
Abstract
Eosinophilic annular erythema (EAE) is a rare entity of unknown etiology that is possibly related to a hypersensitivity reaction and presents as annular erythematous plaques with tissue eosinophilia. It is classified as a figurate erythema with a controversial relationship to Wells syndrome (WS) in the literature, where it is generally considered a separate entity or subset based on clinical and histopathologic differences. EAE typically presents with recurrent, erythematous, arcuate, and annular plaques on the trunk and proximal extremities. The course of the disease is often chronic, recurrent, and relapsing. Responses to treatment are variable but are typically best with systemic steroids and antimalarials. We report a patient refractory to other therapies who had a striking response to dapsone.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
We report a rare case of eosinophilic annular erythema (EAE) and summarize the clinical features and treatments of other cases reviewed in the literature to date.
Case report
Our patient, a 38-year-old Hispanic male with a remote history of Wegener’s granulomatosis and mild chronic leukopenia, presented in February 2014 with a recalcitrant, intensely pruritic, painful, burning, widespread annular eruption (Figs. 1 and 2) affecting the neck, upper thorax, arms, and proximal thighs that had been ongoing for more than two years. The patient provided informed consent to include him in this case report.

Annular plaques on chest

Polycyclic plaques; close-up of chest
Laboratory screening showed a normal comprehensive metabolic panel and a CBC with a chronic mild leukopenia and, in some instances, a mild peripheral eosinophilia. Screening antibodies for ANA, SSA, SSB, RPR, ANCA, hepatitis B and C, and HIV 1/2 were all negative.
Multiple skin biopsies showed a superficial and deep perivascular and interstitial lymphocytic and eosinophilic infiltrate (Figs. 3, 4, and 5), some with focal vacuolar interface changes and prominent pigment incontinence. There were no findings of dermal edema or mucin to suggest cutaneous lupus erythematosus. No signs of vasculitis or flame figures were noted. DIF was negative for discoid lupus and systemic lupus erythematosus, with a negative LE band test reaction observed as well as a negative in vivo ANA test.

Tight perivascular infiltrate H&E (×40)

Dense perivascular infiltrate (×100)

Infiltrate with prominent eosinophils (×200)
The patient was initially temporarily steroid responsive with short courses of oral prednisone, but supplemental monthly intramuscular 60–80 mg kenalog injections were necessary to achieve any noticable control. The dermatitis continued to flare, with recurrent relapses every 3–4 weeks following each injection. Extended patch testing was positive with carba mix but there was a doubtful reaction to Balsam of Peru (Myroxylon pereirae). Balsam of Peru is a marker for a fragrance allergy as well as allergies to certain flavorings, including vanilla, curry, cinnamon, and cloves. When foods containing this allergen are ingested, systemic dermatitis can result. The patient did trial a Balsam-of-Peru-free diet, which involved avoiding chocolate, wine, beer, cola, and tomato-based products such as pizza sauce and salsa. However, he did not experience any significant improvement with this diet, nor any improvement when he was placed on a trial of various antihistamine combinations and topical steroids. He was diagnosed with EAE and placed on hydroxychloroquine 200 mg bid, which prompted only a partial and modest clinical response. Persistent symptomatic annular lesions caused significant pain and itching and affected his quality of life.
Further options were discussed at city-wide grand rounds, including methotrexate, cyclosporine, and dapsone. Given his history of chronic mild leukopenia, cyclosporine was added next, and was incrementally increased to a dose of 200 mg bid (5 mg/kg/day), but there was no response. Oral prednisone was continued and was required to provide relief.
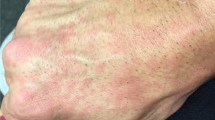
After a hematology evaluation for his leukopenia, dapsone was used in place of the cyclosporine, which led to a prompt response (Fig. 6), with further relief observed on dose escalation to 150 mg daily. The majority of the erythematous annular rings and plaques on the abdomen, chest, and back resolved. While he still had extensive postinflammatory hyperpigmentation, the lesions were flat and macular. He was able to taper off his prednisone completely while maintaining the response to dapsone and hydroxychloroquine. He later relapsed and required some further intermittent oral prednisone courses. However, the response to dapsone was dramatic and, for the first time, provided relief from the severe pruritic symptoms associated with this refractory skin condition, and led to an improved quality of life.

Striking response to dapsone
Discussion
Clinical Presentation
Patients with EAE classically present with recurrent erythematous or urticarial arcuate and annular plaques on the trunk and proximal extremities [1]. Clinically, EAE is described as presenting in one of two patterns: first as a figurate/centrifugum-type pattern reminiscent of deep erythema annulare centrifugum, or second as an urticarial/annular-type pattern [2]. It is devoid of bulla, though there is a single case report of bullous eosinophilic annular erythema with negative direct immunofluorescence and negative indirect IF as well [3].
Histopathology
Histologically, EAE is described as having a dense superficial and deep perivascular and/or interstitial lymphohistiocytic infiltrate with abundant eosinophils. It typically presents without “flame figures,” though they have been noted in some cases and with more longstanding lesions (polymorphism, multicenter). There are some reports of focal interface changes, but generally it is absent of mucin deposition, flame figures, vasculitis, or granulomas. Basal melanosis and pigment incontinence are also reported and may account for clinically observed dusky-toned areas of hyperpigmentation [4]. While still debatable, EAE is generally regarded as being distinct from annular erythema of infancy (AEI) and Wells syndrome (WS) (Table 1). Though it also shows tissue eosinophilia, erythema of infancy predominantly affects children under the age of 1 year and resolves spontaneously [2]. Wells syndrome presents as tender cellulitis-type plaques, though annular and figurate lesions can occur [2]. Many physicians, however, feel that EAE is a subset or variant of WS. In WS, the inflammatory cells, including eosinophils, often involve the dermis and subcutis, whereas in EAE they are generally confined to the dermis [4]. WS also features flame figures that are quite characteristic as well as blood eosinophilia, both of which are not typical of EAE. In addition to WS, the differential diagnosis may also include the deep form of erythema annulare centrifugum, tumid lupus erythematosus, Jessner lymphocytic infiltrate, urticarial bullous pemphigoid, erythema migrans, granuloma annulare, and interstitial granulomatous dermatitis [5].
Pathogenesis and Associated Diseases
The etiology of EAE remains largely unknown, though it is suggested that it may be a hypersensitivity reaction to an unidentified antigen [6]. It has been associated with several chronic conditions, such as autoimmune thyroid disease (both hypothyroidism and hyperthyroidism), chronic borreliosis, chronic H. pylori gastritis, chronic kidney disease, diabetes mellitus, and hepatitis C virus infection [5, 7]. Though suspected to be a hypersensitivity response, it has also been associated with internal malignancy. Specific malignancies have been reported in three cases, including thymoma with complete resolution following thymectomy [7], metastatic prostate adenocarcinoma [8], and renal carcinoma [9].
Treatment
EAE is most commonly reported to respond successfully to systemic steroids and antimalarials, but it typically runs a chronic relapsing and remitting course, with a relapse frequently seen after discontinuing both treatments. It often shows resistance to multiple treatments. Most reports in the literature note the use of antimalarials as the first line for EAE. Response to hydroxychloroquine is typically prompt and usually observed within the first 4 weeks, though a case showing a delayed response with remission at 10 weeks has been reported [10]. Though Wells syndrome has been successfully treated with dapsone, there has been only one report prior to this one of a successful response of EAE to dapsone. That case report noted a complete response to dapsone at 25 mg qd, with the response occurring within 3 weeks and remission sustained at 8 months of follow-up [11]. Another multicenter study of 10 patients with EAE noted a chronic course and resistance to treatment with systemic steroids alone or in combination with hydroxychloroquine and cyclosporine [9]. Researchers reported that EAE is characterized by a chronic course, resistance to treatment, and a high relapse rate [9]. A single case report noted a response to eight sessions of narrow-band UVB following the failure of systemic steroids and hydroxychloroquine; the light therapy cleared the EAE without inducing pigment changes, and no recurrence was noted 9 months after the light therapy had been stopped [1]. There is also a single case report of a dramatic improvement with indomethacin, but repeated syncopal episodes apparently led to the cessation of that therapy [12]. A recent case showed remission with nicotinamide at 900 mg/day dosing [13]. Though the patient had previously required systemic steroids for 7 years, remission was maintained with nicotinamide for a full year after cessation of the steroids. The patient also had a history of autoimmune pancreatitis with elevated immunoglobulin G4 levels (335 mg/dl), but the case report did not comment on the treatment of that or its timing in relation to the EAE.
As one can see from these reports in the literature, EAE commonly runs a chronic relapsing course and is resistant to numerous treatments. Though EAE typically is refractory, it can also be self-limited; treatment may not be required in all instances [6].
Conclusions
Given the rarity of this condition and the lack of a complete response to any particular therapy, dapsone should be considered as a therapeutic option for this often refractory skin condition.
References
Thomas L, Fatah S, Nagarajan S, Natarajan S. Eosinophilic annular erythema: successful response to ultraviolet B therapy. Clin Exp Dermatol. 2015;8:883–6.
Rongioletti F, Fausti V, Kempf W, Rebora A, Parodi A. Eosinophilic annular erythema: an expression of the clinical and pathological polymorphism of Wells syndrome. J Am Acad Dermatol. 2011;4:135–7.
Kato K, Namiki T, Tokoro S, Takayama K, Yokozeki H. Bullous eosinophilic annular erythema. J Dermatol. 2017;44:e42–3.
Nakazato S, Fujita Y, Shinkuma S, Nomura T, Shimizu H. Eosinophilic annular erythema is clinically characterized by central pigmentation reflecting basal melanosis: a clinicopathological study of 10 cases. J Eur Acad Dermatol Venereol. 2017;31:1916–23.
Heymann WR. Eosinophilic annular erythema: attention must be paid. Skinmed. 2014;6:376–8.
Prajapati V, Cheung-Lee M. Spontaneously resolving eosinophilic annular erythema. J Am Acad Dermatol. 2012;67(2):e75–7.
Iga N, Otsuka A, Kaku Y, Miyachi Y, Kabashima K (2015) Eosinophilic annular erythema limited on the palms and the soles and possibly associated with thymoma. J Eur Acad Dermatol Venereol. 2016;30(7):1213–4.
González-López MA, López-Escobar M, Fernández-Llaca H, González-Vela MC, López-Brea M. Eosinophilic annular erythema in a patient with metastatic prostate adenocarcinoma. Int J Dermatol. 2015;3:80–2.
El-Khalawany M, Al-Mutairi N, Sultan M, Shaaban D. Eosinophilic annular erythema is a peculiar subtype in the spectrum of Wells syndrome: a multicentre long-term follow-up study. J Eur Acad Dermatol Venereol. 2013;8:973–9.
Karataş Toğral A, Seçkin D. Eosinophilic annular erythema: a late but complete response to hydroxychloroquine. Australas J Dermatol. 2017;58(3):228–30.
Manriquez J, Berroeta-Mauriziano D, Andino-Navarrete R, Vera-Kellet C. Eosinophilic annular erythema: complete clinical response with dapsone. Int J Dermatol. 2015;4:96–8.
Howes R, Girgis L, Kossard S. Eosinophilic annular erythema: a subset of Well’s syndrome or a distinct entity? Australas J Dermatol. 2008;49:159–63.
Ogawa K, Fukumoto T, Yoshida M, Matsumoto Y, Shobatake C, Asada H. Eosinophilic annular erythema in a patient with autoimmune pancreatitis: nicotinamide therapy may be beneficial for achieving remission. J Dermatol. 2016;43(11):1380–1.
Acknowledgements
Funding
No funding or sponsorship was received for this study or the publication of this article. The article processing charges were funded by the authors.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Luke Wallis, Robert C. Gilson, and Robert T. Gilson have nothing to disclose.
Compliance with Ethics Guidelines
Informed consent was obtained from the patient before they were included in the case report.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to www.medengine.com/Redeem/97FCF0601888C652.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wallis, L., Gilson, R.C. & Gilson, R.T. Dapsone for Recalcitrant Eosinophilic Annular Erythema: A Case Report and Literature Review. Dermatol Ther (Heidelb) 8, 157–163 (2018). https://doi.org/10.1007/s13555-017-0214-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-017-0214-1