
Abstract
Agents which increase intracellular cyclic adenosine monophosphate (cAMP) may have an antagonistic effect on pro-inflammatory molecule production so that inhibitors of the cAMP degrading phosphodiesterases have been identified as promising drugs in chronic inflammatory disorders. Although many such inhibitors have been developed, their introduction in the clinic has been hampered by their narrow therapeutic window with side effects such as nausea and emesis occurring at sub-therapeutic levels. The latest generation of inhibitors selective for phosphodiesterase 4 (PDE4), such as apremilast and roflumilast, seems to have an improved therapeutic index. While roflumilast has been approved for the treatment of exacerbated chronic obstructive pulmonary disease (COPD), apremilast shows promising activity in dermatological and rheumatological conditions. Studies in psoriasis and psoriatic arthritis have demonstrated clinical activity of apremilast. Efficacy in psoriasis is probably equivalent to methotrexate but less than that of monoclonal antibody inhibitors of tumour necrosis factor (TNFi). Similarly, in psoriatic arthritis efficacy is less than that of TNF inhibitors. PDE4 inhibitors hold the promise to broaden the portfolio of anti-inflammatory therapeutic approaches in a range of chronic inflammatory diseases which may include granulomatous skin diseases, some subtypes of chronic eczema and probably cutaneous lupus erythematosus. In this review, the authors highlight the mode of action of PDE4 inhibitors on skin and joint inflammatory responses and discuss their future role in clinical practice. Current developments in the field including the development of topical applications and the development of PDE4 inhibitors which specifically target the subform PDE4B will be discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The clinical symptoms of chronic inflammatory diseases are determined by a number of different inflammatory mediators. In psoriasis, for example, not only the well-recognized tumour necrosis factor (TNF) is an important effector molecule, but interleukin 17 (IL-17), IL-22, interferon γ (IFNγ), IL-2, IL-36, CCL20, IL-8, chemokine CXCL10, IL-23, IL-1, IL-18, IL-12, vascular endothelial growth factor (VEGF), substance P, IFNα, and many others contribute to the inflammatory response both in the joint and skin compartment. Conventional therapies have a broad range of action and inhibit, e.g. preferentially lymphocyte proliferation [cyclosporin (CsA), methotrexate] and lymphokine production (IFNγ, IL-17, IL-22, IL-2) or mainly target the hyperproliferation and abnormal differentiation of keratinocytes (dithranol, tar) or combine the latter with cytokine modifying properties (retinoids, vitamin D, glucocorticoids). Biologics currently used in the clinic target one specific mediator which supposedly plays a key role upstream in the disease-specific cytokine network. An approach which interferes with several inflammatory mediators without the side effects seen with conventional immunosuppressants is of high interest. Interfering with the intracellular levels of cyclic adenosine monophosphate (cAMP) was proposed almost two decades ago as a promising target.
Cyclic Adenosine Monophosphate (cAMP)
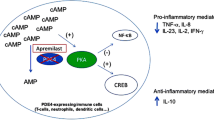
cAMP is a key intracellular second messenger (Fig. 1) [1]. cAMP signalling is activated by a variety of G protein-coupled receptor ligands. The effects of cAMP are transduced by two ubiquitously expressed intracellular cAMP receptors, protein kinase A (PKA) and exchange protein directly activated by cAMP (EPAC). cAMP can also bind to cyclic nucleotide-gated ion channels in certain tissues. The latter cAMP activity plays a role in the clinical symptoms of cholera. Cholera toxin subunit B causes un-leashed cAMP production and consequently chloride secretion through the apical chloride channel into the lumen of the small intestine leading to severe diarrhoea and dehydration [2]. cAMP actions are highly cell type- and context-dependent. cAMP and its downstream signalling are involved in a plethora and large diversity of cellular responses. A key feature of the cAMP/cAMP-dependent PKA transduction system is the compartmentalization of its signalling molecules and effectors. This means that local pools of cAMP expression/PKA activation are generated in distinct subcellular compartments. This allows for precisely regulated activity essential for response specificity. cAMP activates and enables PKA to phosphorylate substrate proteins. PKA activates cAMP response element binding protein (CREB) which is a cAMP-responsive element possessed by several immune-related genes including IL-2, IL-6, IL-10, and TNFα (for review: [3]). cAMP can directly or indirectly (via phosphorylated CREB) inhibit nuclear factor kappa B (NF-κB) pathway activation events. Low intracellular cAMP may thus lead to the preferential expression of proinflammatory mediators. The above mentioned EPAC can activate the Ras-related small guanosine triphosphate (GTP) Rap1 and this activation can lead to biological responses including induction of phagocytosis, and vasorelaxation [4–7].

Cellular pathways involving cyclic adenosine monophosphate (cAMP). Reproduced from Schafer [1], with permission from Elsevier. AC adenylyl cyclase, ATF-1 activating transcription factor, ATP adenosine triphosphate, CBP CREB-binding protein, CREB cAMP response element binding protein, CREM cAMP response element modulator, EPACs exchange protein directly activated by cAMP, Gαs G protein alpha subunit, GPCR G protein coupled receptors, IFN interferon, IKKβ inhibitor of nuclear factor kappa B kinase subunit beta, IL interleukin, IRAK interleukin-1 receptor-associated kinase, IκB inhibitor of NF-κB, NF-κB nuclear factor kappa B, PDE4 phosphodiesterase 4, PG prostaglandin, PKA protein kinase A, TLR4 toll-like receptor 4, TNF tumour necrosis factor, TRAF6 TNF receptor-associated factor
Intracellular concentration of cAMP is determined by the activity of adenylyl cyclases [synthesises cAMP from adenosine triphosphate (ATP)] on the one hand and phosphodiesterases (PDE) on the other. PDEs are also expressed in distinct cellular compartments and functionally coupled to individual receptors—thus providing a way to control sub-compartment cAMP levels in a stimulus-specific manner.
The Role of cAMP in Inflammatory Immune Responses
Substances which increase cAMP in monocytes/macrophages are among the most potent inhibitors of IL-12 family members including IL-12/IL-23 p40. This has been shown for cholera toxin [8–14], histamine [15–19], prostaglandin E2 (PGE2) [20] and other mediators. Another milestone in the investigation of cAMP’s role in immune responses was the finding by Bopp et al. [21] that one of the effector mechanisms underlying T regulatory (Treg) function is the contact-dependent transfer of cAMP via gap-junctions into target cells. Repression of cAMP greatly reduces the suppressive activity of human Treg [22]. cAMP facilitates the functional activity of a transcriptional inhibitor called ICER (inducible cAMP early repressor) and this mechanism seems to be involved in the suppression of the key T cell growth factor IL-2 [23] and other cytokines [24]. In addition, immunosuppressive and anti-inflammatory actions of cAMP have been attributed in part to the ability of cAMP-induced signals to interfere with the function of NF-κB [25]. NF-κB activation is one of the master signalling pathways involved in inflammatory responses and a key target for anti-inflammatory drug design. Important cytokines downstream of NF-κB include TNFα, CCL20, IL-8; IL-1 family members (IL-36, IL-18, IL-1) and (in combination with a priming signal) also IL-12 family members (IL-12, IL-23, IL-27) and many more.
The cAMP system is also involved in a variety of epithelial functions and plays a role in maintenance of the skin barrier. In the keratinocyte cell line HaCat largely suppressed chemokine production (CXCL10, CCL17, and CCL22) has been described [26, 27] in the context of increased cAMP levels.
Phosphodiesterase 4 (PDE4)
There are several PDE families, all isoforms of which are concerned with the intracellular degradation of the phosphodiesterase bonds of cAMP and cyclic guanosine monophosphate (cGMP). PDE4, -7, and -8 degrade cAMP specifically. PDE4 is encoded by four separate genes (PDE4 A–D) and each PDE4 controls non-redundant cellular functions. Inhibition of PDE4 activity leads to elevated levels of intracellular cAMP.
PDE4s are the predominant cAMP degrading isoenzymes in most immune cells including lymphocyte subsets, granulocytes and cells of the monocyte/macrophage lineage [28]. They are also expressed in epithelial cells, vascular endothelium, chondrocytes and smooth muscle cells. The role of PDE4 in immune cells has recently been reviewed by Jin et al. [29], and for respiratory diseases by Page and Spina [30]. In immune cells, the isoforms PDE4A, B and D (but not C) are highly expressed as well as PDE3 and 7 [30]. It is noteworthy, however, that the activity of macrophages may not be significantly inhibited by PDE4 selective inhibitors [31]. The benefit of a combined effect of PDE7 or PDE3 with PDE4 selective inhibitors on macrophage and T cell function has been described [32, 33].
The expression levels of these PDE isoenzymes are regulated by a variety of stimuli. For example, prostaglandin E2 induces PDE3 and 4 activity and PDE3B, 4A4, 4A1, 4D2 and 4D3 expression [34]. T cell receptor stimulation increases the differential expression of PDE4 subtypes in cluster of differentiation 4 (CD4+T) cells [35], and toll-like receptor 4 (TLR4) stimulation acts on PDE4B2 expression in human monocytes [36].
PDE Inhibitors
Non-Selective PDE Inhibitors
Pentoxifylline is a competitive non-selective PDE inhibitor (used in the treatment of peripheral vascular disease) which raises intracellular cAMP levels to inhibit TNF and reduce inflammation. Pentoxifylline is also an adenosine 2 receptor antagonist. It reduces blood viscosity and platelet aggregation. Although suggested by some authors, pentoxifylline is not effective on the activity of psoriasis [37]. Some beneficial effect has been reported in canine atopic dermatitis [38] and human lung sarcoidosis [39].
Theophylline inhibits to some extent PDE1-5 (least effective against PDE4; [40]), is a potent adenosine receptor antagonist and an activator of histone deacetylase 2 (HDAC2) such that it might exert beneficial effects on (allergic) lung inflammation [41].
Selective PDE4 Inhibitors
By increasing cAMP levels, PDE4 inhibitors show anti-inflammatory effects in almost all inflammatory cells. Numerous selective PDE4 inhibitors have been patented in the past two decades and some of them have been evaluated in clinical trials for several inflammatory conditions including asthma, chronic obstructive pulmonary disease (COPD), atopic dermatitis (AD), and rheumatoid arthritis (RA). Inhibitors of different structural classes have been developed but discontinued for most of these compounds because of narrow therapeutic windows. Doses needed for efficacy could not be reached due to dose-limiting adverse events with nausea, diarrhoea, abdominal pain, vomiting, and dyspepsia being the most common. Adverse events of PDE4 inhibitors are evoked through the inhibition of PDE4 in non-target tissue at doses similar to those needed for therapeutic efficacy. It is believed that the inhibition of enzymes encoded by PDE4D in non-target tissues promotes emesis [42]. Thus, the development of PDE4 inhibitors with improved therapeutic indices has been a major focus of pharmaceutical research. Development of PDE4 inhibitors with different delivery routes such as topical application [43] and inhalation (outlined in Page and Spina [30]) are also under development for the treatment of airway inflammation and dermatitis. AN2728 which inhibits PDE4 has been administered topically in phase 2 studies to patients with psoriasis or AD [44, 45].
The first orally active PDE4 inhibitor roflumilast [46] was approved in 2010 by the European Medicines Agency for severe COPD associated with chronic bronchitis in adult patients. In March 2011 the U.S. Food and Drug Administration (FDA) approved it for reducing COPD exacerbations. PDE4 and its inhibition have been studied extensively in the treatment of COPD and asthma [46, 47].
Recent human clinical data on PDE4 inhibitors on skin diseases and in particular on psoriasis are available for apremilast. Apremilast is an orally available PDE4 inhibitor [48] which does not show any marked selectivity among the PDE4 isotypes. It seems to elicit less emetic side effects while also having a wide therapeutic window. The underlying mechanism for this increased tolerability is not known. The effects of apremilast—which are in line with findings reported for increased intracellular cAMP levels—on a range of pro-inflammatory responses in a variety of cells have recently been comprehensively summarized [1].
Unsurprisingly, all PDE4 inhibitors have the potential to reduce the expression of TNFα which is considered a key mediator in a number of inflammatory diseases. Crilly et al. [49] have demonstrated that specific PDE4 inhibitors dose-dependently down regulate the release of TNFα and other cytokines including CCL2, CCL3 (and to a lesser extent IL-1ß) from primary RA synovial digest suspensions. McCann et al. [50] have demonstrated TNFα inhibition in human rheumatoid synovial membrane cultures for apremilast. It is of interest that some PDE4 subtypes such as PDE4B seem to be more concerned with the inhibition of TNF production in murine monocyte/macrophages [51, 52]. Apremilast has inhibitory activity on TNFα release by ultraviolet B (UVB) activated (50 mJ/cm2) keratinocytes [53].
PDE4 Inhibitors in Dermatologic Diseases
Data suggest a promising therapeutic effect for selective PDE4 inhibitors on inflammatory skin diseases [54]. Of note, a PDE7A inhibitor was also successful in suppressing dermatitis and TNF expression in mice studies [55]. In a humanised [severe combined immunodeficiency (SCID) mice, grafted human psoriasis skin triggered with psoriatic natural killer (NK) cells] psoriasis model oral apremilast led to significant reduction in epidermal lesion thickness [54]. The psoriasiform histology was clearly reduced with regard to parakeratosis, hyperkeratosis, lymphocytic and neutrophilic infiltration. Clinical studies for psoriasis are summarized below.
One study points to a potentially beneficial effect of apremilast in cutaneous sarcoidosis [56] and it will be interesting to further explore the activity of PDE4 inhibitors in granulomatous skin diseases including Melkerson Rosenthal syndrome for which the therapeutic options are limited at present. Although PDE4 selective inhibitors inhibit IL-12 and TNF a mixed PDE4/3/7 preparation may have improved activity on macrophages which are key cells in granulomatous diseases. PDE4 inhibitors may be of benefit in lupus erythematosus (LE) [57]. For example, a recently published open-label, single arm pilot study with apremilast showed favourable results of a 20 mg twice daily (bd) dose regime in cutaneous discoid lupus erythematosus [58]. Apremilast was well tolerated in these patients.
For skin diseases, the availability of topical preparations (as mentioned above) is of high interest and ongoing trials are exploiting the potency of topical PDE4 inhibition [44, 45]. The anti-fibrotic effect makes PDEs potential drugs for the treatment of scleroderma. However, PDE5 inhibitors seem more promising in this disease as well as in the treatment of secondary Raynaud’s phenomenon (improvement of endothelial dysfunction and prevention of vascular remodelling) [59].
PDE4 inhibitors including apremilast have beneficial effects in animal models of dermatitis, in particular allergic contact dermatitis (ACD, for review [60]). The elicitation phase of ACD follows a Th1 like dominated response pattern where contact allergens impact on TLR activation, reactive oxygen species (ROS) and NLRP3 inflammasome activation which are key mechanisms in the induction phase of ACD [61–63]. As mentioned above, inhibition of ROS production may be better achieved in vitro by combined PDE inhibitors (PDE4 and 3 or 7).
Two clinical studies on AD have recently been published [64, 65]. Samrao et al. [64] used apremilast at 2 doses (20 and 30 mg bd, for 3 months, 6 months) in an open-label study with 16 adult AD patients. They found a reduced Eczema Area and Severity Index (EASI) and Dermatology Life Quality Index (DLQI) for the 30 mg group at 3 months and a reduction in baseline pruritus and DLQI in the 20 mg group after 3 and 6 months time. Volf et al. [65] performed a phase 2, open-label study with apremilast in patients suffering from severe ACD or AD. A dose of 20 mg bd was given for 3 months in 10 patients with AD and/or ACD. Apremilast was well tolerated but was only minimally effective in this small study with a heterogeneous study population.
From what is known on PDE4 action on lymphocytes, macrophages/dendritic cells subtypes, eosinophils and mast cells (for review, [29]) the overall net effect of PDE4 inhibitors seems more prominent for IFNγ or IL-17 dominated immune responses than IL-4/5/13 one [66]. Interestingly, a better effect on IFNγ dominated inflammation has been described for Treg in vivo studies [67]. Indeed, the effect of IL-4 on B cell function can even be accentuated. This leads to the notion that PDE4 inhibitors may be more potent in the treatment of IL-12/IL-23, thus IFNγ/IL-17 dominated responses than Th2 ones. Based on this consideration, apremilast may be effective in the effector phase of ACD, psoriasis and in the very chronic phases of AD in which the initial Th2 pattern has switched to a more Th1 dominated phenotype in the skin compartment [68]. In chronic AD the topical application may be the desirable way of application as the Th2 dominated response pattern in the blood of atopic individuals remains unaltered.
Clinical Studies in Psoriasis
A small (19 patients) single arm, open-label pilot study was performed in subjects with moderate to severe plaque psoriasis. Patients were treated for 29 days with 20 mg od of apremilast [69]. CD11c cells, T cells and epidermal thickness were reduced. Immunohistologic analysis of lesional-skin biopsies showed reduction in epidermal thickness and reduced infiltration of T cells and CD11c cells in responder patients. Psoriasis Area and Severity Index (PASI) was improved in 14 out of 19 patients.
The efficacy of apremilast in psoriasis has been assessed in a phase 2b study using doses of 10, 20, and 30 mg bd with a placebo comparator [70]. In this study, 352 patients were enrolled with active psoriasis of moderate severity [PASI of more than or equal to 12 or a body surface area affected by psoriasis of more than or equal to 10%, although mean baseline scores for PASI and body surface area (BSA) were 18.5% and 22%, respectively] who were candidates for phototherapy or systemic therapy. The primary target was the proportion of subjects achieving 75% improvement in PASI (PASI75) at 16 weeks (the placebo controlled phase). At 16 weeks patients on placebo could be re-randomised to active treatment but the dose was still concealed to both patient and physician. Further outcomes were assessed at 24 weeks. At 16 weeks PASI75 was achieved by 6% of patients on placebo, 11% of those on 10 mg bd, 29% of those on 20 mg bd, and 41% of those on 30 mg bd. The results for apremilast 20 mg bd and 30 mg bd were significantly different from placebo. The median number of days to achieve PASI75 was 57 for placebo and 70, 83, and 44 for 10, 20 and 30 mg bd, respectively. At week 16 13% of patients on placebo were ‘clear or almost clear’ on the physicians global assessment; the corresponding figures for apremilast were 10%, 24%, and 33% for 10, 20, and 30 mg bd, respectively. Adverse events were largely mild to moderate: upper respiratory tract infections, gastrointestinal symptoms (diarrhoea and nausea), and headache were the most frequent of these in the active treatment groups. No opportunistic infections were seen [70].
Clinical Studies in Psoriatic Arthritis
In psoriatic arthritis there is only one published study of the efficacy of apremilast—a phase 2 randomized placebo controlled study [71]. The results of the phase 3 PALACE-I study were presented at the American College of Rheumatology (ACR) meeting in Washington DC in November 2012 [72].
The phase II study enrolled 204 patients with active psoriatic arthritis, defined by more than or equal to 3 tender and 3 swollen joints. Only co-prescription with a stable dose of methotrexate or oral glucocorticoids was allowed: all other disease modifying drugs had to be discontinued before enrolment. The usual restrictions on major co-morbid conditions applied. Patients were randomized equally to placebo, apremilast 20 mg bd or apremilast 40 mg once daily (od), stratified by baseline methotrexate use. After 12 weeks of treatment patients could stop treatment or enter a further 12 week extension phase, the latter option occurring as an amendment to the original protocol design, and re-randomisation of placebo to one of the active treatment groups. The primary efficacy endpoint was the proportion of patients achieving a modified (by joint count) ACR 20% improvement at 12 weeks (ACR20). The primary endpoint was achieved by 43.5% of patients in the apremilast 20 mg bd group, 35.8% of patients in the 40 mg od group, and 11.8% of patients on placebo, the differences between active drug and placebo being highly significant (see Table 1) [71, 72]. In the extension phase, where patients who had initially taken placebo were transferred to an active drug, a similar improvement was seen in the people who transferred, and the initial improvements in the active treatment groups were maintained. Stratified for methotrexate use there was no difference in primary outcome between the two groups, although more people on combination had gastro-intestinal side effects. No assessments of skin, enthesitis, dactylitis, or axial involvement were made in this study. Overall safety data were good with diarrhoea and headache being the major, albeit no more than moderate, side effects. Abnormal laboratory results, including liver enzyme elevations, were infrequent.
The PALACE-I study has only been reported in abstract form [72]. This study enrolled 504 patients with active psoriatic arthritis (more than three tender and swollen joints) who were randomized in an equal ratio to placebo, apremilast 20 mg bd and apremilast 30 mg bd. The patients were stratified by previous disease modifying drug use and about three quarters were TNF inhibitor naive. The primary outcome measure was again the ACR20 at 16 weeks which was achieved by 19.4%, 31.3%, and 41% of the placebo, 20 and 30 mg bd groups, respectively. At 24 weeks the corresponding figures for per protocol treatment (i.e. those still taking placebo) were ACR20 of 13%, 36%, and 45%. Patients on placebo had the chance to re-randomise to active drug at 16 weeks and a long-term extension for all patients is underway. As expected, patients who had previously taken biologics had less impressive responses, the ACR20 rates for the 20 and 30 mg bd groups at 16 weeks being 31% and 28%, respectively. Those taking disease modifying drugs (mostly methotrexate) had rather blunted responses (ACR20 rates of 31% and 35% for 20 and 30 mg bd, respectively). Skin responses were also reported: in patients with a skin surface area of greater than 3% at baseline the PASI75 rates at week 24 were 5%, 18% and 21% for placebo, 20 mg bd and 30 mg bd, respectively. Serious adverse events were rare and, again, adverse events were mainly gastrointestinal (diarrhoea and nausea) and headache, but a small increase in upper respiratory infections was also seen [72].
Discussion
In summary PDE4 inhibitors are orally active agents with a good short-term safety that have therapeutic possibilities in a variety of inflammatory disorders. In psoriasis, apremilast has moderate efficacy in psoriasis and the associated psoriatic arthritis. What is the likely use of this drug in clinical practice? It is worth considering the current treatment algorithms in use in this disease. Psoriasis and psoriatic arthritis will be considered separately and then as a combined approach.
From the data available so far PDE4 inhibitors such as apremilast may be a valuable addition to the psoriasis treatment portfolio. Their place may be similar to fumaric acid and methotrexate as systemic monotherapy in mild to moderate psoriasis not sufficiently responsive to topical glucocorticoids and vitamin D derivatives. Apremilast shares functional properties with fumaric acid with respect to suppression of IL-12, IL-23, and TNF. Although drugs such as apremilast seem to have a favourable side effect profile, both direct comparison with other drugs and long-term studies are needed to complete the picture. Apremilast may also have an advantage in women of child-bearing potential in whom acitretin (and to a certain extent methotrexate) is contra-indicated. It might also be worth noting that PDE4 inhibitors could have a beneficial effect on depressive disorders, a common finding in patients with moderate to severe psoriasis and psoriatic arthritis. PDE4 inhibitors appear less effective than TNFi in psoriasis and they are also probably less effective than CsA. However, combination therapy with other immunomodulators may be an attractive proposition both to reduce the dose of the other immunomodulator and to reduce the side effects of PDE4 inhibition. Drugs such as apremilast may also be used as maintenance therapy once remission has been induced by another drug and it may help prevent relapses often seen after withdrawal of, for example, CsA. There are as yet no data on the safety profile of PDE inhibitors with UV therapy but it would be assumed that their safety profile would be favourable when compared to drugs such as methotrexate and CsA. In conclusion, from a purely cutaneous perspective PDE4 inhibition is probably similar to treatment with fumaric acid and methotrexate, and probably less effective than cyclosporin and TNF inhibitors. Combination therapy may be the way forward and novel applications such as the topical route need exploration.
Psoriatic arthritis is a heterogeneous disease with diverse clinical manifestations. From a rheumatic point of view it is appropriate to consider the condition as peripheral and axial arthritis [73]. Peripheral arthritis can be considered as either oligoarticular (less than 4 joints) or polyarticular, although it should be accepted that this division is somewhat arbitrary. There is little other data to support the split and, by use of sophisticated imaging techniques, many cases of oligoarthritis are found to be polyarticular. For this reason it is difficult to design a single treatment algorithm to cover all aspects of the disease. The situation is complicated by the lack of evidence supporting the use of many of the so called ‘disease modifying drugs’ for use in psoriatic arthritis. Indeed, the drug that is the mainstay of treatment of psoriatic arthritis and the one that most rheumatologists first turn at disease onset, methotrexate, has little support from randomized controlled trials [74]. Further, methotrexate has no efficacy on the axial disease [75]. Nevertheless, there is sufficient evidence from both observational studies [76], uncontrolled trials [77] and physicians own experience for methotrexate to maintain a pivotal role in the treatment of peripheral psoriatic arthritis. Methotrexate is not without problems: patients often complain of nausea, hair thinning, and both physicians and patients worry about hepatotoxicity, particularly in the overweight patients and those who consume moderate amounts of alcohol. If methotrexate fails many physicians will be looking to use TNFi, particularly, if there are adverse prognostic factors. However, many European countries advise the use of a second agent, such as sulfasalazine or leflunomide, before moving onto biologics.
Given this scenario how will apremilast fit into such an algorithm? Although no head to head trials have been conducted, from an efficacy point of view it is likely that apremilast is less effective than TNFi in the treatment of both axial and peripheral arthritis. From the point of view of peripheral arthritis an ACR20 rate of 41% at 12 weeks does not compare well with TNFi (59% for etanercept, and 58% for adalimumab, for example [78, 79]) although the data currently available suggest that more patients will achieve ACR20 with continued exposure to apremilast. It is difficult to compare the ACR rates with methotrexate although the RESPOND [77] study, which was effectively open label, found an ACR20 rate of 67% at 16 weeks. The Methotrexate In Psoriatic Arthritis (MIPA) study, in which doses of methotrexate were modest, absolute rates of achieving ACR20 were 34% and 21% for methotrexate and placebo, respectively, a difference that was not statistically significant. Taken together these two studies probably overestimate (RESPOND) and underestimate (MIPA) the effect of methotrexate on psoriatic arthritis.
Apremilast also has efficacy in the cutaneous component of the disease, and, unlike methotrexate, may have efficacy in the axial component, present in about 40% of cases of psoriatic arthritis. It is also worth noting that there were no safety concerns of hepatotoxicity in the short-term studies with apremilast so this might confer advantages over methotrexate if a physician was considering treatment in a patient with risk factors for liver disease. However, it is difficult to see physicians making major changes to their prescribing habits given the current lack of clear cut evidence for superiority of apremilast and the concerns about the initial gastrointestinal tolerability issues. Long-term familiarity and safety concerns will also play a part in prescribing patterns. And finally, the cost at which the drug is marketed will have a major impact on its position in the prescribing hierarchy, particularly in cash strapped economies and countries with ‘guidance’ mechanisms in place. Will apremilast be positioned after TNFi in psoriatic arthritis? This seems unlikely although it is possible to envisage a scenario where a patient may have failed a TNFi, for whatever reason, and be offered another oral therapy for their disease, although it would have to be made clear that improvement rates after failure of a TNFi are only moderate. It is worth remembering though that achieving an ACR20, although the yardstick by which drugs have been tested of late in psoriatic arthritis, is not a very good result for a patient—ACR rates of 50 or 70 are needed for the patient to feel there has been real improvement in their condition and these rates were disappointingly low for apremilast.
Conclusion
PDE4 inhibitors are a class of drugs which act intracellularly to down regulate inflammatory pathways and to promote innate anti-inflammatory pathways. They have a potentially wide range of therapeutic uses in chronic inflammatory diseases. In particular, apremilast has already proven effective in psoriasis and the peripheral arthritis of psoriatic arthritis. Efficacy in psoriasis is probably equivalent to methotrexate but less than TNFi. In psoriatic arthritis efficacy is probably similar to methotrexate but less than TNFi. Apremilast appears to have a good safety profile and this, together with the oral dosing are likely to be major factors in the decision to use the drug. However, much will depend on the cost and long-term tolerability and safety.
References
Schafer P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem Pharmacol. 2012;83:1583–90.
Cassel D, Pfeuffer T. Mechanism of cholera toxin action: covalent modification of the guanyl nucleotide-binding protein of the adenylate cyclase system. Proc Natl Acad Sci USA. 1978;75:2669–73.
Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J Immunol. 2010;185:6413–9.
Jeyaraj SC, Unger NT, Eid AH, Mitra S, Paul El-Dahdah N, Quilliam LA, Flavahan NA, Chotani MA. Cyclic AMP-Rap1A signaling activates RhoA to induce α(2c)-adrenoceptor translocation to the cell surface of microvascular smooth muscle cells. Am J Physiol Cell Physiol. 2012;303:C499–511.
Zieba BJ, Artamonov MV, Jin L, et al. The cAMP-responsive Rap1 guanine nucleotide exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity. J Biol Chem. 2011;286:16681–92.
Stokman G, Qin Y, Genieser HG, et al. Epac-Rap signaling reduces cellular stress and ischemia-induced kidney failure. J Am Soc Nephrol. 2011;22:859–72.
Kim JG, Moon MY, Kim HJ, et al. Ras-related GTPases Rap1 and RhoA collectively induce the phagocytosis of serum-opsonized zymosan particles in macrophages. J Biol Chem. 2012;287:5145–55.
la Sala A, He J, Laricchia-Robbio L, et al. Cholera toxin inhibits IL-12 production and CD8alpha+ dendritic cell differentiation by cAMP-mediated inhibition of IRF8 function. J Exp Med. 2009;206:1227–35.
Coccia EM, Remoli ME, Di Giacinto C, et al. Cholera toxin subunit B inhibits IL-12 and IFN-{gamma} production and signaling in experimental colitis and Crohn’s disease. Gut. 2005;54:1558–64.
Lavelle EC, Jarnicki A, McNeela E, et al. Effects of cholera toxin on innate and adaptive immunity and its application as an immunomodulatory agent. J Leukoc Biol. 2004;75:756–63.
Burkart V, Kim YE, Hartmann B, et al. Cholera toxin B pretreatment of macrophages and monocytes diminishes their proinflammatory responsiveness to lipopolysaccharide. J Immunol. 2002;168:1730–7.
Boirivant M, Fuss IJ, Ferroni L, De Pascale M, Strober W. Oral administration of recombinant cholera toxin subunit B inhibits IL-12-mediated murine experimental (trinitrobenzene sulfonic acid) colitis. J Immunol. 2001;166:3522–32.
Cong Y, Oliver AO, Elson CO. Effects of cholera toxin on macrophage production of co-stimulatory cytokines. Eur J Immunol. 2001;31:64–71.
Braun MC, He J, Wu CY, Kelsall BL. Cholera toxin suppresses interleukin (IL)-12 production and IL-12 receptor beta1 and beta2 chain expression. J Exp Med. 1999;189:541–52.
Gschwandtner M, Bunk H, Köther B, et al. Histamine down-regulates IL-27 production in antigen-presenting cells. J Leukoc Biol. 2012;92:21–9.
Gutzmer R, Diestel C, Mommert S, et al. Histamine H4 receptor stimulation suppresses IL-12p70 production and mediates chemotaxis in human monocyte-derived dendritic cells. J Immunol. 2005;174:5224–32.
Caron G, Delneste Y, Roelandts E, et al. Histamine polarizes human dendritic cells into Th2 cell-promoting effector dendritic cells. J Immunol. 2001;167:3682–6.
van der Pouw Kraan TC, Snijders A, Boeije LC, et al. Histamine inhibits the production of interleukin-12 through interaction with H2 receptors. J Clin Invest. 1998;102:1866–73.
Elenkov IJ, Webster E, Papanicolaou DA, et al. Histamine potently suppresses human IL-12 and stimulates IL-10 production via H2 receptors. J Immunol. 1998;161:2586–93.
Wu CY, Wang K, McDyer JF, Seder RA. Prostaglandin E2 and dexamethasone inhibit IL-12 receptor expression and IL-12 responsiveness. J Immunol. 1998;161:2723–30.
Bopp T, Becker C, Klein M, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med. 2007;204:1303–10.
Klein M, Vaeth M, Scheel T, et al. Repression of cyclic adenosine monophosphate upregulation disarms and expands human regulatory T cells. J Immunol. 2012;188:1091–7.
Bodor J, Bopp T, Vaeth M, et al. Cyclic AMP underpins suppression by regulatory T cells. Eur J Immunol. 2012;42:1375–84.
Bodor J, Habener JF. Role of transcriptional repressor ICER in cyclic AMP-mediated attenuation of cytokine gene expression in human thymocytes. J Biol Chem. 1998;273:9544–51.
Gerlo S, Kooijman R, Beck IM, et al. Cyclic AMP: a selective modulator of NF-kappaB action. Cell Mol Life Sci. 2011;68:3823–41.
Baumer W, Kietzmann M. Effects of cyclosporin A and cilomilast on activated canine, murine and human keratinocytes. Vet Dermatol. 2007;18:107–14.
Qi XF, Kim DH, Yoon YS, et al. The adenylyl cyclase-cAMP system suppresses TARC/CCL17 and MDC/CCL22 production through p38 MAPK and NF-kappaB in HaCaT keratinocytes. Mol Immunol. 2009;46:1925–34.
Torphy TJ. Phosphodiesterase isozymes: molecular targets for novel antiasthma agents. Am J Respir Crit Care Med. 1998;157:351–70.
Jin SL, Ding SL, Lin SC. Phosphodiesterase 4 and its inhibitors in inflammatory diseases. Chang Gung Med J. 2012;35:197–210.
Page CP, Spina D. Selective PDE inhibitors as novel treatments for respiratory diseases. Curr Opin Pharmacol. 2012;12:275–86.
Hatzelmann A, Schudt C. Anti-inflammatory and immunomodulatory potential of the novel PDE4 inhibitor roflumilast in vitro. J Pharmacol Exp Ther. 2001;297:267–79.
Smith SJ, Brookes-Fazakerley S, Donnelly LE, et al. Ubiquitous expression of phosphodiesterase 7A in human proinflammatory and immune cells. Am J Physiol Lung Cell Mol Physiol. 2003;284:L279–89.
Milara J, Navarro A, Almudéver P, Lluch J, Morcillo EJ, Cortijo J. Oxidative stress-induced glucocorticoid resistance is prevented by dual PDE3/PDE4 inhibition in human alveolar macrophages. Clin Exp Allergy. 2011;41:535–46.
Seybold J, Newton R, Wright L, et al. Induction of phosphodiesterases 3B, 4A4, 4D1, 4D2, and 4D3 in Jurkat T-cells and in human peripheral blood T-lymphocytes by 8-bromo-cAMP and Gs-coupled receptor agonists. Potential role in beta2-adrenoreceptor desensitization. J Biol Chem. 1998;273:20575–88.
Peter D, Jin SL, Conti M, Hatzelmann A, Zitt C. Differential expression and function of phosphodiesterase 4 (PDE4) subtypes in human primary CD4+ T cells: predominant role of PDE4D. J Immunol. 2007;178:4820–31.
Ma D, Wu P, Egan RW, Billah MM, Wang P. Phosphodiesterase 4B gene transcription is activated by lipopolysaccharide and inhibited by interleukin-10 in human monocytes. Mol Pharmacol. 1999;55:50–7.
Magela Magalhaes G, Coelho da Silva Carneiro S, Peisinodo Amaral K, et al. Psoriasis and pentoxifylline: a clinical, histopathologic, and immunohistochemical evaluation. Skinmed. 2006;5:278–84.
Singh SK, Dimri U, Saxena SK, Jadhav RK. Therapeutic management of canine atopic dermatitis by combination of pentoxifylline and PUFAs. J Vet Pharmacol Ther. 2010;33:495–8.
Park MK, Fontana Jr, Babaali H, et al. Steroid-sparing effects of pentoxifylline in pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2009;26:121–31.
Boswell-Smith V, Cazzola M, Page CP. Are phosphodiesterase 4 inhibitors just more theophylline? J Allergy Clin Immunol. 2006;117:1237–43.
Spina D, Landells LJ, Page CP. The role of theophylline and phosphodiesterase4 isoenzyme inhibitors as anti-inflammatory drugs. Clin Exp Allergy. 1998;28:24–34.
Robichaud A, Stamatiou PB, Jin SL, et al. Deletion of phosphodiesterase 4D in mice shortens alpha(2)-adrenoceptor-mediated anesthesia, a behavioral correlate of emesis. J Clin Invest. 2002;110:1045–52.
Kagayama K, Morimoto T, Nagata S, et al. Synthesis and biological evaluation of novel phthalazinone derivatives as topically active phosphodiesterase 4 inhibitors. Bioorg Med Chem. 2009;17:6959–70.
Nazarian R, Weinberg JM. AN-2728, a PDE4 inhibitor for the potential topical treatment of psoriasis and atopic dermatitis. Curr Opin Investig Drugs. 2009;10:1236–42.
Akama T, Baker SJ, Zhang YK, et al. Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis. Bioorg Med Chem Lett. 2009;19:2129–32.
Michalski JM, Golden G, Ikari J, Rennard SI. PDE4: a novel target in the treatment of chronic obstructive pulmonary disease. Clin Pharmacol Ther. 2012;91:134–42.
Fabbri LM, Beghe B, Yasothan U, Kirkpatrick P. Roflumilast. Nat Rev Drug Discov. 2010;9:761–2.
Man HW, Schafer P, Wong LM, et al. Discovery of (S)-N-[2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihy dro-1H-isoindol-4-yl] acetamide (apremilast), a potent and orally active phosphodiesterase 4 and tumor necrosis factor-alpha inhibitor. J Med Chem. 2009;52:1522–4.
Crilly A, Robertson SE, Reilly JH, et al. Phosphodiesterase 4 (PDE4) regulation of proinflammatory cytokine and chemokine release from rheumatoid synovial membrane. Ann Rheum Dis. 2011;70:1130–7.
McCann FE, Palfreeman AC, Andrews M, et al. Apremilast, a novel PDE4 inhibitor, inhibits spontaneous production of tumour necrosis factor-alpha from human rheumatoid synovial cells and ameliorates experimental arthritis. Arthritis Res Ther. 2010;12:R107.
Jin SL, Lan L, Zoudilova M, Conti M. Specific role of phosphodiesterase 4B in lipopolysaccharide-induced signaling in mouse macrophages. J Immunol. 2005;175:1523–31.
Jin SL, Conti M. Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-alpha responses. Proc Natl Acad Sci USA. 2002;99:7628–33.
Schafer PH, Parton A, Gandhi AK, et al. Apremilast, a cAMP phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br J Pharmacol. 2010;159:842–55.
Baumer W, Hoppmann J, Rundfeldt C, Kietzmann M. Highly selective phosphodiesterase 4 inhibitors for the treatment of allergic skin diseases and psoriasis. Inflamm Allergy Drug Targets. 2007;6:17–26.
Kadoshima-Yamaoka K, Goto M, Murakawa M, et al. ASB16165, a phosphodiesterase 7A inhibitor, reduces cutaneous TNF-alpha level and ameliorates skin edema in phorbol ester 12-O-tetradecanoylphorbol-13-acetate-induced skin inflammation model in mice. Eur J Pharmacol. 2009;613:163–6.
Baughman RP, Judson MA, Ingledue R, Craft NL, Lower EE. Efficacy and safety of apremilast in chronic cutaneous sarcoidosis. Arch Dermatol. 2012;148:262–4.
Keravis T, Monneaux F, Yougbaré I, et al. Disease progression in MRL/lpr lupus-prone mice is reduced by NCS 613, a specific cyclic nucleotide phosphodiesterase type 4 (PDE4) inhibitor. PLoS ONE. 2012;7:e28899.
De Souza A, Strober BE, Merola JF, Oliver S, Franks AG. Apremilast for discoid lupus erythematosus: results of a phase 2, open-label, single-arm, pilot study. J Drugs Dermatol. 2012;11:1224–6.
Shenoy PD, Kumar S, Jha LK, et al. Efficacy of tadalafil in secondary Raynaud’s phenomenon resistant to vasodilator therapy: a double-blind randomized cross-over trial. Rheumatology (Oxford). 2010;49:2420–8.
Alase A, Wittmann M. Therapeutic strategies in allergic contact dermatitis. Recent Pat Inflamm Allergy Drug Discov. 2012;6:210–21.
Martin SF. Contact dermatitis: from pathomechanisms to immunotoxicology. Exp Dermatol. 2012;21:382–9.
Martin SF, Esser PR, Weber FC, et al. Mechanisms of chemical-induced innate immunity in allergic contact dermatitis. Allergy. 2011;66:1152–63.
Martin SF, Dudda JC, Bachtanian E, et al. Toll-like receptor and IL-12 signaling control susceptibility to contact hypersensitivity. J Exp Med. 2008;205:2151–62.
Samrao A, Berry TM, Goreshi R, Simpson EL. A pilot study of an oral phosphodiesterase inhibitor (apremilast) for atopic dermatitis in adults. Arch Dermatol. 2012;148:890–7.
Volf EM, Au SC, Dumont N, Scheinman P. Gottlieb, A.B. A phase 2, open-label, investigator-initiated study to evaluate the safety and efficacy of apremilast in subjects with recalcitrant allergic contact or atopic dermatitis. J Drugs Dermatol. 2012;11:341–6.
Yamaki K, Li X, Uchida H, et al. Effects of the phosphodiesterase IV inhibitor rolipram on Th1 and Th2 immune responses in mice. J Pharm Pharmacol. 2004;56:877–82.
Dehzad N, Bopp T, Reuter S, et al. Regulatory T cells more effectively suppress Th1-induced airway inflammation compared with Th2. J Immunol. 2011;186:2238–44.
Werfel T, Wittmann M. Regulatory role of T lymphocytes in atopic dermatitis. Chem Immunol Allergy. 2008;94:101–11.
Gottlieb AB, Strober B, Krueger JG, et al. An open-label, single-arm pilot study in patients with severe plaque-type psoriasis treated with an oral anti-inflammatory agent, apremilast. Curr Med Res Opin. 2008;24:1529–38.
Papp K, Cather JC, Rosoph L, et al. Efficacy of apremilast in the treatment of moderate to severe psoriasis: a randomised controlled trial. Lancet. 2012;380:738–46.
Schett G, Wollenhaupt J, Papp K, et al. Oral apremilast in the treatment of active psoriatic arthritis. Arthritis Rheum. 2012;64:12.
Kavanaugh A, Mease PJ, Gomez-Reino JJ, et al. et al. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis: results of a phase 3, randomised, controlled trial. Paper presented at: American College of Rheumatology, annual meeting, vol. 64 supplement. Arthritis and Rheumatism, Washington DC; 2012.
Taylor WJ, Zmierczak HG, Helliwell PS. Problems with the definition of axial and peripheral disease patterns in psoriatic arthritis. J Rheumatol. 2005;32:974–7.
Kingsley GH, Kowalczyk A, Taylor H, et al. Methotrexate is not disease modifying in psoriatic arthritis: the MIPA trial. Rheumatology. 2012;51:1368–77.
Haibel H, Sieper J. Use of methotrexate in patients with ankylosing spondylitis. Clin Exp Rheumatol. 2010;28:S128–31.
Chandran V, Schentag CT, Gladman DD. Reappraisal of the effectiveness of methotrexate in psoriatic arthritis: results from a longitudinal observational cohort. J Rheumatol. 2008;35:469–71.
Baranauskaite A, Raffayová H, Kungurov NV, et al. Infliximab plus methotrexate is superior to methotrexate alone in the treatment of psoriatic arthritis in methotrexate naive patients: the RESPOND study. Ann Rheum Dis. 2012;71:541–8.
Mease PJ, Kivitz AJ, Burch FX, et al. Etanercept treatment of psoriatic arthritis: safety, efficacy, and effect on disease progression. Arthritis Rheum. 2004;50:2264–72.
Mease PJ, Gladman DD, Ritchlin CT, Adalimumab Effectiveness in Psoriatic Arthritis Trial Study Group, et al. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005;52:3279–89.
Acknowledgments
Prior to peer review Celgene were offered the opportunity to review this paper for scientific accuracy. No writing assistance, other editorial involvement, or financial support was provided by the manufacturer in the production of this manuscript. This article does not necessarily reflect the opinions, policies, or recommendations of Celgene or any of its employees. Dr. Helliwell is the guarantor for this article, and takes responsibility for the integrity of the work as a whole.
Conflict of interest
Philip Helliwell has received honoraria from Celgene. Miriam Wittmann declares no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wittmann, M., Helliwell, P.S. Phosphodiesterase 4 Inhibition in the Treatment of Psoriasis, Psoriatic Arthritis and Other Chronic Inflammatory Diseases. Dermatol Ther (Heidelb) 3, 1–15 (2013). https://doi.org/10.1007/s13555-013-0023-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-013-0023-0