
Abstract
The perennial herbaceous plant Euphorbia jolkinii (Euphorbiaceae) is a noxious weed widely distributed in the grasslands of northwestern Yunnan and has greatly threatened the local biodiversity. Phytochemical investigation on the fresh roots of E. jolkinii afforded six new diterpenoids 1, 2, 4–6, and 8, together with fifteen known diterpenoids. Their structures were elucidated on the basis of 1D and 2D NMR and other spectroscopic methods. Casbane, lathyrane, abietane, and ent-kaurane diterpenoids were reported from this plant for the first time. Selected compounds were evaluated for their antifeedant and anti-RSV (respiratory syncytial virus) activities. Compound 2 and ingenol (3) exhibited moderate antifeedant activity against a generalist insect herbivore, Spodoptera exigua, with EC50 values of 17.88 and 17.71 μg/cm2 respectively. Compound 19 showed significant anti-RSV activity, with 50 % inhibition (IC50) value of 10.0 μM and selective index of 8.0. Compounds 1 and 2 were less active against RSV virus, both with IC50 value of 25 μM, and with selective indices of 1.0 and 3.2 respectively. These findings provided new evidence for the biological functions and utilization of the diversified diterpenoid metabolites in the roots of this rich but harmful plant.
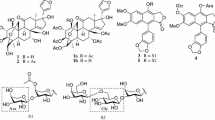
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Euphorbia is the largest genus of Euphorbiaceae which contains more than 2000 species, mainly distributing in the tropical and subtropical regions of Africa and America [1]. Extensive phytochemical investigations of different Euphorbia species have been conducted, therefore it has been proved to be a rich source of diterpenoids with diverse structures and interesting biological activities [2–4].
There are more than 80 Euphorbia species in China [5], widely distributed throughout the whole country especially in the southwestern Hengduan Mountains and northwestern arid regions. Euphorbia jolkinii (or Euphorbia nematocypha), a perennial herbaceous plant, is indigenous to Southwest of China and particularly rich in the grasslands of northwestern Yunnan. This plant has a highly developed root system that could extend 1–2 m far away from where they thrived. It is harmful to local agriculture and animal husbandry, which has caused serious grassland degradation and ecological disasters in its distribution area. Previous phytochemical investigations on E. jolkinii have led to the isolation and identification of a number of terpenoid secondary metabolites [6–8], most of which exhibited only weak or no biological activities. To understand the biological functions of the secondary metabolites of E. jolkinii, we have carried out a detailed investigation on the chemistry of the fresh roots of this plant. Herein, we described the isolation and structural elucidation of six new and 15 known diterpenoids from E. jolkinii, and the antifeedant and antiviral activities of some of these compounds.
2 Results and Discussion
Compound 1 has a molecular formula C26H32O7, as deduced from the HR-EI-MS (found: m/z 456.2169; calcd. for 456.2148), requiring eleven degrees of unsaturation. The presence of hydroxyl groups (3434 cm−1), carbonyl groups (1719 and 1686 cm−1), and olefinic groups (1637 cm−1) were clearly shown in the IR spectrum of 1. The UV maximum at 267 nm and MS fragmental peak at m/z 330 [M-CHO(CH=CH)2COOH]− indicated the existence of a (2E,4E)-6-oxo-2,4-hexadienoyloxy group, which was supported by five sequentially coupled protons at δH 6.48 (d, J = 14.3 Hz), 7.46 (dd, J = 14.3, 11.2 Hz), 7.44 (dd, J = 14.4, 11.2 Hz), 6.51 (dd, J = 14.4, 7.8 Hz), and 9.70 (d, J = 7.8 Hz) in the 1H NMR spectrum of 1 (Table 1). In addition, the 1H NMR signals suggested the presence of a secondary methyl at δH 0.92 (d, J = 7.0 Hz) and three tertiary methyls at δH 1.04, 1.10, and 1.80, two olefinic protons at δH 5.81 (s) and 6.04 (d, J = 3.9 Hz), two oxy-methines at δH 3.62 (s) and 4.43 (br. s), and one oxy-methylene at δH 4.63 (ABd, J = 12.8 Hz) and 4.84 (ABd, J = 12.8 Hz). In the relatively high field, two protons at δH 0.66 and 0.85 were indicative of a cyclopropyl residue. The 13C NMR spectrum of 1 exhibited 26 carbons. Besides the signals for the above-mentioned (2E,4E)-6-oxo-2,4-hexadienoyloxy moiety, there were still 20 carbons which were classified as four methyls, two methylenes including an oxymethylene, eight methines including two oxymethines and two olefinic methines, and six quaternary carbons including an oxygenated one, two olefinic ones, and a keto. The two methines at δC 23.9 and 24.1 and the quaternary carbon at δC 24.4 further supported the existence of a cyclopropyl residue. The molecular formula of 1 still needed three degrees of unsaturation, indicating three additional rings were available in 1. These spectral evidences indicated that compound 1 was a tetracyclic diterpenoid containing a cyclopropane in its skeleton and a six-carbon ester side chain as its substituent. Comparison of the 1H and 13C NMR spectra of 1 with those of 20-O-(2′E,4′E-decadienoyl) ingenol [9] and sikkimenoid E [10], which were isolated from Euphorbia kansui and Euphorbia sikkimensis respectively, indicated that 1 was very similar to these two compounds, with the only difference in their C-20 ester side chains. The decadienoyl group in 20-O-(2′E,4′E-decadienoyl) ingenol and sikkimenoid E was found to have been replaced by a (2E,4E)-6-oxo-2,4-hexadienoyl group in 1, which was confirmed by the 2D NMR experiments (1H–1H COSY, HSQC, and HMBC) of 1 (for selected HMBC correlations, see Fig. 2). The ROESY spectrum of 1 indicated that the configurations of the chrial centers in the skeleton of 1 were identical to those of 20-O-(2′E,4′E-decadienoyl) ingenol and sikkimenoid E. Therefore, compound 1 was concluded to be 20-O-[(2′E,4′E)-6-oxo-2′,4′-hexadienoyl] ingenol (Fig. 1).

Chemical structures of compounds 1-21
The molecular formula of compound 2 was established as C28H34O7 through an analysis of its HR-EI-MS (found: m/z 482.2301; calcd. for 482.2305). The 1H and 13C NMR spectral data attributable to the diterpenoid skeleton of 2 (Table 1) were almost same with those of 1. The main difference was that two additional olefinic methines appeared at the downfield in the 13C NMR spectrum of 2, suggesting that the 6-oxo-2,4-hexadienoyloxy group in 1 was replaced by a 8-oxo-2,4,6-octatrienoyloxy group in 2. The coupling constants of those olefinic protons (J2′,3′ = 15.3 Hz; J6′,7′ = 15.2 Hz) indicated that both the configurations of Δ2′,3′ and Δ6′,7′ were E. Although H-4′ and H-5′ were partially overlapped and their couplings were complicated, the E configuration of Δ4′,5′ could be determined by the ROESY interactions between H-3′ and H-5′ and between H-4′ and H-6′ (Fig. 3). Ultimately, the structure of 2 was identified as 20-O-[(2′E,4′E,6′E)-8-oxo-2′,4′,6′-octatrienoyl] ingenol (Fig. 1).
Compound 4 was isolated as colorless needles, and its molecular formula was determined to be C20H30O3 on the basis of the HR-EI-MS (found: m/z 318.2194; calcd. for 318.2195). Its IR absorption at 3442, 1716, and 1632 cm−1 showed the presence of hydroxyl group, carbonyl group, and olefinic group. In the 1H NMR spectrum of 4, four tertiary methyls at δH 0.75, 0.84, 0.88, and 1.13 were clearly shown (Table 1). Two proton signals resonated from oxygenated methines were present at δH 3.52 (ddd, J = 11.6, 4.4, 4.0 Hz) and 4.05 (d, J = 5.2 Hz). In addition, an ABX system for a vinyl group at δH 4.96 (dd, J = 10.8, 1.4 Hz), 5.01 (dd, J = 17.6, 1.4 Hz), and 5.94 (dd, J = 17.6, 10.8 Hz) and an additional olefinic proton at δH 5.46 (brs) were observed. In the 13C NMR spectrum of 4, twenty carbon signals were exhibited, which were further classified by DEPT-90 and DEPT-135 experiments as four methyls, five methylenes including a terminal olefinic methylene (δC 112.0), six methines including two oxygenated methines (δC 74.6 and 82.6) and two olefinic methines (δC 122.4 and 148.3), and five quaternary carbons including an olefinic one (δC 135.0) and a keto (δC 210.9). The above spectral evidences, in combination with a comparison of the NMR spectra of 4 with those of a known isopimarane diterpenoid that was isolated from E. jolkinii, (3β,12α)-3,12-dihydroxyisopimara-7,15-dien-2-one [8], revealed that the two compounds were extremely similar. The 1H–1H COSY, HSQC, and HMBC (Fig. 2) experiments of 4 confirmed that its planar structure was actually identical to (3β,12α)-3,12-dihydroxyisopimara-7,15-dien-2-one. The only difference between these two compounds was that the oxygenated methine (C-12) at δC 72.3 in (3β,12α)-3,12-dihydroxyisopimara-7,15-dien-2-one [8] shifted downfield to δC 74.6 in 4, and concurrent chemical shift changes of those carbons surrounding C-12 also occurred. In the ROESY spectra of 4 (Fig. 3), the correlations of H-3 with Me-18 and H-5 indicated that H-3 was remained to be α-oriented. Moreover, the correlation of Me-17 with H-11β elucidated a β-orientation for Me-17, thus confirmed the isopimarane skeleton of 4. However, the correlations of H-12 with H-9 and H-11α indicated that H-12 was in α-orientation, which was on the contrary to that of (3β,12α)-3,12-dihydroxyisopimara-7,15-dien-2-one [8]. Finally, compound 4 was characterized as (3β,12β)-3,12-dihydroxyisopimara-7,15-dien-2-one (Fig. 1).

Selected HMBC (arrow) correlations of compounds 1, 4, and 8

Key ROESY correlations of 2 and 4
Compound 5 was also obtained as colorless needles. The molecular ion peak at m/z 304.2401 in HR-EI-MS showed a molecular formula of C20H32O2 (calcd. for 304.2402). The close resemblance between the NMR spectra of 5 (Table 2) and those of 4 indicated that 5 was also an isopimarane diterpenoid. The major difference between them was that the keto group in 4 disappeared while one more methylene carbon (δC 28.4) appeared in 5. Based on 1H-1H COSY correlations of H2-1 and H-3 with the methylene protons, it was elucidated that this menthylene was ascribable to C-2. The relative configuration of all chiral centers in 5 remained unchanged, as was established by a 2D ROESY experiment. The ROEs of H-3 with Me-18 and H-5, of 3-OH with Me-19, and of H-14β with 12-OH, indicated that both H-3 and H-12 were in α-orientation. Accordingly, the structure of compound 5 was identified as (3β,12β)-3,12-dihydroxyisopimara-7,15-diene (Fig. 1).
Compound 6 was attributed a molecular formula of C20H28O3 by its HR-EI-MS (found: m/z 316.2038; calcd. for 316.2038), corresponding to seven degrees of unsaturation. Analysis of its NMR spectral data (Table 2) disclosed that 6 was again an isopimarane diterpenoid whose structure was similar to 4. However, it was noteworthy that an additional tri-substituted double bond was found to have replaced a methylene and an oxygenated methine in 6. In addition, the keto carbon in 6 was shifted dramatically upfield by 10.4 ppm, suggesting that this keto group was conjugated. In the HMBC spectrum of 6, correlations from Me-19 (δH 1.16) and Me-18 (δH 1.20) to the keto carbon at δC 200.5 were observed, which allowed the location of this keto group to be at C-3, thus also suggested that the additional tri-substituted double bond should be formed between C-1 and C-2. The HMBC cross-peaks from the deshielded olefinic singlet δH 6.21 (H-1) to C-20 (δC 15.8), C-10 (δC 36.3), C-5 (δC 48.0), C-3 (δC 200.5), and C-2 (δC 143.6), and from the broad singlet at δH 6.05 (2-OH) to C-1 (δC 123.6) indicated that C-2 of 6 was hydroxylated. The ROESY spectrum of 6 showed identical correlations to those for 4. Consequently, compound 6 was determined as (12β)-2,12-dihydroxy isopimara-1,7,15-trien-3-one (Fig. 1).
Compound 8 was assigned a molecular formula of C23H36O3, as determined by its HR-EI-MS (found: m/z 360.2682; calcd. for 360.2664), indicating six degrees of unsaturation. The IR spectrum showed the existence of carbonyl group at 1706 cm−1. The 1H NMR spectrum (Table 2) showed five tertiary methyls at δH 1.01, 1.03, 1.14, 1.28, and 1.29, and a pair of AB doublets at δH 3.62 and 3.95 owing to an oxymethylene. The 13C NMR and DEPT spectra (Table 2) indicated the presence of a keto carbon (δC 215.6) and 22 sp3 carbons separately for five methyls, nine methylenes including an oxy-methylene (δC 75.0), three methines, and six quaternary carbons including an oxygenated one (δC 83.0) and a ketal carbon (δC 109.1). These NMR data were very similar to those of ent-l6α,17-dihydroxyatisan-3-one that was isolated from E. fidjiana [11], implying that 8 was also an ent-atisane diterpenoid but containing an acetonide. This acetonide could only be formed between C-17 and C-16, as was supported by the downfield chemical shifts of C-16 (δC 83.0) and C-17 (δC 75.0) and the HMBC correlations from H2-17 to the ketal carbon (Fig. 2). Thus, the structure of compound 8 was assigned as ent-3-oxoatis-16α,17-acetonide (Fig. 1).
Fifteen known diterpenoids, namely, ingenol (3) [12], (3β,12α)-3,12-dihydroxyisopimara-7,15-dien-2-one (7) [8], ent-l6α,17-dihydroxyatisan-3-one (9) [11], ent-(3α,5β,8α,9β,10α,12α)-3-hydroxyatis-16-en-14-one (10) [8], ent-(5β,8α,9β,10α,11α,12α)-11-hydroxyatis-16-ene-3,14-dione (11) [8], ent-(5β,8α,9β,10α,11α,12α)-3,14-dioxoatis-16-en-11-yl acetate (12) [8], jolkinolide E (13) [13, 14], 7β-hydroxy-ent-abieta-8(14),13(15)-dien-12α,16-olide (14) [15], pekinenal (15) [16], pekinenin A (16) [17], pekinenin D (17) [18], ent-l6β,17-isopropylidenedioxy kauran-3β-ol (18) [19], jolkinol A (19) [20], jolkinol A′ (20) [20], and 3β,5α,20-trihydroxy-15β-cinnamoyloxy-14-oxolathyra-6Z,12E-diene (21) [21], were also isolated and identified by comparison of their spectroscopic data with those reported values in the literatures.
The antifeedant activity of compounds 2, 3, and 15 against the larvae of a generalist insect herbivore, the beet armyworm (Spodoptera exigua), were tested as previously described [22, 23]. It was found that compounds 2 and 3 showed moderate deterrence against the insect, with EC50 values of 17.88 and 17.71 μg/cm2, respectively (Table 3), which were less active than the commercial neem oil (1 % azadirachtin, EC50 = 3.71 μg/cm2) by Kunming Rixin Dachuan Technology Co., Ltd. Compounds 1–3, 10, 11, 13, 15, and 19, were assayed for their anti-RSV activity according to the reported method [24, 25]. Compound 19 showed significant anti-RSV activity, with IC50 value of 10.0 μM and selective index of 8.0. Compounds 1 and 2 were less active against RSV, both having IC50 value of 25.0 μM, and SI of 1.0 and 3.2 respectively (Table 4). However, the other compounds were basically inactive. Compounds 10, 13, and 15 were also evaluated for their antiviral activities against Herpes simplex type one (HSV-1) and Coxsackie B3 (Cox B3) viruses as described in the literature [24], but none of them showed obvious activity.
In summary, we have isolated and identified six new and fifteen known diterpenoids from the fresh roots of E. jolkinii. These compounds belong to seven different types of skeletons, including ingenane, isopimarane, ent-atisane, casbane, lathyrane, abietane, and ent-kaurane. Among them, the diterpenoids with casbane, lathyrane, abietane, and ent-kaurane skeletons were reported from this plant for the first time, which further broadened our knowledge of the diversified diterpenoid metabolites in E. jolkinii. The antifeedant, antiviral, and cytotoxic activities of these diterpenoids implied their possible roles in the plant as constitutive defense compounds against insects, animals, and microbes, and provided new evidence for the exploitation of this rich but harmful plant, which are worthy of more future investigations.
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were determined on a Horiba-SEAP-300 spectropolarimeter. UV spectral was obtained on a Shimadzu-210A double-beam spectrophotometer. IR spectra were recorded on a Bruker-Tensor-27 spectrometer with KBr pellets. 1H NMR, 13C NMR, and 2D NMR spectra were taken on either a Bruker AM-400, DRX-500 or Bruker Avance III 600 spectrometers with TMS as internal standard. EI-MS and HR-EI-MS data were measured on a VG-Auto-Spec-3000 spectrometer. Semipreparative HPLC was carried out on an Agilent 1200 series instrument equipped with a quaternary pump, a vacuum degasser, an autosampler, a thermostatic column compartment, and a diode array detector. Column chromatography was performed with silica gel (200–300 mesh, Qingdao Marine Chemical Factory, P. R. China), Sephadex LH-20 (GE Healthcare Bio-Xciences AB), and MCI gel CHP-20P (75–150 μm, Mitsubishi Chemical Corp., Tokyo, Japan). TLC was carried out on precoated silica gel GF254 glass plates. Spots were visualized under UV light and by dipping with 15 % H2SO4 in ethanol followed by heating. All solvents including petroleum ether (60–90 °C) were distilled prior to use.
3.2 Plant Material
The fresh roots of Euphorbia jolkinii (or Euphorbia nematocypha) were collected from the alpine grasslands of Zhongdian county, Yunnan province, China, in June 2012, and were identified by Dr. Jian Liu. A voucher specimen (KIB20120620) has been deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and Isolation
The fresh roots of E. jolkinii (27 kg) were chopped and extracted with MeOH (25 L × 3, each time 24 h) at room temperature. The combined extracts were evaporated to dryness under reduced pressure to obtain a crude extract (3470 g), which was suspended in H2O (4 L) and then extracted with petroleum ether (PE, 4 L × 4) and EtOAc (4 L × 4), successively, to yield a PE-soluble fraction (67.1 g) and an EtOAc-soluble fraction (61.6 g). The PE-soluble fraction (67.1 g) was subjected to silica gel column chromatography eluting with a gradient solvent system of chloroform/acetone (from 10:0 to 0:10, v/v) to afford six fractions (A–F) on the basis of TLC detection. Fraction A (30.8 g, CHCl3/Me2CO, 10:0) was chromatographed on silica gel columns, and then purified using a Sephadex LH-20 column (CHCl3/MeOH, 1:1, v/v) to yield compounds 13 (10 mg), 15 (25 mg), and 17 (6 mg). Fraction C (6.6 g, CHCl3/Me2CO, 8:2) was further chromatographed over a silica gel column using PE/Me2CO (9:2, v/v) as eluents to give four subfractions C1–C4. Subfraction C2 (560 mg) was purified by silica gel (PE/2-propanol, 30:1, v/v) and Sephadex LH-20 (CHCl3/MeOH, 1:1, v/v) columns to afford 3 (34 mg) and 10 (40 mg). Subfraction C3 (3.7 g) was repeatedly chromatographed on a Sephadex-LH20 column (Me2CO) and purified by reversed-phase semipreparative HPLC using 69 % MeOH in water as eluents (flow rate: 3 mL/min; column: ZORBAX SB-C18, 5 μm, 9.4 × 250 mm; detection: UV 280 nm; retention times: 12.1 and 15.3 min respectively) to obtain 1 (16 mg) and 2 (12 mg). Similarly, the EtOAc-soluble fraction (61.6 g) was separated by column chromatography over silica gel with a mixture of chloroform and acetone (from 10:0 to 0:10, v/v) to give six fractions a-f. Fraction b (5.0 g, CHCl3/Me2CO, 9:1) was chromatographed on silica gel and Sephadex-LH20 columns (CHCl3/MeOH, 1:1, v/v) to yield 6 (6 mg), 9 (75 mg), 19 (33 mg), 20 (4 mg), and 21 (2 mg). Fraction c (1.4 g) was subjected to silica gel column eluted with PE/Me2CO (40:1, v/v) to afford 8 (3 mg) and 18 (2 mg). The remaining subfractions were enriched (19.6 g) and were passed through a MCI gel column (MeOH/H2O, from 6:4 to 10:0) to give six subfractions (a1–a6). Subfraction a2 (2.3 g) was further separated with silica gel columns with PE and isopropyl alcohol (20:1, v/v) as eluents to afford 5 (4 mg), 7 (6 mg), 11 (6 mg), 12 (8 mg), 14 (4 mg), 16 (7 mg), and 37 mg of crude sample mainly containing 4. Compound 4 (6.3 mg) was achieved by further purification with reversed-phase semipreparative HPLC using 80 % MeOH in water as eluents.
3.4 20-O-[(2′E,4′E)-6-oxo-2′,4′-Hexadienoyl] ingenol (1)
White soild; [α] 17D + 11.1 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 267 (3.15), and 202 (3.05) nm; IR (KBr) νmax 3434, 2954, 2940, 2871, 1719, 1686, 1637, 1450, 1382, 1226, 1177, 1103, 1015 cm−1; 1H and 13C NMR: see Table 1; EIMS m/z 456 [M]+ (6), 411 (13), 368 (13), 330 (77), 135 (43), 121 (89), 91 (100), 77 (100), 67 (88), 55 (99); HREIMS m/z 456.2169 (calcd for C26H32O7, 456.2148).
3.5 20-O-[(2′E,4′E,6′E)-8-oxo-2′,4′,6′-octatrienoyl] ingenol (2)
White soild; [α] 17D + 18.2 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 280 (3.03), and 203 (3.16) nm; IR (KBr) νmax 3433, 2955, 2925, 2872, 1716, 1638, 1381, 1149, 1016 cm−1; 1H and 13C NMR: see Table 1; EIMS m/z 482 [M]+ (10), 464 (12), 312 (15), 241 (18), 135 (55), 121 (64), 91 (72), 77 (100); HREIMS m/z 482.2301 (calcd for C28H34O7, 482.2305).
3.6 (3β,12β)-3,12-Dihydroxyisopimara-7,15-dien-2-one (4)
Colorless needles; [α] 17D + 29.4 (c 0.3, MeOH); UV (MeOH) λmax (log ε) 203 (2.75) nm; IR (KBr) νmax 3442, 2968, 2924, 1716, 1632, 1391, 1112, 1059 cm−1; 1H and 13C NMR: see Table 1; EIMS m/z 318 [M]+ (38), 300 (100), 285 (23), 227 (35), 185 (43), 164 (44), 105 (54), 55 (50); HREIMS m/z 318.2194 (calcd for C20H30O3, 318.2195).
3.7 (3β,12β)-3,12-Dihydroxyisopimara-7,15-diene (5)
Colorless needles; [α] 17D – 26.0 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 203 (2.74) nm; IR (KBr) νmax 3441, 3425, 2962, 2928, 2853, 1635, 1384, 1088, 1039, 997, 915 cm−1; 1H and 13C NMR: see Table 2; EIMS m/z 304 [M]+ (8), 286 (100), 268 (43), 253 (65), 146 (51), 105 (41), 55 (47); HREIMS m/z 304.2401 (calcd for C20H32O2, 304.2402).
3.8 (12β)-2,12-Dihydroxyisopimara-1,7,15-trien-3-one (6)
Colorless oil; [α] 17D + 9.0 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 266 (2.64), and 203 (2.84) nm; IR (KBr) νmax 3433, 2967, 2925, 2873, 1714, 1673, 1651, 1408, 1221, 1052 cm−1; 1H and 13C NMR: see Table 2; EIMS m/z 316 [M]+ (6), 288 (7), 152 (100), 124 (98), 109 (51), 83 (73), 55 (58); HREIMS m/z 316.2038 (calcd for C20H28O3, 316.2038).
3.9 Ent-3-oxoatis-16α,17-acetonide (8)
Colorless needles; [α] 17D – 44.7 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 228 (2.51), and 200 (2.42) nm; IR (KBr) νmax 2982, 2935, 2869, 1706, 1674, 1451, 1381, 1370, 1261, 1212, 1053 cm−1; 1H and 13C NMR: see Table 2; positive ESIMS m/z 383 [M + Na]+; HREIMS m/z 360.2682 (calcd for C23H36O3, 360.2664).
3.10 Antifeeding Assay
Beet armyworms (Spodoptera exigua) were purchased from the Pilot-Scale Base of Bio-Pesticides, Institute of Zoology, Chinese Academy of Sciences. A modified dual-choice bioassay as previously described was performed for antifeedant test [22, 23] The larvae were reared on an artificial diet in our laboratory under controlled photoperiod (light: dark, 12:8 h) and temperature (25 ± 2 °C). Larvae were starved 3–4 h prior to each bioassay. Fresh leaf discs were cut from Brassica chinensis, using a cork borer (1.1 cm in diameter). Treated leaf discs were painted with 10 μL of acetone solution containing the test compound, and control leaf discs with the same amount of acetone. After air drying, two tested leaf discs and two control ones were set in alternating position in the same Petri dish (90 mm in diameter), with moistened filter paper at the bottom. Two thirds of instars were placed at the center of the Petri dish. Five replicates were run for each treatment. After feeding for 24 h, areas of leaf discs consumed were measured. The insect antifeedant potency of the test compound was evaluated in terms of the EC50 value which was determined by Probit analysis for each insect species.
3.11 Cytotoxicity Assay
Cytotoxicity assay was kindly carried out according to the established protocols [24, 25]. The Hep-2 cells (human larynx epidermoid carcinoma cell line) and Vero cells (African green monkey kidney cell line) were grown in 96-well plates (8 mm diameter; Falcon Plastics, Oxnard, CA) and incubated at 37 °C in a humidified atmosphere supplied with 5 % carbon dioxide. When the cell cultures were confluent, the culture medium was removed from each well and replenished with 0.1 mL of the maintenance medium. To test for cytotoxicity, 0.1 mL of the maintenance medium containing the tested samples in serial twofold dilutions was added to each of the wells. The well with 0.1 mL maintenance medium but without sample was prepared to act as a cell control. All the cultures were then incubated at 37 °C for 3 days. The morphology of the cells was inspected daily and observed microscopically for any detectable alterations. The 50 % cytotoxicity concentration (CC50), the concentration required to cause visible changes in 50 % of the intact cells, was evaluated. The maximal non-cytotoxic concentration (MNCC) was estimated as the maximal concentration of the samples that did not exert toxic effects by microscopic monitoring.
3.12 Antivirus Assay
RSV (respiratory syncytial virus, long strain), Cox B3 (Coxsackie B3), and HSV-1 (Herpes simplex virus type 1, 15577 strain) were used for antivirus assay as methods described in the literature [24, 25]. A cytopathic effect (CPE) reduction assay was adopted for screening the antiviral activities of the pure compounds isolated from the roots of E. jolkinii in the present study. In brief, to confluent cell monolayers in a 96-well plate, 100 TCID50 (tissue culture medium infective dose) virus suspension and serial two-fold dilutions of compounds were added to each test well simultaneously. The virus suspension and dilution medium without samples were added, respectively, to the cell cultures to serve as the virus control and cell control. The plates were then incubated at 37 °C in a humidified CO2 atmosphere for 3–6 days. The RSV induced CPE was scored under light microscopy (score 0, 0 %; score 1, 0–25 %; score 2, 35–50 %; score 3, 50–75 %; and score 4, 75–100 %). The concentration that reduced 50 % of CPE in respect to the virus control was estimated from the plots of the data and was defined as the 50 % inhibitory concentration (IC50). The selective index (SI) was calculated from the ratio CC50/IC50 [26].
References
A.R. Jassbi, Phytochemistry 67, 1977–1984 (2006)
J.R. Hanson, Nat. Prod. Rep. 20, 70–78 (2003)
Q.W. Shi, X.H. Su, H. Kiyota, Chem. Rev. 108, 4295–4327 (2008)
J.R. Hanson, Nat. Prod. Rep. 29, 890–898 (2012)
B.B. Zhang, X.L. Han, Q. Jiang, Z.X. Liao, C. Liu, Y.B. Qu, Fitoterapia 83, 1242–1247 (2012)
D. Cao, Y.L. Su, J.S. Yang, Acta Pharm. Sin. 27, 445–451 (1992)
D.G. Wu, B. Sorg, W. Adolf, E.H. Seip, E. Hecker, Phytother. Res. 6, 237–240 (1992)
F. He, J.X. Pu, S.X. Huang, W.L. Xiao, L.B. Yang, X.N. Li, Y. Zhao, J. Ding, C.H. Xu, H.D. Sun, HeIv. Chim. Acta 91, 2139–2147 (2008)
L.Y. Wang, N.L. Wang, X.S. Yao, S. Miyata, S. Kitanaka, J. Nat. Prod. 65, 1246–1251 (2002)
D.S. Yang, W.B. Peng, Z.L. Li, X. Wang, J.G. Wei, K.C. Liu, Y.P. Yang, X.L. Li, Nat. Prod. Bioprospect. 3, 112–116 (2013)
A.R. Lal, R.C. Cambie, P.S. Rutledge, P.D. Woodgate, Phytochemistry 29, 1925–1935 (1990)
Z. Jia, Y. Ding, Planta Med. 57, 569–571 (1991)
D. Uemura, Y. Hirata, Chem. Lett. 7, 819–822 (1974)
N. Crespi-Perellino, L. Garofano, E. Arlandini, V. Pinciroli, A. Minghetti, F.F. Vincieri, B. Danieli, J. Nat. Prod. 59, 773–776 (1996)
F. Jeske, J. Jakupovic, W. Berendsohn, Phytochemistry 40, 1743–1750 (1995)
Q.L. Liang, C.C. Dai, J.H. Jiang, Y.P. Tang, J.A. Duan, Fitoterapia 80, 514–516 (2009)
F.G. Shao, R. Bu, C. Zhang, C.J. Chen, J. Huang, J.H. Wang, J. Asian Nat. Prod. Res. 13, 805–810 (2011)
W.W. Tao, J.A. Duan, Y.P. Tang, N.Y. Yang, J.P. Li, Y.F. Qian, Phytochemistry 94, 249–253 (2013)
R.M. Dawson, P.R. Jefferies, J.R. Knox, Phytochemistry 14, 2593–2597 (1975)
C. Valente, M. Pedro, J.R. Ascenso, P.M. Abreu, M.S. Nascimento, M.J.U. Ferreira, Planta Med. 70, 244–249 (2004)
H. Wang, X.F. Zhang, Y. Zhou, S.L. Peng, D.W. Zhou, L.S. Ding, Fitoterapia 79, 262–266 (2008)
S.H. Luo, Q. Luo, X.M. Niu, M.J. Xie, X. Zhao, B. Schneider, J. Gershenzon, S.H. Li, Angew. Chem. Int. Ed. 49, 4471–4475 (2010)
S.H. Luo, L.H. Weng, M.J. Xie, X.N. Li, J. Hua, X. Zhao, S.H. Li, Org. Lett. 13, 1864–1867 (2011)
Y.L. Li, P.P. But, V.E. Ooi, Antivir. Res. 68, 1–9 (2005)
Y.L. Li, L.S. Ooi, H. Wang, P.P. But, V.E. Ooi, Phytother. Res. 18, 718–722 (2004)
A. Kujumgiev, I. Tsvetkova, Y. Serkedjieva, V. Bankova, R. Christov, S. Popov, J. Ethnopharmacol. 64, 235–240 (1999)
Acknowledgments
This research was supported financially by the National Natural Science Foundation of China (31200263), the Youth Innovation Promotion association of Chinese Academy of Sciences (awarded to Shi-Hong Luo), the “Western Light” Program of Chinese Academy of Sciences (awarded to Shi-Hong Luo), and the “Hundred Talents Program” of Chinese Academy of Sciences (awarded to Sheng-Hong Li).
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Huang, CS., Luo, SH., Li, YL. et al. Antifeedant and Antiviral Diterpenoids from the Fresh Roots of Euphorbia jolkinii. Nat. Prod. Bioprospect. 4, 91–100 (2014). https://doi.org/10.1007/s13659-014-0009-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-014-0009-3