopyranosyl (Fig. 1).
The total alkaloids of S. spirale have been screened for the protective anti-tussive effect against ammonia liquor induced cough and the protective expectorant activity used by phenol red secretion test in mice. The results showed that the total alkaloids exhibited an inhibited tendency on antitussive in mice (Tables 2, 3).
Compounds 1–3 were evaluated for their cytotoxicity against five human cancer cell lines using the MTT method as reported previously [23]. Cisplation (sigma, USA) was used as the positive control. Unfortunately, the results showed that all compounds were inactive (IC50 values >40 µM).
3 Experimental Section
3.1 Plant Material
Air-dried fruits of S. spirale were collected in December 2014 from Shuangbai county, Yunnan province, P.R china, and identified by one of the author Yun-Li Zhao, Kunming Institute of Botany, Chinese Academy of Sciences. A voucher specimen (No. 20141225) was deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.2 Extraction and Isolation
The air-dried fruits from S. spirale (5 kg) were crushed and extracted with 20 L 90 % MeOH for five times under reflux for a total 3 h, and then combined extract was concentrated under reduced pressure to afford an extract. The extract was partitioned between EtOAc and 0.5 % HCl solution, getting a nor-alkaloid fraction. The acidic water-soluble, adjusted pH to 9–10, was extracted with EtOAc to give total alkaloids and water-soluble fraction. The total alkaloid fraction (84 g) was subjected to silica gel column (200–300 mesh; gradient CHCl3–MeOH 20:1, 10:1, 8:1, 6:1, 4:1, 3:1, 1:1, 0:1 v/v) to afford fractions 1–6, respectively. Fr.3 (5 g) was chromatographed over silica gel CC (200–300 mesh), eluted with repeated CHCl3:MeOH:H2O (10:1:0.1 to 0:1:1) to obtain three subfractions (Fr.3-1 to Fr.3-3). Fr.3-1 (66 mg) was then separated by repeated RP-18 CC with MeOH:H2O (5:5 to 10:0) and Sephdex LH-20 (MeOH) to give 3 (9 mg). Fr.4 (8.4 g) was submitted to RP-18 CC and elicited with MeOH:H2O (1:9 to 8:2) to afford five subfractions (Fr.4-1 to Fr.4-5). Fr.4-3 (900 mg) was chromatographed over silica gel CC (200–300 mesh), eluted with CHCl3:MeOH:H2O (8:2:0.2 to 0:1:1) to obtain three subfractions (Fr.4-3-1 to Fr.4-3-3). Fr.4-3-2 (35 mg) was further purified by Sephdex LH-20 (MeOH/H2O, 1:9) to obtain 2 (10 mg). Fr.6 (6 g) was separated by RP-18 CC and elicited with MeOH:H2O (1:9 to 10:0) to yield five subfractions (Fr.6-1 to Fr.6-5). Fr.6-3 (1.5 g) was further separated by repeated silica gel with CHCl3:MeOH:H2O (7:3:0.3 to 0:1:1), then followed by Sephdex LH-20 with MeOH:H2O (1:9 to 0:1) to obtained three subfractions (Fr.6-3-1 to Fr.6-3-3), Fr.6-3-2 (25 mg) was further purified by semi-preparative HPLC (28 % CH3CN) to yield 1 (13 mg).
Spiraloside A (1): White amorphous powder; \([\alpha ]_{\text{D}}^{23}\) −46.6 (c 0.03, MeOH); IR (KBr) v max 3441, 2934, 2870, 1623, 1442, 1383, 1343, 1266, 1127, 1056, 1038, 809 cm−1; 1H, 13C-NMR spectroscopic data see Table 1; Positive ESIMS m/z 788 [M+Na]+; HREIMS m/z 788.4553 [M+Na]+ (calcd for C41H67NO12, 788.4561).
Spiraloside B (2): white amorphous powder; \([\alpha ]_{\text{D}}^{23}\) −43.9 (c 0.03, MeOH); IR (KBr) v max: 3440, 2969, 2928, 1630, 1553, 1445, 1383, 1267, 1197, 1079, 1046 cm−1; 1H, 13C-NMR spectroscopic data see Table 1; Positive ESIMS m/z 642 [M+Na]+; HREIMS m/z 642.3975 [M+Na]+ (calcd for C35H57NO8, 642.3976).
Spiraloside C (3): white amorphous powder;\([{{\alpha }}]_{\text{D}}^{24}\) −30.1 (c 0.09, MeOH); IR (KBr) v max: 3427, 2928, 2858, 1727, 1619, 1444, 1382, 1075, 1046 cm−1; 1H, 13C-NMR spectroscopic data see Table 1; Positive ESIMS m/z 644 [M+Na]+; HREIMS m/z 621.4231 [M+H]+ (calcd for C35H59NO8, 621.4241).
3.3 Acid Hydrolysis of compounds 1–3 and GC Analysis
Compounds 1–3 (each 3 mg) were refluxed with 2 M HCl (1,4 dioxane/H2O 1:1, 2 mL) on water bath for 2 h. After cooling, the reaction mixture was neutralized with 1 M NaOH. The reaction mixture was extracted with CHCl3 (3 × 5 mL). The aqueous layer was evaporated to dryness. The dried residue was dissolved in 1 mL anhydrous pyridine and treated with l-cysteine methyl ester hydrochloride (1.5 mg) stirred at 60 °C for 1 h. Trimethylsilylimidazole (1.0 mL) was added to the reaction mixtures, and they were kept at 60 °C for 30 min. The supernatants (4 μL) were analyzed by GC, respectively, under the following conditions: H2 flame ionization detector. Column: 30QC2/AC-5 quartz capillary column (30 m × 0.32 mm). Column temperature: 180–280 °C with the rate of 3 °C/min, and the carrier gas was N2 (1 mL/min) injector temperature: 250 °C; and split ratio: 1/50. Peaks of the hydrolysate were detected by comparison with retention times of authentic samples of d-glucose and L-rhamnose after treatment with trimethyl–chlorosilane (TMCS) in pyridine. The absolute configurations of the compounds 1–3 were determined by comparison of the retention times of the corresponding derivatives with those of standard d-glucose and L-rhamnose giving a single peak at 19.01 and 15.43 min, respectively.
3.4 Animals
ICR mice of either sex (20–22 g) were purchased from Kunming Medical College (License number SYXK2014-0004). All animals were housed at room temperature (20–25 °C) and constant humidity (40–70 %) under a 12 h light–dark cycle in SPF grade laboratory. The animal study was performed according to the international rules considering animal experiments and the internationally accepted ethical principles for laboratory animal use and care.
3.5 Anti-tussive Activity Assay
ICR mice of either sex weighing 21–24 g were divided randomly, 10 mice per group. The negative control of animals was treated with distilled water orally, and the positive control was treated with codeine phosphate, the remaining groups treated were with test samples respectively. Anti-tussive activity was investigated on a classical mouse cough model induced by ammonia liquor [24]. Briefly, each mouse was placed in a 300 mL special glass chamber and exposed to 40μL 25 % NH4OH. The cough frequency produced during 2 min exposure period was counted. In the second assay for alkaloids, cough frequency and latent period of cough were recorded.
3.6 Expectorant Effect Assessment
The procedures were performed as described previously [25]. Male and female mice were randomly allotted and treated with a single dose 30 min before intraperitoneal injection of phenol red solution (5 % in saline solution, w/v, 0.1 mL/10 g body weight). Mice were sacrificed by cervical dislocation 30 min after application of phenol red. After dissected free from adjacent organs, the trachea was removed from the thyroid cartilage to the main stem bronchi and put into 2 mL normal saline immediately. After ultrasonic for 5 min, 0.1 mL of 1 M NaOH solution was added to the saline and optical density of the mixture were measured at 546 nm using enzyme standard instrument.
3.7 Cytotoxicity Assay
Five human cancer cell lines, lung cancer A-549, human myeloid leukemia HL-60, hepatocellular carcinoma SMMC-7721, breast cancer MCF-7, and colon cancer SW480 cells, were used in the cytotoxic assay. All the cells were cultured in RPMI-1640 or DMEM medium (Hyclone, USA), supplemented with 10 % fetal bovine serum (Hyclone, USA) in 5 % CO2 at 37 °C. The cytotoxicity assay was performed according to the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) method in 96-well microplates [26]. Briefly, 100 μL of adherent cells was seeded to each well of a 96-well cell culture plates and allowed to adhere for 12 h before drug addition, while suspended cells were seeded just before drug addition with an initial density of 1 × 105 cells/mL in 100 μL of medium. Each tumor cell line was exposed to the test compound dissolved in DMSO at concentrations of 0.064, 0.32, 1.6, 8, and 40 μM in triplicates for 48 h, with cisplatin (Sigma, USA) as a positive control. After compound treatment, cell viability was detected, and the cell growth curve was graphed. The IC50 value was calculated by Reed and Muench’s method [27].
References
The Editorial Board of Flora of China, Flora, Reipublicae Popularis Sinicae, (Tomus 67, Science Press, Beijing, 1978), p. 73
State Administration of Traditional Chinese Medicine of China, ZhongHuaBenCao-Dai Ethnopharmacy, (Science Press, Shanghai, 2005), p. 231
P. Temin, A.K. Das, R. Shankar, C. Tamuly, M. Hazarika, Am. J. Pharm Tech Res. 5, 307–314 (2015)
K. Sukanya, N. Surapol, L. Saisunee, T. Aphiwat, T. Kongkiat, C. Patchanee, Chiang Mai J. Sci. 39, 445–454 (2012)
K. Sukanya, L. Boonsom, L. Saisunee, T. Aphiwat, G.P. Stephen, Nat. Prod. Commun. 7, 955–958 (2012)
X.F. Teng, Y.J. Zhang, C.R. Yang, Yunnan Zhiwu Yanjiu 30, 239–242 (2008)
R. Helmut, Phytochemistry 43, 705–707 (1996)
M.P. Boruah, J.G. Handique, IJAPBC 3, 426–433 (2014)
M. Friedman, K.R. Lee, H.J. Kim, I.S. Lee, N. Kozukue, J. Agric. Food Chem. 53, 6162–6169 (2005)
K.R. Lee, N. Kozukue, J.S. Han, J.H. Park, E.Y. Chang, E.J. Baek, J.S. Chang, M. Friedman, J. Agric. Food Chem. 52, 2832–2839 (2004)
R.J. Bushway, S.A. Savage, B.S. Ferguson, Am. Potato J. 64, 409–413 (1987)
J.M. Wierenga, R.M. Hollingworth, Nat. Toxins 1, 96–99 (1992)
M. Friedman, T.E. Fitch, W.H. Yokoyama, Food Chem. Toxicol. 38, 549–553 (2000)
M. Friedman, T.E. Fitch, W.H. Yokoyama, J. Food Sci. 65, 897–900 (2000)
A.M. Fewell, J.G. Roddick, M. Weissenberg, Phytochemistry 37, 1007–1011 (1994)
P.K. Agrawal, Phytochemistry 31, 3307–3330 (1992)
R. Kasai, M. Okihara, J. Asakawa, K. Mizutani, O. Tanaka, Tetrahedron 35, 1427–1432 (1979)
C.N. Lin, K.H. Gan, Planta Med. 55, 48–50 (1989)
S. Ito, M. Miyashita, Y. Fukazawa, A. Mori, I. Iwai, M. Yoshimura, Tetrahedron Lett. 29, 2961–2964 (1972)
D.M. Xu, M.L. Xu, S.Q. Wang, E.X. Hung, X.G. Wen, J. Nat. Prod. 53, 549–552 (1990)
K. Ohyama, A. Okawa, Y. Moriuchi, Y. Fujimoto, Phytochemistry 89, 26–31 (2013)
Y. Tezuka, T. Kikuchi, W. Zhao, J. Chen, Y. Guo, J. Nat. Prod. 61, 1078–1081 (1998)
T. Feng, X.H. Cai, Y.P. Liu, Y. Li, Y.Y. Wang, X.D. Luo, J. Nat. Prod. 73, 22–26 (2010)
S.Y. Xu, R.L. Bian, X. Chen, Pharmacological Experiment Methodology (People’s Medical Publishing House, Beijing, 1991), p. 1167
H. Engler, I. Szelenyi, J Pharmacol Methods 319, 151–157 (1984)
T. Mosmann, J. Immunol. Methods 65, 55–63 (1983)
L.J. Reed, H. Muench, Am. J. Hyg. 27, 493–497 (1938)
Acknowledgments
The authors are grateful to the Natural Science Foundation of China (81225024), and to the analytical group of the Laboratory of Phytochemistry, Kunming Institute of Botany for spectral measurements.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
13659_2016_103_MOESM1_ESM.pdf
Supporting Information Available: bioassay, 1D, 2D-NMR, MS and IR spectra of spiralosides A–C (1–3), these supplementary materials are available in the online version of this article and is accessible for authorized users (PDF 1341 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, D., Zhao, YL., Qin, XJ. et al. Spiralosides A–C, Three New C27-Steroidal Glycoalkaloids from the Fruits of Solanum spirale . Nat. Prod. Bioprospect. 6, 225–231 (2016). https://doi.org/10.1007/s13659-016-0103-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-016-0103-9