
Abstract
Although the mitochondrial molecule of the honey bee is completely sequenced, the control region is rarely used for genetic inferences as in other invertebrates, due to several constraints mainly a biased A plus T content and extensive variable length repeats. Herein, we have analyzed the control region of honey bees from the Iberian Peninsula and North Atlantic islands. The information retrieved when comparing individuals from different populations was crucial to understand and characterize how the control region is organized in this species. As expected, this region in Apis mellifera appears to contain valuable although limited genetic information at the population level. Furthermore, the comparisons of the A. mellifera control region with other species of the same genus highlight the structural role of particular sequences within the A+T rich control region as proposed here.
Similar content being viewed by others

1 Introduction
The mitochondrial molecule of the honeybee, Apis mellifera L., was fully sequenced in 1993 by Crozier and Crozier. Since then, several regions from the mitochondrial molecule have been used for phylogeography and molecular diversity studies of A. mellifera subspecies and populations, particularly the tRNAleu-cox2 intergenic region (Evans et al. 2013; Meixner et al. 2013). This intergenic region shows length and sequence variation allowing a grouping of subspecies largely congruent with the morphometrical classification that subdivide the variation of A. mellifera into four different evolutionary lineages: West (M) and North Mediterranean (C) lineages; an African lineage (A) which groups the African honey bee subspecies; and the Oriental lineage (O) located geographically in the Middle East (Ruttner 1988). Several further divisions have been recognized within the aforementioned lineages via sequencing of the intergenic region (Franck et al. 2001; De la Rúa et al. 2001; Pinto et al. 2012). Based on these molecular markers, Iberian honey bee populations have been recognized as part of a cline from North Africa to Europe (Cánovas et al. 2008; Pinto et al. 2013), while honey bee populations from the Atlantic coast including the Macaronesian region have been distinguished as an African sublineage with Atlantic distribution (De la Rúa et al. 2006, 2009; Muñoz et al. 2013, Murray et al. 2009).
For most organisms, the main noncoding region of the mitochondrial genome is the control region (CR). It is widely used for genetic inferences especially in mammals where it is also known as the D-loop (for displacement-loop), a three-stranded structure that forms during replication of the molecule. At least in vertebrates, functional structures within this region have been identified across a wide number of species namely highly conserved sequences associated with the molecule replication, conserved and variable blocks, as well as palindromic and stable cloverleaf structures that may be involved in the replication of the molecule as well as transcription initiation sites for each strand (Chang and Clayton 1984; Saccone et al. 1991; Clayton 1982, 1996). In invertebrates, the control region is much less studied and understood. Its use as a genetic marker is very scarce and there are good reasons for this already pointed out by Zhang et al. (1995) and Zhang and Hewitt (1997). Particularly in insects, the control region is known as A+T rich region due to its high content in adenine and thymine nucleotides that can make up to 96 % of the base content as is the case of Apis and Drosophila. Most of this content is due to length polymorphisms, tandem repetitions of (T)n(A)n motifs, stretches of Ts and As and innumerous insertion/deletions of nucleotides. The overall sequence similarity of the A+T rich region, even among closely related sequences, is problematic and its use to infer evolutionary relationships is limited (Brehm et al. 2003; Vila and Björklund 2004; Kim et al. 2007; Wan et al. 2011a, b). Attempts to use the A+T rich region in insects have been focused on its structure and evolution with particular emphasis on functional conserved structures and putative regulatory sequences rather than intraspecific comparisons (Simon et al. 1994; Brehm et al. 2001; Sugihara et al. 2006; Kim et al. 2007; Zhang et al. 2013). Zhang and Hewitt (1997) divided the control region of insects into two groups according to their structural conservation. One of the groups included Drosophila with conserved and variable domains, and a second group, where such division is much less perceivable, was formed by grasshoppers, mosquitoes, and butterflies (Zhang and Hewitt 1997; Kim et al. 2007). Alignment of sequences could be possible only for those conserved blocks that kept some degree of homology, since the lack of variability in the conserved blocks is also a handicap for lower taxa discrimination. On the other hand, the high mutation rate that characterizes the variable blocks is certainly the cause for the high number of parallel or recurrent mutations in different lineages producing poorly resolved phylogenetic networks or unsupported topologies. In relation to the genus Apis, only three complete mitochondrion genomes containing the control region (A. mellifera, Apis cerana, and Apis florea) exist in databases (Crozier and Crozier 1993; Tan et al. 2011; Wang et al. 2013), but further characterization of the sequence variation of the A+T rich region has not been performed.
The aim of the present work is to present a deeper analysis of the A+T rich region in A. mellifera by looking at sequences from individuals belonging to different populations of the Iberian Peninsula and Macaronesian islands. Such an analysis should provide us with an accurate picture of the structure of the A+T rich region and the way it is organized in terms of conserved/variable domains. As far as we know, this is the first time a comparison of sequences from the control region has been done in an otherwise well-studied species. We also intend to evaluate the usefulness of the A+T rich region as a genetic marker particularly to address intraspecific variation.
2 Material and methods
2.1 Samples
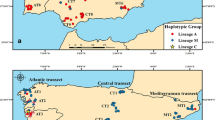
A. mellifera honey bee workers (n = 54) were taken from the inner frames of colonies (one worker per colony, from two to six colonies per locality) in three localities of the Iberian Peninsula [Cádiz (Spain), n = 4; Málaga (Spain), n = 4; Viseu (Portugal), n = 5], the Canary Islands [Tenerife, n = 5; La Palma, n = 5; La Gomera, n = 4; Gran Canaria, n = 5], Madeira (n = 7), Azores Islands (São Miguel, n = 5), Morocco [Tan Tan, n = 2; South Rabat, n = 2], and Cabo Verde archipelago (n = 6) and preserved in ethanol until analysis. All the samples were previously identified as belonging to the African evolutionary lineage through the determination of their tRNAleu-cox2 haplotype (De la Rúa et al. 2001, 2006, 2007, unp. results; Cánovas et al. 2008). We have not performed a morphometric analysis of these samples to determine their subspecies status but from the geographic location it can be inferred that Iberian samples corresponded to Apis mellifera iberiensis and Moroccan samples to Apis mellifera intermissa (South Rabat) and Apis mellifera sahariensis (Tan Tan). Island samples were included in the African evolutionary sublineage of Atlantic distribution.
2.2 DNA isolation and PCR amplification
Total DNA was extracted from thoraces and legs of single individuals of A. mellifera following the E. Z. N. A. Insect DNA Isolation kit protocol (Omega Bio-Tek). PCR amplification of the mitochondrial DNA (mtDNA) control region was performed in a 50 μL reaction containing 10 ng of DNA, 10 pmol of each primer [12SAIR: 5′-AGGGTATCTAATCCTAGTTT-3′ and METAPIS: 5′-GACAGGGTATGAACCTGTTAGCTT-3′], 50 mM MgCl2, 5 μL 10X PCR Buffer, 2 U Taq DNA Polymerase (both from Invitrogen Life Technologies), and 10 mM of each dNTP. After initial heating at 95 °C for 180 s, 35 cycles of PCR amplification were performed, consisting of three steps: denaturation at 94 °C for 60 s, annealing at 50 °C for 90 s, and extension at 72 °C for 120 s, and a final extension at 72 °C for 240 s was included. A single fragment of about 1.2 kb was obtained. Amplified products were visualized after electrophoretic separation on 1 % agarose gels, stained with ethidium bromide, and photographed under UV illumination. As expected, a slight size variation of these primary fragments occurred in different individuals so the amplification of each product was performed in triplicate for each specimen ensuring that each individual always yielded fragments of the same size.
2.3 Sequencing of mtDNA
Direct sequencing of 54 PCR products was performed in an ABI310 Genetic Analyzer (Applied Biosystems). Four internal primers were used [INTF1: 5′-ATCTTAAAAACTACAACATGA-3′, INT2F: 5′-CATTGTTTCAGAATTCTCT-3′, INT2R: 5′-TTATTATGCTTATTTATTC-3′, and INTR1: 5′-CGAATCTAGAGTTAAAGTTAGA-3′, see Figure 1]. All reactions were done in triplicate for each individual to ensure that no mistakes were done in the cycle process of PCR amplification and sequencing. Sequences were aligned using the program DNASTAR (Lasergene) to produce complete fragments from all partial sequences obtained using all of forward and reverse primer pairs.


The complete sequence of the control region from AZ04 and its main features mentioned in the text. The partial sequence of the 23S rRNA small subunit (green and underlined) and both transfer RNAs (tRNAglu and tRNAser, both colored and underlined) are also included and correspond to the sequences depicted in supplementary Tables S1 and S2. Length and position of three of the internal primers used are also depicted above the sequence. Boundaries of the control region are shown by dark square brackets. Variable blocks 1–4 are limited by arrows linked by solid lines. The sequence limited by arrows and a dashed line that includes the T-stretch refers to conserved block 1 (CB1) mentioned in the text. The sequence highlighted in pink is block B proposed by Zhang et al. (1995) discussed in the text. The putative STOP signal is underlined. The sequence wave-underlined corresponds to the long stem-loop structure discussed in the text and depicted in detail in Figure 2b.
2.4 Structural and phylogenetic sequence analysis
DNA sequences reported here are available on the European Nucleotide Archive at www.ebi.ac.uk/ena/ under accession numbers LN775355-LN775408. Multiple sequence alignments of the complete fragments were performed with CLUSTAL X (Thompson et al. 1997) implemented in MEGA 5 (Tamura et al. 2011) and maximized for sequence similarity by a careful visual inspection. Potential secondary structures of the A+T rich region were identified using the MFOLD program (Mathews et al. 1999; Zuker et al. 1999).
Genetic relationships of haplotypes were depicted using the software NETWORK v. 4.6.1.1 (http://www.fluxus-engineering.com/) using two strategies: firstly, we used all individuals to perform a network of haplotypes using the combined conserved blocks of the A+T rich region, and secondly, we performed the same analysis separately for each population using the whole region (conserved and variable blocks). To perform a network analysis with all the individuals, we followed the specifications of the program and applied the following constraints: transversions were weighted three times more than transitions, individual characters were weighted 10 (default value) except where runs of Ts or As (as the case of the T-stretch) produced individual variation as an insertion or deletion. In these cases, bases inserted or deleted in the runs were weighted zero. Other hypervariable characters characterized by an indel across individuals were weighted 5. The Sstar contraction algorithm (SC, Forster et al. 2001) was applied to get a network with the minimum possible complexity. After this, the median-joining (Bandelt et al. 1999) option was applied to clean up the contracted network. Finally, we ran the maximum parsimony option (Polzin and Daneschmand 2003) to further clear the topology from unnecessary median vectors and links. In the case of population-specific networks, we applied the MJ option alone.
Haplotype diversity and nucleotide composition of the whole A+T region and of conserved blocks combined were obtained using Arlequin V3.0 (Excoffier et al. 2005). Genetic distances among sequences were computed using the conserved blocks combined. To assess the population differentiation among populations, the corrected average pairwise differences, population pairwise Ф st (similar to F st) and analysis of molecular variance (AMOVA) were also computed using Arlequin for which the three populations from the Iberian Peninsula were considered as a group.
As supporting files, we provide the complete aligned sequences as a FASTA file (additional Table S1, remove the .txt terminus) as well as an Excel file with all variable positions including indels (additional Table S2).
3 Results
The complete sequences of the A+T rich region including flanking regions comprising a partial segment of a srRNA and two complete transfer RNAs (tRNAs) (tRNAglu and tRNAser) were obtained for 54 individuals. Identical sequences (in the triplicate repeats from each individual) from genes surrounding the control region were always obtained reducing the chance of being dealing with numts of this region.
3.1 Base composition and size
The average nucleotide composition of the A+T rich region was 45.9 % T, 3.0 % C, 50.2 % A, and 0.9 % G. Thus on average, an AT base composition of 96.1 % was found reflecting an extremely high bias towards A and T. The transition/transversion bias (R) is 3.85 under a Kimura (1980) two-parameter model. The size of the A+T rich region varied across the 54 individuals studied and ranged from 897 bp (TF56) to 984 bp (HP8). Most of the length variation observed was due to three separate regions encompassing tandem repetitions of short sequences as described below. Other small insertions/deletions also occurred along the A+T rich region and did not contribute significantly to the overall size variation of this region. The sequence and main features of a complete control region and its flanking regions are presented in Figure 1 (in this case, we chose AZ04 as the model to which all comparisons were done; see also additional complementary Tables S1 and S2).
3.2 Domains and tandem repetitions
To a large extent, size variation of the A+T rich region was due to three regions of different lengths containing a variable number of tandem repeat units. The first of these regions encompasses a variable stretch of mainly TAA tandem repeats which is located exactly at the beginning of the A+T rich region (variable block 1—VB1, Figure 1). This block can have 3–14 repeats of TAA and is interrupted at the beginning by a TATA motif which is common to all individuals analyzed. VB1 can thus vary from 16 to 49 nucleotides. A second block (variable block 2—VB2, Figure 1) lies within a region of TA repeats. Nevertheless, this TA region shows a high number of transversions of the type A↔T. VB2 is the longest region of the A+T rich region with variable number of repeats and mutated bases. The length of variable block 2 ranges from 118 nts (as in LP51, LP52) to 187 nts (as in HP8). This region is a type of [T(T)A(A)]n sequence which is also apparent in similar positions in other insects (Zhang and Hewitt 1997) though a definite proof of homology is still lacking. Variable block 3 (VB3) is situated towards the end of CR (variable block 3—VB3, Figure 1). It ranges from 98 nts (LP51 and LP52) to 146 nts (AZ2, 3, 4). This block includes a subregion, ranging from 34 to 36 bp with an extremely high content of adenines punctuated here and there by thymines. Three of the individuals analyzed (AZ2, 3, 4) show a partial repetition of this variable block (Figure 1, see Supplementary additional files). Finally, at the very end of the A+T rich region and close to the tRNAglu, lies VB4, a small variable region whose sequence varies from 9 to 12 nts.
The high conserved domain between VB1 and VB2, called here conserved block 1 or CB1, harbors a sequence which is a putative secondary structure forming a stem and loop described by Clary and Wolstenholme (1987) and Zhang et al. (1995) and is possibly implicated in the replication of circular DNA molecules. This sequence is read on the complementary strand (Clary and Wolstenholme 1987), and with a few nucleotide changes, it is the one highlighted in the pink box of Figure 1 and the grey box in Figure 2a. Interestingly, within this structure, there is an almost exact match of a stop signal described in humans and mice by Doda et al. (1981) which has the motif 5′-ACATTAAAYYAAT-3′ and is present on A. mellifera complementary strand as 5′-ATATTAAAATAAT-3′ (bold letters denote mutated nucleotides, see sequence underlined in CB1 of Figure 1 and also Figure 2a). This sequence, highlighted in pink in Figure 2, is conserved in all individuals analyzed but never forms a single stem and loop structure contrary to the Apis model proposed by Zhang et al. (1995, see their Figure 4c) unless at a high energetic cost. In Drosophila, it has been more or less established that a conserved stem and loop structure is a common feature to all species (Monforte et al. 1993; Zhang et al. 1995; Andrianov et al. 2010), and that it may be related to the replication of mtDNA particularly the site of initiation of the second strand synthesis. In A. mellifera, this region apparently exists but not as the stem and loop structure described for Drosophila. It bears other features that resemble signals for initiation of the L-strand replication. For example, the conserved motif 5′….G(A) n T…3′ (n can vary from 1 to 4) which is a flanking attribute of this region in mammals, insects, chickens, etc., is also apparently present in the honey bee but in the complementary strand, read as 3′….GAAT……5′. The consensus sequence at the 5′ flanking region of this sequence does not have a TATA motif that is apparent in other organisms although closely related sequences have been proposed (like TAATA, Zhang et al. 1995). In view of the description of this new STOP signal mentioned above, we propose alternative locations for the TATA box which are those highlighted in blue in Figure 1: one TATA box is the already mentioned included in VB1 and a second location is overlapping the STOP signal we just described. Further experimental approaches will allow infer the proper locations although the second seems much more plausible since it is located in a conserved domain. As to structural elements generally found in other insects, a poly-thymidine stretch was found just before block 2 and was composed of 16–18 Ts (poly-T, Figure 1). Positive identification of this structure is further confirmed because the (T)n stretch is flanked by two guanines. As proposed by Clary and Wolstenholme (1987), this sequence represents the site of initiation of the second-strand synthesis (rather than first-strand synthesis). The complete CB1 consistently forms five hairpin structures (Figure 2a), one of them carrying the T-stretch just mentioned as the terminal loop of one of the stems. The other hairpin structures are quite stable and with the exception of a single loop-out, all of them produce stems with almost complete paired nucleotides.

a Secondary structure model predicted for the conserved block 1 (CB1) showing the five stem-loops mentioned in the text. Arrows and accompanying letters refer to polymorphisms found in the 54 individuals analyzed. Asterisks denote insertions of a thymine. A white triangle denotes a deletion. The grey area corresponds to the putative STOP signal described in the text. b The most stable secondary structure of a long stem-loop existing in the conserved block 2 (CB2). An arrow denotes the only polymorphism found in all individuals analyzed.
The second highly conserved region (CB2) is the longest of the control region. It ranges from 413 to 442 nucleotides. Excluding all variable positions and indels, this region is 82 % conserved among all individuals studied. Second structure elements (a 30-bp long stem with only four mismatches and a terminal loop of just five nucleotides, see Figure 1 for its location) within CB2 consistently appear varying in the degree of energy (∆G) required which points to a folding structural stability in this region. This stem-loop structure that never changes in all folding alternatives for this region is depicted in detail in Figure 2b. The existence of stem and loop structures is to be expected with sequences that are 97 % biased to A+T. Stability of such secondary structures improve with the number of repeats. The present case seems to be a very stable segment that is always present within CB2, a fragment that has few polymorphic sites with the stem and loop structure admitting just a single singleton. Finally, after VB3, the remaining sequence can be considered also highly conserved among the individuals studied except for a very small stretch imbedded in it that in Figure 1 is named variable block 4 (VB4).
3.3 The tRNAs
Figure 3 depicts the most probable secondary structure of tRNAglu and tRNAser. Both tRNA genes fully agree with the structures inferred by Crozier and Crozier (1993). The glutamine tRNA presents a polymorphism across the individuals studied represented by deletions of two adenines in the dihydrouridine (DHU) arm. tRNAser lacks the typical cloverleaf structure because of the absence of the DHU arm which is typical of most other insects (Yang et al. 2011, Zhang et al. 2013) and presented no intraspecific polymorphisms. Anticodons of both tRNAs are TTC and TCT, respectively.

Most stable secondary structures of tRNAglu and tRNAser. Asterisks with lambda symbol represent polymorphisms since the adenine stretch may have 8–10 nucleotides.
3.4 Genetic variability indices and phylogenetic value of the A+T rich region
There are no significant differences between base composition of the complete A+T rich region and that of the conserved blocks combined (49.2 % T, 3.5 % C, 46.1 % A, and 1.2 % G). This is equally true if we compare base composition among the populations studied. This “homogeneity” is just apparent because the several conserved blocks are extremely different in relation to base composition. Excluding CB4, the other three conserved blocks show statistically significant different base composition: CB1 (54.3 T, 1.4 C, 43.1 A, 1.3 G), CB2 (48.0, 3.7, 47.2, 1.1), and CB3 (32.1 T, 11.3 C, 54.7 A, 1.9 G) (Kruskal-Wallis test, P < 0.008). Such differences are mainly due to CB3 that is diverging from the average showing an increment in Cs and detriment of Ts. Finally, we performed an analysis of molecular variance based solely on the conserved regions combined to see if a substructuring of the populations is perceivable, independently of a phylogeographic link among them. The overall F st was 0.489 (P < 0.0001). Most of the existing variability is due to within populations variation (51 %), variation among populations within groups was 48 % (in this case, the only group formed was composed of the three populations from the Iberian Peninsula, all other populations formed separately) and only 1 % could be attributed to among groups’ differences. The fact that no common haplotypes exist is certainly the cause for these results, a feature common to other insects (Wan et al. 2011a, b). Pairwise differences (as F st) varied from 0.0 (AZ vs. MAL) to 0.83 (CV vs. VIS) and were all statistically significant except for the pair AZ/MAL. The AMOVA performed using all the populations separately as groups did not change much. In fact, most of the variance is still attributed to within population variability and the remaining attributed to among populations differences (data not shown).
The final topology depicted from network (Figure 4) using the constraints described in the methods section showed one of the three shortest trees found differing from each other by a couple of minor branch swapping. Even using just the conserved blocks with only 54 variable sites, it is possible to perceive that some geographically related groups retain close links as it is the case of Iberian VIS and CA groups and CV. The Cape Verde honey bees (CV) which subspecies classification remains unknown (but see Pedersen 2001) are all related to the Moroccan ones (A. m. intermissa from SR and A. m. sahariensis from TT). In congruence with previous results based on nuclear (microsatellites) variation (De la Rúa et al. 2001; Muñoz et al. 2013), honey bees from the Canary Islands (yellow in Figure 4) are not a homogeneous group since at least two distinct clades emerge in the topology. A more or less complex group occupies a central position in the topology and connects most populations from Madeira (MD), Azores (AZ), and the Iberian Peninsula (VI, ML) which reflects the dispersion of the African sublineage with Atlantic distribution in the prospected area (Pinto et al. 2012).

Median-joining networks topologies of mtDNA control region haplotypes observed in eight populations from the Iberian Peninsula and northwestern Atlantic Islands. Circle sizes are proportional to haplotype frequencies. Black dots represent median vectors that connect related haplotypes. Line sizes are proportional to mutational positions connecting haplotypes.
4 Discussion
In A. mellifera, the almost absence in the A+T rich region of G/C nucleotide pairs makes more difficult the search for homologous positions since GC sites are most probably less affected by mutations due to a possible functional role. This is particularly true when looking for similarities among distant taxa, but it is not the case when looking at intraspecific variation. Actually, it has been the comparison of individual honey bees belonging to different locations that allowed the detection of highly conserved blocks that have kept a severe constraint both on point mutations or length variation events. The conserved blocks are thus prone to be used in phylogenetic studies given their low mutational rate. In this sense, the obtained network topology demonstrates that using a few variable characters from conserved blocks will allow us to recover a plausible connection among individuals. If one performs a network analysis for each population separately, the topology becomes even clearer and almost free from redundant sites or complex relationships among haplotypes which usually appear as highly complex reticulations (data not shown). This means that within populations the relationships among haplotypes are straightforward making each population a solid and discrete unit. The substitution rate of these conserved blocks is probably much slower than that of third position of coding regions something that was already found in Drosophila and mosquitoes (Caccone et al. 1996; Brehm et al. 2001). Such constraint on mutation rates further points to a possible functional role of these regions. It will be interesting to verify if this mutation rate in other Apis species agrees with previous observations in other organisms.
The length variable segments for phylogenetic inferences should be used in caution since it may obscure relationships due to recurrent variation in the number of repeats and equally important, the occurrence of mutations within the repeat units. Zhang and Hewitt (1997) claimed that the length variation at intraspecific level is mainly due to high mutation rate, and in spite of the fact that we did not find any signs of heteroplasmy in the individuals studied, the obvious pattern of these tandem repetitions among populations could well be the result of a phylogenetic relationship. Until now, length variation in the genus Apis has been observed in the intergenic region between mtDNA cox2 and the tRNAleu genes but not in the A+T rich region (Cornuet et al. 1991). The present data outlines the importance of looking at intraspecific variation in order to detect such variations. The fact is that the control region in A. mellifera has important segments of variable size due solely to variable number of tandem repeats. Moreover, mutations occurring within these variable repeats are in some cases individual specific (e.g., VB2). Other models have been proposed to explain such observations (for a revision, see Zhang and Hewitt 1997), but most probably the length variation typically observed in the A+T rich region could be better explained by replication slippage. Apart from the limited variable blocks in A. mellifera, the reduced variability of most of the A+T rich region may be due to high mutational pressure towards AT, TA, or GC/AT substitutions.
In conclusion, the A+T rich region of A. mellifera appears to contain valuable information especially at the population level. The region has important conserved blocks with a limited number, if any, of parsimoniously informative positions preventing its use to infer phylogenetic relationships. Nevertheless, the information retrieved from the comparison of individuals belonging to different populations was crucial to understand and characterize how the control region is organized in A. mellifera. The comparison of A. mellifera A+T rich region with other species of the same genus will be most valuable in order to confirm the suggestions made in the present study regarding the role of particular sequences within the A+T rich region of this insect.
References
Andrianov, B., Goryacheva, I., Mugue, N., Sorokina, S., Gorelova, T., Mitrofanov, V. (2010) Comparative analysis of the mitochondrial genomes in Drosophila virilis species group (Diptera: Drosophilidae). Trends Evol. Biol. 2, e4. doi:10.4081/eb.2010.e4
Bandelt, H.-J., Forster, P., Röhl, A. (1999) Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48
Brehm, A., Harris, D., Hernández, M., Cabrera, V., Larruga, J., Pinto, F., González, A. (2001) Structure and evolution of the mitocondrial DNA complete control región in the Drosophila subobscura subgroup. Insect Mol. Biol. 10, 573–578
Brehm, A., Harris, D., Hernández, M., Perez, J., Larruga, J., Pinto, F., González, A. (2003) Phylogeography of Drosophila subobscura from north Atlantic islands inferred from mtDNA A+T rich region sequences. Mol. Phylogenet. Evol. 30, 829–834
Caccone, A., Garcia, B.A., Powell, J. (1996) Evolution of the mitochondrial DNA control region in the Anopheles gambiae complex. Insect Mol. Biol. 5, 51–59
Cánovas, F., De la Rúa, P., Serrano, J., Galián, J. (2008) Geographic patterns of mitochondrial DNA variation in Apis mellifera iberiensis (Hymenoptera: Apidae). J. Zool. Syst. Evol. Res. 46, 24–30
Chang, D., Clayton, D. (1984) Precise identification of mitochondrial promoters for transcription of each strand of human mitochondrial DNA. Cell 36, 635–643
Clary, O., Wolstenholme, D. (1987) Drosophila mitochondrial DNA: conserved sequences in the A+T rich region and supporting evidence for a secondary structure model of the small ribosomal RNA. J. Mol. Evol. 25, 116–125
Clayton, D. (1982) Replication of animal mitochondrial DNA. Cell 28, 693–705
Clayton, D. (1996) Mitochondrial DNA replication. In: DePamphilis, M.L. (ed.) DNA replication in eukaryotic cells, pp. 1015–1027. Cold Spring Harbor Laboratory Press, Plainview, N.Y
Cornuet, J.M., Garnery, L., Solignac, M. (1991) Putative origin and function of the intergenic region between COI and COII of Apis mellifera L. mitochondrial DNA. Genetics 128, 393–403
Crozier, R.H., Crozier, Y.C. (1993) The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics 133, 97–117
De la Rúa, P., Galián, J., Serrano, J., Moritz, R.F.A. (2001) Genetic structure and distinctiveness of Apis mellifera L: populations from the Canary Islands. Mol. Ecol. 19, 1733–1742
De la Rúa, P., Galián, J., Pedersen, B.V., Serrano, J. (2006) Molecular characterization and population structure of Apis mellifera from Madeira and the Azores. Apidologie 37, 699–708
De la Rúa, P., Radloff, S.E., Hepburn, H.R., Serrano, J. (2007) Do molecular markers support morphometric and pheromone analyses? A preliminary case study in Apis mellifera populations of Morocco. Arch. Zootec. 213, 33–42
De la Rúa, P., Jaffé, R., Dall’Olio, R., Muñoz, I., Serrano, J. (2009) Biodiversity, conservation and current threats to European honeybees. Apidologie 40, 263–284
Doda, J., Wright, C., Clayton, D. (1981) Elongation of displacement-loop strands in human and mouse mitochondrial DNA is arrested near specific template sequences. Proc. Natl. Acad. Sci. USA 78, 6116–6120
Evans, J.D., Chen, Y.P., Cornman, R.S., De la Rúa, P., Foret, S., et al. (2013) Standard methods for molecular research in Apis mellifera. In: Dietemann, V.; Ellis. J.D.; Neumann, P. (Eds) The COLOSS BEEBOOK, Volume I: standard methods for Apis mellifera research. J. Apic. Res. doi:10.3896/IBRA.1.52.4.11
Excoffier, L., Laval, G., Schneider, S. (2005) Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinforma. 1, 47–50
Forster, P., Torroni, A., Renfrew, C., Röhl, A. (2001) Phylogenetic star contraction applied to Asian and Papuan mtDNA evolution. Mol. Biol. Evol. 18, 1864–1881
Franck, P., Garnery, L., Loiseau, A., Oldroyd, B.P., Hepburn, H.R., Solignac, M., Cornuet, J.M. (2001) Genetic diversity of the honeybee in Africa: microsatellite and mitochondrial data. Heredity 86, 420–430
Kim, I., Cha, S., Kim, M., Lee, S., Lee, K., Choi, Y., Hwang, J., Jin, B., Han, Y. (2007) Polymorphism and genomic structure of the A+T rich region of mitochondrial DNA in the oriental mole cricket, Gryllotalpa orientalis (Orthopera: Gryllotalpidae). Biochem. Genet. 45, 589–610
Kimura, M. (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120
Mathews, D., Sabina, J., Zuker, M., Turner, D. (1999) Expanded Sequence Dependence of Thermodynamic Parameters Improves Prediction of RNA Secondary Structure. J. Mol. Biol. 288, 911–940
Meixner, M.D., Pinto, M.A., Bouga, M., Kryger, P., Ivanova, E., Fuchs, S. (2013) Standard methods for characterising subspecies and ecotypes of Apis mellifera. In: V Dietemann; J D Ellis; P Neumann (Eds) The COLOSS BEEBOOK, Volume I: standard methods for Apis mellifera research. J. Apic. Res. 52(4): doi:10.3896/IBRA.1.52.4.05
Monforte, A., Barrio, E., Latorre, A. (1993) Characterization of the length polymorphism in the A + T-rich region of the Drosophila obscura group species. J. Mol. Evol. 36, 214–23
Muñoz, I., Pinto, M.A., De la Rúa, P. (2013) Temporal changes in mitochondrial diversity highlights contrasting population events in Macaronesian honey bees. Apidologie 44, 295–305
Murray, T.E., Kuhlmann, M., Potts, S.G. (2009) Conservation ecology of bees: populations, species and communities. Apidologie 40, 211–236
Pedersen, B.V. (2001) Identification of honeybees from Cape Verde. Beekeeping and Development 60 (retrieved from http://www.beesfordevelopment.org/portal/article.php?id=2358 on the 17/12/2013)
Pinto, M.A., Muñoz, I., Chávez-Galarza, J., De la Rúa, P. (2012) The Atlantic side of the Iberian Peninsula: a hot-spot of novel African honey bee maternal diversity. Apidologie 43, 663–673
Pinto, M.A., Henriques, D., Neto, M., Guedes, H., Muñoz, I., Azevedo, J., De la Rúa, P. (2013) Maternal diversity patterns of Ibero-Atlantic populations reveal further complexity of Iberian honey bees. Apidologie 44, 430–439
Polzin, T., Daneschmand, S.V. (2003) On Steiner trees and minimum spanning trees in hypergraphs. Oper. Res. Lett. 31, 12–20
Ruttner, F. (1988) Biogeography and taxonomy of honeybees. Springer, Berlin
Saccone, C., Pesole, G., Sbisá, E. (1991) The main regulatory region of mammalian mitochondrial DNA: structure-function model and evolutionary pattern. J. Mol. Evol. 33, 83–91
Simon, C., Frati, F., Beckenbach, A., Crespi, B., Liu, H., Flook, P. (1994) Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701
Sugihara, T., Yui, R., Ibaragi, Y., Matsuura, E. (2006) Complete nucleotide sequence of the A+T-rich region of Drosophila mauritiana mitochondrial DNA. Genes Genet. Syst. 81, 21–28
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., Kumar, S. (2011) MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 28, 2731–2739
Tan, H.W., Liu, G.H., Dong, X., Lin, R.Q., Song, H.Q., Huang, S.Y., Yuan, Z.G., Zgao, G.H., Zhu, X.Q. (2011) The complete mitochondrial genome of the Asiatic cavity-nesting honeybee Apis cerana (Hymenoptera: Apidae). PloS one 6(8), e23008
Thompson, J., Gibson, T., Plewniak, F., Jeanmongin, F., Higgins, D. (1997) The ClustalX windows surface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl. Acids Res. 24, 4876–4882
Vila, M., Björklund, M. (2004) The utility of the neglected mitochondrial control region for evolutionary studies in Lepidoptera (Insecta). J. Mol. Evol. 58, 280–290
Wan, X., Li, J., Kim, M., Park, H., Kim, S., Kim, I. (2011a) DNA sequence variation of the tobacco cutworm, Spodoptera litura (Lepidoptera: Noctuidae), determined by mitochondrial A+T – rich region and nuclear ITS2 sequences. Biochem. Genet. 49, 760–787
Wan, X., Li, J., Kim, M., Kang, T., Jin, B., Kim, I. (2011b) Population genetic structure of the migratory rice leaf roller, Cnaphalocrocis medinalis (Lepidoptera: Pyralidae), inferred from the mitochondrial A+T – rich region and nuclear ITS2 sequences. Gen. Mol. Res. 10, 273–294
Wang, A.R., Kim, M.J., Park, J.S., Choi, Y.S., Thapa, R., Lee, K.Y., Kim, I. (2013) Complete mitochondrial genome of the dwarf honeybee, Apis florea (Hymenoptera: Apidae). Mitochondrial DNA 24, 208–210
Yang, F., Du, Yu-zhou, Wang, L., Cao, J., Yu, W. (2011) The complete mitochondrial genome of the leafminer Liriomyza sativae (Diptera: Agromyzidae): Great difference in the A+T -rich region compared to Liriomyza trifolii. Gene 485, 7–15
Zhang, D., Hewitt, G.M. (1997) Insect mitochondrial control region: a review of its structure, evolution and usefulness in Evolutionary studies. Biochem. Syst. Ecol. 25, 99–120
Zhang, D., Szymura, J., Hewitt, G.M. (1995) Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 40, 382–391
Zhang, H., Zeng, H., Huang, Y., Zhang, Z. (2013) The complete mitochondrial genomes of three grasshoppers, Asiotmethis zacharjini, Filchnerella helanshanensis and Pseudotmethis rubimarginis (Orthoptera: Pamphagidae). Gene 517, 89–98
Zuker, M., Mathews, D., Turner, D. (1999) Algorithms and Thermodynamics for RNA Secondary Structure Prediction: A Practical Guide. In: Barciszewski, J., Clark, B.F.C. (eds.) RNA Biochemistry and Biotechnology. NATO ASI Series, pp. 11–43. Kluwer Academic Publishers, Dordrecht, NL
Acknowledgments
We are very grateful to the beekeepers for generously providing the samples analyzed here and to two reviewers for their helpful comments. This research has been financed by projects “Acción Integrada Hispano-Lusa HP-2004-088” to A. Brehm and P. De la Rúa and Fundación Séneca (project 11961/PI/09) to P. De la Rúa.
Author information
Authors and Affiliations
Corresponding author
Additional information
Structure et variation génétique de la région de contrôle de l’ADN mitochondrial chez l’abeille Apis mellifera
Apidae / région riche en A et T / phylogéographie / Péninsule ibérienne / Macaronésie
Aufbau und genetische Variation der mitochondrialen Kontrollregion der Honigbiene, Apis mellifera
Apidae / Kontrollregion / A+T-reiche Regionen / Struktur / Phylogeographie / Iberische Halbinsel / Makaronesische Inseln
Manuscript editor: Bernd Grünewald
Rights and permissions
About this article

Cite this article
Gonçalves, R., Freitas, A.I., Jesus, J. et al. Structure and genetic variation of the mitochondrial control region in the honey bee Apis mellifera . Apidologie 46, 515–526 (2015). https://doi.org/10.1007/s13592-014-0341-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-014-0341-y