
Abstract
Male honey bees trapped at Drone Congregation Areas (DCAs) can be used to infer the number of colonies from which drones were derived, and thereby colony density in the environment. Crucial to the accuracy of this method is precise grouping of males into brother groups based on genetic markers, and a sample size that is sufficient so that all colonies in the area are included in the sample. The optimal sample size is a trade-off between cost and accuracy and cannot be known prior to sampling. Therefore, follow-up surveys may be necessary if the data indicate that the first sample was too small. However, the effect of multiple sampling on allele frequency estimates and the accuracy of the method is poorly understood. Here we trapped drones from two independent DCAs every month over 2 and 2.5 years. We analysed our data using the sibship grouping programme COLONY in three ways: (i) using data from the entire year, and counting the number of colonies identified in each month; (ii) using monthly data with allele frequencies from the entire season; and (iii) using data from each month separately. Although there were significant changes in allele frequencies over the year, these changes had no material effect on classifications of drones into families. Therefore, multiple samples can provide more robust estimates of family groupings due to the larger sample size and can be used with confidence where required.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
It is sometimes necessary or desirable to obtain estimates of the density of honey bee colonies in an area, either as an instantaneous estimate, or across time (Utaipanon et al. 2019b). Circumstances in which such information may be needed include planning a biosecurity response to an incursion of an exotic pest or disease, assessing the impacts of feral honey bees on native flora and fauna, quantifying any declines in honey bee abundance over time, or determining whether there are sufficient colonies present in an agricultural area to provide an adequate pollination service (Breeze et al. 2011; Potts et al. 2016; Nolan and Delaplane 2017; Utaipanon et al. 2019b).
Assessing the density of wild honey bee colonies by direct surveys is almost impossible. First, the density can be as high as 150 colonies/km2, with a non-uniform distribution of nests that is influenced by hollow availability and the distribution of food resources (McNally and Schneider 1996; Oldroyd et al. 1997). Second, honey bee colonies mostly build their nests in lofty locations in trees (Seeley and Morse 1976), human-built structures, or caves and rock faces (Oldroyd and Wongsiri 2006) where they can be difficult to spot. For the cavity-nesting species (Apis mellifera, A. cerana, A. koschevnikovi, A. nuluensis and A. nigrocincta), the nest entrance is often small and cryptic, making it even more difficult to identify nests by direct observation than it is with the open-nesting species (A. dorsata, A. laboriosa, A. florea and A. andreniformis).
Unlike most other insects, sampling foraging bees is an inappropriate method for estimating honey bee densities. Foragers recruit their nestmates to the best food sources (Visscher and Seeley 1982), and their foraging range can exceed 10 km (Eckert 1933; Visscher and Seeley 1982; Beekman and Ratnieks 2000; Steffan-Dewenter and Kuhn 2003). This means that the density of honey bees in an area can be drastically over- or under-estimated depending on when and where the physical sampling takes place (Utaipanon et al. 2019b).
An alternative to direct searches for colonies or foragers is to sample male bees at their mating aggregations (Moritz et al. 2008; Jaffé et al. 2010; Arundel et al. 2014; Hinson et al. 2015; Utaipanon et al. 2019b). Mature honey bee drones go on mating flights each afternoon, weather permitting. The exact timing of these flights depends on the species, the season and the region (Koeniger and Koeniger 1991; Loper et al. 1992; Yoshida et al. 1994; Otis et al. 2000). Once they leave their nest, drones fly to mating leks called Drone Congregation Areas (DCAs) where they search for a virgin queen with which to copulate (Ruttner 1974; Ruttner 1976). Each species has its own preferences for the location of a DCA. The DCAs of the western honey bee, A. mellifera, tend to be in an open space surrounded by tree lines such that there is a reduced horizon. (Suburban sports fields are often sites for DCAs.) These characteristics of DCAs allow human investigators to locate likely sites, often by using satellite maps such as Google Earth (Mortensen and Ellis 2014).
Once a DCA has been located, drones can be lured into a Williams drone trap (Williams 1987; Moritz et al. 2007; Hinson et al. 2015; Utaipanon et al. 2019a). The Williams trap is a tapered cylinder about 1 m long and 500 mm in diameter at the base, made up of three metal loops covered by thin insect netting (Scheiner et al. 2013). The trap utilizes dummy queens as lures to attract males. The lures are blackened cigarette filters infused with synthetic 9-oxo-decanoic acid (9-ODA) (Williams 1987), a major component of the queen’s sex attractant, queen mandibular pheromone (Butler et al. 1962; Brockmann et al. 2006). The lures are hung both inside and slightly beneath the trap where they serve as both visual and chemical cues for males. The trap is normally lifted by a Helium-filled weather balloon about 10–30 m above the ground (Dietemann et al. 2013; Mortensen and Ellis 2014). Drones are attracted by the movements of the queen lures. Once a drone approaches the lure, he tends to fly upward and gets caught inside the net. During good weather conditions, it is easy to collect several hundred males in 30 min.
Males are assumed to be drawn from a circular area surrounding the DCA (Utaipanon et al. 2019a). Microsatellite genotypes of trapped drones can be used to group brother drones into families and to infer the number of families (colonies) in the trapping area that contributed drones to the sample (Kraus et al. 2005b; Moritz et al. 2007; Jaffé et al. 2010; Arundel et al. 2013; Moritz et al. 2013; Arundel et al. 2014; Utaipanon et al. 2019a). Honey bee males are haploid, greatly facilitating the assignment of males to individual families using maximum likelihood. This calculation approach is implemented in COLONY (Wang 2005), along with algorithms for minimizing the effects of genotyping and miscellaneous errors (Wang 2004; Wang 2013). The number of colonies inferred using this technique has acceptable accuracy provided that the sample size is adequate (Utaipanon et al. 2021). After the number of colonies present in a sample has been determined, this count can be converted into a colony density estimate based on the assumption that drones fly a maximum of 3.75 km from their colony to a DCA (Utaipanon et al. 2019a). Therefore, a sample of drones obtained at a DCA samples the colonies in an area of 44 km2.
One of the limitations of the drone-sampling technique arises as a consequence of honey bee reproductive biology; colonies do not produce drones all year round (Allen 1958; Allen 1963; Free and Williams 1975; Seeley and Mikheyev 2003). Generally, mature drones are abundant during the period from spring to autumn, but largely absent from late autumn to early spring when they are rejected from their colony and die (Free and Williams 1975; Currie and Jay 1988). In addition, even during the breeding season, drone flight activity is strongly affected by temperature and wind speed (Reyes et al. 2019). Hence, the sampling period can be critical to the quality of the sample in terms of its size and the proportion of colonies that are successfully sampled. Further, the larger the sample size, the more accurate the grouping of drones into families will be. Larger sample sizes also minimize non-sampling error—the failure to sample drones from a colony within the DCA’s catchment. Utaipanon et al. (2021) showed that provided the average number of sampled drones per family is equal or greater than six, the number of colonies represented in a sample is estimated with less than 10% error from all sources: non- sampling and misclassification.
COLONY utilizes population allele frequencies to obtain the most likely grouping of genotypes into families. In practice, allele frequencies are calculated directly from the genotyping data using a group-likelihood approach. This approach helps correct the allele frequencies for the unequal numbers of offspring from each family (Wang 2004). However, it is also possible to use independent estimates of allele frequency obtained by other means—for example as a result of repeated sampling at the same site. In theory, the better the allele frequency estimate, the more accurate the inference of family membership. However, there is a caveat to this generality because allele frequency is likely to fluctuate somewhat over time (Kraus et al. 2005a; Jaffé et al. 2009). Thus, using repeated samples from the same site may introduce its own set of problems.
In this study, we quantify the significance of sampling at different times of year on the number of inferred colonies at the same sites. To do this, we repeatedly collected drones at two highly populous DCAs every month during the drone-flight season for 2–2.5 years using a Williams drone trap (Williams 1987). We then analysed drone genotypes by year and by month for each DCA to infer the number of colonies represented at the DCA. Based on these analyses, we explore the effects of sample size and time of sampling on the estimate of the number of colonies present in the area. We also calculate independent estimates of allele frequency based on various groups of samples, using the estimates generated by COLONY itself during its calculations. We then explore the effects of using the different estimates of allele frequency on the reconstructed queen populations. Finally, we make recommendations about handling data sets that have been collected over time, or at different times of the year.
2 Methods
2.1 Sample collection
We sampled two DCAs in Sydney Australia. The first site was number 1 oval at the University of Sydney’s (USYD) Camperdown campus (33° 53′ 16.1″ S, 151° 11′ 6.1″ E). This site is on the fringe of the CBD, and the DCA has been established since before 1980 (personal observations of BPO). The second site was Barra Brui oval (BB), St. Ives, NSW, Australia (33° 44′ 36.3″ S, 151° 10′ 3.8″ E) on the north east urban fringe of Sydney, which is surrounded by remnant bushland. Each site is a large sports field surrounded by trees. The two sites are 16 km apart, much further than the typical drone flight distance (Utaipanon et al. 2019a).
Drones were caught every month using Williams drone traps except during winter when we confirmed that no drones were present. Drones were preserved in 95% ethanol once trapped. If the weather on the sampling day was not suitable for drone flight, i.e. cloudy or windy, or if ‘comets’ of drones (i.e. 10–100 drones chasing each other) were not present during the sampling, we resampled the site during the following week. At the USYD site, we sampled from September 2017 to April 2019. At the BB site, we sampled from January 2018 to April 2020 (Table I).
2.2 Microsatellite genotyping
DNA of sampled drones was extracted from one hind leg using the Chelex protocol (Walsh et al. 1991). Extracted DNA was then used as template for nine pairs of microsatellite primers: A8, A24, A29, A79, A88, A107, A113, B124 and HbThe3 (Estoup et al. 1994; Solignac et al. 2003). These loci are unlinked either to each other or to centromeres. Reactions were performed in two multiplexed PCRs using fluorescently labelled primers. PCR products were electrophorized in a 3130xl Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, USA). The output from the analyzer was collated and corrected using GeneMapper v5 software (Applied Biosystems®), and final microsatellite genotypes were assembled manually. We ensured that we minimized the effects of genotyping errors and unequal information among datasets by removing any drone genotype in which more than one locus failed to amplify. Information on allelic diversity for each locus and population is given in Table S1. The genotype of each drone is given in Tables S2 and S3.
2.3 Inference of number of colonies
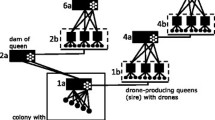
Drone genotypes were used to group individuals into families of brothers using COLONY (Wang 2004; Yang et al. 2013). To explore the effects of sample size and the method used to estimate allele frequency on sibship assignment, we analysed the entire data set from 2 years for USYD and 2 and a half years for BB in three alternative ways to determine which is best (Figure 1).

Diagram of the inference methods used in this study.
First, drones were grouped together based on the pooled sample for each site-year combination, that is, the combined September-April samples in each of the 2 years were treated as a single sample. Then, the number of colonies present in each month was determined by assigning each individual drone to the month in which it was caught (method (i) in Figure 1).
Second, we explored the effects of the alternative methods of estimating population allele frequencies (method (ii) in Figure 1). Here we used COLONY to calculate the number of families present in each monthly sample, but used the allele frequencies calculated by COLONY from the entire year’s data, rather than just the month in question.
Finally, we analysed each month’s genotype data separately on a monthly basis. From these monthly data inputs, we obtained numbers of colonies per month using allele frequencies estimated by COLONY for each individual month by default (method (iii) in Figure 1). This kind of analysis (a single sample from a single site) is what is typically done in practice (Moritz et al. 2007; Utaipanon et al. 2019a).
Data were analysed using COLONY 2.0.6.5 with the following settings: long length runs, haplodiploid mating system, 3 replicate runs, updated allele frequency and full likelihood. We ran the analysis twice: once with sibship scaling and once without sibship scaling.
2.4 Data analysis
We compared the number of families estimated with and without sibship scaling and found that the numbers of families inferred from the second-year data at USYD and first-year data at BB changed by less than 2.6%, while the other two samples returned similar numbers of colonies. ‘Scaling-On’ is commonly applied when analysing data with COLONY because it is thought to provide the greater precision (Wang 2013; Utaipanon et al. 2021). We therefore report our results below with the sibship scaling setting ‘on’.
We used Wilcoxon paired signed-rank tests to determine whether there were significant differences in the number of families inferred from each month using the three alternative sampling schemes. In addition, we evaluated the adequacy of sample size for each method using the criteria that the average of numbers of drones (Di) per number of inferred families (NC) should be equal to or greater than 6 (Utaipanon et al. 2021). Finally, we used a Monte Carlo version of Fischer’s exact test to perform pairwise comparisons of allele frequencies of DCA populations across months. The tests were performed in R 3.6.2 (R Core Team 2016) using MASS (Venables and Ripley 2002) and stats packages (R Core Team 2016).
3 Results
We collected 5074 drones at USYD over a 2-year period, and 4944 from BB over 2.5 years. Drone mating seasons ended and began at similar times at both locations. Generally, drones visited DCAs from the beginning of spring (September–October) until mid-autumn (March–April) each year. The number of drones caught each month was highly variable—a minimum of 3 drones to a maximum of 574 drones. This was despite the fact that we repeated our sampling whenever the number of caught drones was < c.a. 300 within 1 week for each month (Table I). This suggests that the population of mature drones present in the area was very low during those months when we failed to catch a lot of drones, and that our low sample size during these months was not a consequence of abiotic factors, but an actual absence of drones.
The estimated number of colonies that contributed drones to the trap varied according to the season and the data set used in the analysis (Table I). When we used the annual dataset of drone genotypes (method (i) in Figure 1), there were 243 and 246 unique colonies found at USYD in the first (2017–2018) and second (2018–2019) years respectively. At BB, we inferred 101 colonies from the January–April 2018 samples, and 274 (September 2018–April 2019) and 257 (September 2019–April 2020) colonies from the full-year samples.
The number of inferred colonies present in each month’s samples based on the data from individual months (method (iii), Figure 1) was significantly smaller than those inferred using annual data sets (V=0, df= 34, p<0.001, Table II). In addition, the average number of drones per inferred family (Di:NC ) (Utaipanon et al. 2021) in each month based on the monthly data was relatively low (Table II), whereas the Di:NC ratios were above six for all analyses based on yearly data. When allele frequencies based on the entire year’s data were used for the analysis of the monthly data (methods ii, Figure 1), the number of inferred colonies per month remained similar to those inferred based on monthly allele frequencies alone (methods iii, Figure 1) (Table III).
Allele frequencies of sampled drones at each DCA varied significantly between months within seasons (Fischer’s exact test, p<0.001 (111 pairs)), though most likely this was due to lower sample size per month rather than actual changes in allele frequency.
3.1 Estimates of non-sampling error
The number of non-sampled colonies can be estimated by fitting the distribution of family sizes to a truncated Poisson distribution (Baudry et al. 1998; Utaipanon et al. 2019b). We can calculate number of total colonies including the missing colonies (NT) from:
where \(\hat{\lambda}\) is the maximum likely estimate of the expected number of colonies per family (i.e. the observed mean number of drones per family), and Nd is the total number of sampled drones (Baudry et al. 1998; Utaipanon et al. 2019b). Based on this calculation, there were only 0 or 1 non-sampled colonies per month when the monthly data were used (Table I and Table S4). We report the number of colonies following the Poisson correction (NT) (Table I). However, since the distribution of family sizes was a poor fit to the Poisson distribution in all cases (P < 0.001, see Table S4), the reliability of this correction is unclear.
4 Discussion
A risk of using drone samples that have been collected over a period of time is that allele frequencies in the drone population in the sampled area will have changed over time (Kraus et al. 2005a; Chapman et al. 2019). Variance in allele frequency over time could potentially lead to inaccurate family reconstruction (Kraus et al. 2005a). Here we have shown that the benefits to be gained from high sample size as a result of repeated samplings outweighs the possible drawback of temporal fluctuations in allele frequencies arising from real changes in DCA population structure over time. (If allele frequency fluctuations derive from low sample size, pooling is always advantageous.) Our results show that the number of families inferred using monthly data sets alone was lower than when the number of families was inferred from longer-term studies. Only when we used multiple samples did the average number of drones per inferred family (Di:NC) exceed 6, indicating that sample sizes had been adequate (Table II) (Utaipanon et al. 2021). We therefore conclude that the higher number of families detected per month using the annual data set was a result of an improvement in inference due to higher sample size and reduced non-sampling error (NSE) (Baudry et al. 1998; Utaipanon et al. 2019b). NSE arises when drones from a particular colony are visiting a sampled DCA, but by chance were not sampled on the sampling day. We know that non-sampling error was a real problem, since some colonies were identified months apart, without being seen in the intermediate months (data not shown). This emphasises that repeated samples at the same site improve the estimate of colony density, by increasing the sample size. Our analysis also suggests that fitting the distribution of family sizes to the Poisson distribution is unlikely to solve the problem of NSE if the sample size has been inadequate.
Small sample size can mean that some families are represented by just a few males, which can be erroneously allocated by COLONY into existing families with which they share alleles, especially when families are highly related, or the available genetic information is limited. This problem can be ameliorated by increasing the sample size. Alternatively, the precision of allocation of individuals to family groups can be improved by using more microsatellite or SNP markers.
Allele frequencies estimated from a DCA population can fluctuate significantly over a period of just 2–3 days (Moritz et al. 2007; Collet et al. 2009). Whether these differences are attributable to sampling error or real changes in population allele frequencies is an open question. We detected significant differences in allele frequency over the seasons evaluated. However, our data show that when allele frequencies estimated from the annual drone population are used a priori for the analysis of monthly data (method (ii) in Figure 1), the number of inferred families remained similar to those based on allele frequencies estimated from each month’s data (method (iii) in Figure 1 ) (Table III). This indicates that fluctuations in allele frequencies over time have no material impact on the numbers of colonies inferred. Thus, if repeated sampling is required because the sample size is low, data from the repeated samples can be pooled with confidence. In this case, the research question should be taken into an account to assess whether repeated sampling is warranted, and how long the study period should be.
Our study shows that drone sampling should not be conducted until at least 1 month after drones first appear at a DCA after winter, and at least 1 month before drones cease flying in winter. Although the number of families found in each month was variable (Table I), comets of drones were present from October to March at both locations in each year, whereas the presence of comets was uneven in September and April. Therefore, the time of the year that sampling is conducted is likely to have a material influence on the accuracy of the estimates of colony density that are obtained.
5 Conclusions
In an environment where the density of colonies is unknown, an a priori goal for sample size is unknown, as it depends on the number of colonies present in the environment (Utaipanon et al. 2021). (Of course, sample size should always be as large as is practicable, within the constraints of a field trip.) Where the actual colony density is very low, small sample sizes (c.a. 200 drones) are likely to be adequate. If the density of colonies is high, higher sample sizes are necessary (> 3000 drones) (Utaipanon et al. 2021). Our study suggests that there is no harm in collecting drones over a period of a week or even months; allele frequencies, real or due to sampling error, do not fluctuate sufficiently to be cause for concern. This property of drone populations and maximum likelihood estimates of allele frequencies means that researchers can analyse an initial sample and return to the field to collect additional samples if the average number of drones per family turns out to be inadequate. Such a protocol will maximise the efficiency of the method. It is also possible to fit the data to a truncated Poisson distribution to estimate the number of non-sampled colonies (Baudry et al. 1998), but because the distribution of family size is often a poor fit to the Poisson distribution, we suggest that additional samples should be obtained wherever possible.
References
Allen M.D. (1958) Drone brood in honey bee colonies. J. Econ. Entomol. 51(1): 46-48.
Allen M.D. (1963) Drone production in honey-bee colonies (Apis mellifera L.). Nature 199(4895): 789-790.
Arundel J., B.P. Oldroyd, S. Winter. (2013) Modelling estimates of honey bee (Apis spp.) colony density from drones. Ecol. Model. 267: 1-10.
Arundel J., P.R. Oxley, A. Faiz, J. Crawford, S. Winter, et al. (2014) Remarkable uniformity in the densities of feral honey bee Apis mellifera Linnaeus, 1758 (Hymenoptera: Apidae) colonies in South Eastern Australia. Aust. Entomol. 53(3): 328-336.
Baudry E., M. Solignac, L. Garnery, M. Gries, J.M. Cornuet, et al. (1998) Relatedness among honeybees (Apis mellifera) of a drone congregation. Proc. R. Soc. B Biol. Sci. 265(1409): 2009-2014.
Beekman M., F.L.W. Ratnieks. (2000) Long-range foraging by the honey-bee, Apis mellifera L. Funct. Ecol. 14(4): 490-496.
Breeze T.D., A.P. Bailey, K.G. Balcombe, S.G. Potts. (2011) Pollination services in the UK: how important are honeybees? Agric. Ecosyst. Environ. 142(3): 137-143.
Brockmann A., D. Dietz, J. Spaethe, J. Tautz. (2006) Beyond 9-ODA: sex pheromone communication in the european honey bee Apis mellifera L. J. Chem. Ecol. 32(3): 657-667.
Butler C.G., R.K. Callow, N.C. Johnston. (1962) The isolation and synthesis of queen substance, 9-oxodec-trans-2-enoic acid, a honeybee pheromone. Proc. R. Soc. B Biol. Sci. 155(960): 417-432.
Chapman N.C., R. Dos Santos Cocenza, B. Blanchard, L.M. Nguyen, J. Lim, et al. (2019) Genetic diversity in the progeny of commercial Australian queen honey bees (Hymenoptera: Apidae) produced in autumn and early spring. J. Econ. Entomol. 112(1): 33-39.
Collet T., A.S. Cristino, C.F.P. Quiroga, A.E.E. Soares, M.A. Del Lama. (2009) Genetic structure of drone congregation areas of Africanized honeybees in southern Brazil. Genet. Mol. Biol. 32(4): 857-863.
Currie R.W., S.C. Jay. (1988) The influence of a colony's queen state, time of year, and drifting behaviour, on the acceptance and longevity of adult drone honeybees (Apis mellifera L.). J. Apic. Res. 27(4): 219-226.
Dietemann V., J. D Ellis, P. Neumann. (2013) The COLOSS BEEBOOK Volume I, standard methods for Apis mellifera research: introduction.
Eckert J.E. (1933) The flight range if the honeybee. J. Agric. Res. 47(5): 257-285.
Estoup A., M. Solignac, J.-M. Cornuet. (1994) Precise assessment of the number of patrilines and of genetic relatedness in honeybee colonies. Proc. R. Soc. B Biol. Sci. 258(1351): 1-7.
Free J.B., I.H. Williams. (1975) Factors determining the rearing and rejection of drones by the honeybee colony. Anim. Behav. 23(3): 650-675.
Hinson E.M., M. Duncan, J. Lim, J. Arundel, B.P. Oldroyd. (2015) The density of feral honey bee (Apis mellifera) colonies in South East Australia is greater in undisturbed than in disturbed habitats. Apidologie 46(3): 403-413.
Jaffé R., V. Dietemann, R. Crewe, R. Moritz. (2009) Temporal variation in the genetic structure of a drone congregation area: an insight into the population dynamics of wild African honeybees (Apis mellifera scutellata). Mol. Ecol. 18(7): 1511-1522.
Jaffé R., V. Dietemann, M.H. Allsopp, C. Costa, R.M. Crewe, et al. (2010) Estimating the density of honeybee colonies across their natural range to fill the gap in pollinator decline censuses. Conserv. Biol. 24(2): 583-593.
Koeniger N., G. Koeniger. (1991) An evolutionary approach to mating behaviour and drone copulatory organs in Apis. Apidologie 22(6): 581-590.
Kraus F.B., N. Koeniger, S. Tingek, R.F.A. Moritz. (2005a) Temporal genetic structure of a drone congregation area of the giant Asian honeybee (Apis dorsata). Naturwissenschaften 92(12): 578-581.
Kraus F.B., N. Koeniger, S. Tingek, R.F.A. Moritz. (2005b) Using drones for estimating colony number by microsatellite DNA analyses of haploid males in Apis. Apidologie 36(2): 223-229.
Loper G.M., W.W. Wolf, O.R. Taylor. (1992) Honey bee drone flyways and congregation areas: radar observations. J. Kansas Entomol. Soc. 65(3): 223-230.
McNally L.C., S.S. Schneider. (1996) Spatial distribution and nesting biology of colonies of the african honey bee Apis mellifera scutellata (Hymenoptera: Apidae) in Botswana, Africa. Environ. Entomol. 25(3): 643-652.
Moritz R.F.A., F.B. Kraus, P. Kryger, R.M. Crewe. (2007) The size of wild honeybee populations (Apis mellifera) and its implications for the conservation of honeybees. J. Insect Conserv. 11(4): 391-397.
Moritz R.F.A., V. Dietemann, R. Crewe. (2008) Determining colony densities in wild honeybee populations (Apis mellifera) with linked microsatellite DNA markers. J. Insect Conserv. 12(5): 455-459.
Moritz R.F.A., F.B. Kraus, A. Huth-Schwarz, S. Wolf, C.A.C. Carrillo, et al. (2013) Number of honeybee colonies in areas with high and low beekeeping activity in Southern Mexico. Apidologie 44(1): 113-120.
Mortensen A.N., J.D. Ellis. (2014) Scientific note on a single-user method for identifying drone congregation areas. J. Apic. Res. 53(4): 424-425.
Nolan M., K. Delaplane. (2017) Distance between honey bee Apis mellifera colonies regulates populations of Varroa destructor at a landscape scale. Apidologie 48(1): 8-16.
Oldroyd B.P., S. Wongsiri. (2006) Asian honey bees: biology, conservation, and human interactions. Harvard University Press, Cambridge.
Oldroyd B.P., E.G. Thexton, S.H. Lawler, R.H. Crozier. (1997) Population demography of Australian feral bees (Apis mellifera). Oecologia 111(3): 381-387.
Otis G., N. Koeniger, T. Rinderer, S. Hadisoesilo, T. Yoshida, et al. (2000) Comparative mating flight times of Asian honey bees., 7th International conference on tropical bees: management and diversity and 5th Asian apicultural association conference, IBRA, Chaingmai, Thailand, pp. 137-141.
Potts S., V.L. Imperatriz-Fonseca, H.T. Ngo, J. Biesmeijer, T. Breeze, et al. (2016) Summary for policymakers of the assessment report of the intergovernmental science-policy platform on biodiversity and ecosystem services on pollinators, pollination and food production. IPBES: 1-36.
R Core Team. (2016) R: A language and environment for statistical computing, Vienna, Austria.
Reyes M., D. Crauser, A. Prado, Y. Le Conte. (2019) Flight activity of honey bee (Apis mellifera) drones. Apidologie 50(5): 669-680.
Ruttner H. (1974) Drohnensammelplätze ein beispiel von paarungsverhalten bei Insekten. Anz. Schälingskde. Pflanzen-Umweltschutz 47: 39-42.
Ruttner H. (1976) Untersuchungen über die flugaktivität und das paarungsverhalten der drohnen. VI.—Flug auf und über höhenrücken. Apidologie 7(4): 331-341.
Scheiner R., C.I. Abramson, R. Brodschneider, K. Crailsheim, W.M. Farina, et al. (2013) Standard methods for behavioural studies of Apis mellifera. J. Apic. Res. 52(4): 1-58.
Seeley T.D., A.S. Mikheyev. (2003) Reproductive decisions by honey bee colonies: tuning investment in male production in relation to success in energy acquisition. Insect. Soc. 50(2): 134-138.
Seeley T.D., R.A. Morse. (1976) The nest of the honey bee (Apis mellifera L.). Insect. Soc. 23(4): 495-512.
Solignac M., D. Vautrin, A. Loiseau, F. Mougel, E. Baudry, et al. (2003) Five hundred and fifty microsatellite markers for the study of the honeybee (Apis mellifera L.) genome. Mol. Ecol. Notes 3(2): 307-311.
Steffan-Dewenter I., A. Kuhn. (2003) Honeybee foraging in differentially structured landscapes. Proc. R. Soc. B Biol. Sci. 270(1515): 569-575.
Utaipanon P., M.J. Holmes, N.C. Chapman, B.P. Oldroyd. (2019a) Estimating the density of honey bee (Apis mellifera) colonies using trapped drones: area sampled and drone mating flight distance. Apidologie 50: 578–592.
Utaipanon P., T.M. Schaerf, B.P. Oldroyd. (2019b) Assessing the density of honey bee colonies at landscape scales. Ecol. Entomol. 44: 291–304.
Utaipanon P., T.M. Schaerf, M.J.C. Holmes, N. Chapman, B.P. Oldroyd. (2021) Using trapped drones to assess the density of honey bee colonies — a simulation and empirical study to evaluate the accuracy of the method. Ecol. Entomol. 46:128–137.
Venables W.N., B.D. Ripley. (2002) Modern applied statistics with S, Springer, New York.
Visscher P.K., T.D. Seeley. (1982) Foraging strategy of honeybee colonies in a temperate deciduous forest. Ecology 63(6): 1790-1801.
Walsh P.S., D.A. Metzger, R. Higuchi. (1991) Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. BioTechniques 10(4): 506-513.
Wang J. (2004) Sibship reconstruction from genetic data with typing errors. Genetics 166(4): 1963-1979.
Wang J. (2005) Estimation of effective population sizes from data on genetic markers. Philos. Trans. R. Soc. B 360(1459): 1395-1409.
Wang J. (2013) An improvement on the maximum likelihood reconstruction of pedigrees from marker data. Heredity 111(2): 165.
Williams J.L. (1987) Wind-directed pheromone trap for drone honey-bees (Hymenoptera, Apidae). J. Econ. Entomol. 80(2): 532-536.
Yang W., H. Kuang, S. Wang, J. Wang, W. Liu, et al. (2013) Comparative sucrose responsiveness in Apis mellifera and A. cerana foragers. PLoS ONE 8(10): e79026.
Yoshida T., J. Saito, N. Kajigaya. (1994) The mating flight times of native Apis cerana japonica Radoszkowski and introduced Apis mellifera L. in sympatric conditions. Apidologie 25(4): 353-360.
Acknowledgements
This project was supported by AgriFutures Australia, through funding from the Australian Government, Department of Agriculture, Water and the Environment as part of its Rural R&D for Profit programme.
Data and materials availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Code availability
Not applicable
Funding
This project was funded by AgriFutures Australia, through funding from the Australian Government, Department of Agriculture, Water and the Environment as part of its Rural R&D for Profit programme.
Author information
Authors and Affiliations
Contributions
BPO and PU designed an experiment and wrote the manuscript. PU and MH conducted fieldworks. PU, MH and GB conducted laboratory works. PU analysed the data. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
Conflict of interest
The authors declare no competing interests.
Additional information
Handling Editor: Klaus Hartfelder
Séparer ou combiner? Effets de l'échantillonnage répété et de la mise en commun des données sur l'estimation du nombre de colonies à partir du génotypage des mâles.
Echantillonnage écologique / insecte social / densité de la colonie / microsatellite / estimation de la taille de la population.
Trennen oder kombinieren? Auswirkungen wiederholter Probennahme und dem Poolen von Daten auf die Abschätzung von Völkerzahlen anhand der Genotypierung von Drohnen.
Ökologische Probennahme / soziales Insekt / Völkerdichte / Mikrosatelliten / Abschätzung der Populationsgrösse.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Table S1.
Haplotype diversity and numbers of alleles of each microsatellite marker for the drone populations at USYD and BB (XLSX 12 kb)
Table S2.
Genotypes of drones sampled at USYD during 2017-2019. Table S3. Genotypes of drones sampled at BB during 2018-2020 (XLSX 535 kb)
Table S4.
Maximum likelihood of mean family size, \(\hat{\lambda}\), and number of colonies (NC) and total number of colonies including missing colonies (NT) in DCA populations each month base on the assumption of Poisson distribution (XLSX 13 kb)
Rights and permissions
About this article

Cite this article
Utaipanon, P., Holmes, M.J., Buchmann, G. et al. Split or combine? Effects of repeated sampling and data pooling on the estimation of colony numbers obtained from drone genotyping. Apidologie 52, 620–631 (2021). https://doi.org/10.1007/s13592-021-00848-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-021-00848-8