
Abstract
Skeletal muscle innervation is a multi-step process leading to the neuromuscular junction (NMJ) apparatus formation. The transmission of the signal from nerve to muscle occurs at the NMJ level. The molecular mechanism that orchestrates the organization and functioning of synapses is highly complex, and it has not been completely elucidated so far. Neuromuscular junctions are assembled on the muscle fibers at very precise locations called end plates (EP). Acetylcholine receptor (AChR) clusterization at the end plates is required for an accurate synaptic transmission. This review will focus on some mechanisms responsible for accomplishing the correct distribution of AChRs at the synapses. Recent evidences support the concept that a dual transcriptional control of AChR genes in subsynaptic and extrasynaptic nuclei is crucial for AChR clusterization. Moreover, new players have been discovered in the agrin–MuSK pathway, the master organizer of postsynaptical differentiation. Mutations in this pathway cause neuromuscular congenital disorders. Alterations of the postynaptic apparatus are also present in physiological conditions characterized by skeletal muscle wasting. Indeed, recent evidences demonstrate how NMJ misfunctioning has a crucial role at the onset of age-associated sarcopenia.
Similar content being viewed by others


1 Introduction
Motor neuron innervation is the most relevant maturation event in skeletal muscle development since muscle physiology, also accounting for locomotion and breath, depends on it. Innervation implies sophisticated interactions between motor neuron axons and end plates on the muscle fibers, thus resulting in a development of highly elaborated synapses called neuromuscular junctions (NMJs). As soon as the action potential reaches the axon terminals, it induces the opening of the voltage-gated Ca2+ channels on the presynaptic nerve membrane. This allows a Ca2+ influx that induces synaptic vescicles to fuse with the presynaptic membrane and to release the neurotransmitter acetylcholine (ACh) in the synaptic cleft. From here, ACh diffuses and binds to acetylcoline receptors (AChRs) localized on the postsynaptic membrane on the muscle fiber. This binding makes AChRs permeable to both Na+ and K+ and opens the associated voltage-gated Na+ channels on the muscle membrane which, in turn, initiate an action potential causing Ca2+ release from the sarcoplasmic reticulum into the cytosol and muscle contraction. Acetylcholinesterase, located on the synaptic portion of the basal lamina that envelops muscle fibers, quickly inactivates ACh released from the presynaptic membrane so that the ACh concentration in the synaptic cleft decreases rapidly and neurotransmission stops. The correct nerve–muscle impulse transmission requires an intricate network of interacting signaling pathways displaying a certain rate of redundancy, which is, however, necessary to ensure the process’s spatial and temporal accuracy.
Defects in the signaling pathways that regulate NMJ differentiation and functioning lead to a variety of congenital neuromuscular disorders termed congenital myasthenic syndromes (CMS), typified by muscle weakness and fatigue [1]. Loss of the nerve signaling caused by the degeneration of motor neurons leads to a debilitating loss of muscle mass, atrophy, and paralysis. Also, aging and diseases such as cancer, AIDS, and chronic heart failure are physical conditions in which NMJ normal activity, muscle mass, and function are highly compromised. For these reasons, understanding the molecular basis of neuromuscular synapse functioning is fundamental to such disorders’ therapeutic approach. This review will focus on the development of the postsynaptic apparatus and will highlight the role of some defects of this process which are at the onset of neuromuscular disease and muscle wasting-associated pathologies.
2 NMJ formation
NMJs do not develop at random locations in muscles; rather, they are assembled in a narrow central region of the muscle fiber, so that many NMJs are located in a row, forming an end plate across the fiber. Each NMJ consists of an area of apposition between a motoneuron axon branch and a single multinucleated muscle fiber. AChRs localize on the postsynaptic muscle membrane, and their spatial distribution is critical for synaptic function. A high density of AChRs at synapses is required to initiate a synaptic action potential in the myofiber. Vice versa, around synapses and in the rest of the fiber, the density of AChRs has to be kept low in order to allow a complete maturation of the NMJ. When myoblasts fuse to form myotubes, AChR subunits are assembled into the membrane at a very low density (1,000 μm−2). In mature synapses, AChRs accumulate at a density >10,000 μm−2directly beneath the motor nerve terminal while they drop to <10 μm−2 in the extrasynaptic membrane [2, 3].
A puzzling question about the developing postsynaptic structure is whether the motoneurons or the muscle fibers determine where and how the NMJs are formed. The motor nerve might initiate AChR cluster formation (neurocentric model) or clusters might form aneurally and then recognized by the nerve (myocentric model) [4]. Some studies indicate that the primitive AChR clusterization is nerve independent, while the nerve induces only maturation and enlargement of some of the primitive aneural AChR clusters. During early mouse embryogenesis, between embryonic day 12.5 (E12.5) and E14.5, aneural muscle fibers begin spontaneously to accumulate AChRs in a central region where innervation will occur (prepatterning) [5–9]. This phenomenon also occurs in mutant mice where motor axons fail to contact skeletal muscle [10]. Following such nerve-independent AChR clustering, nerve terminals overlap some prepatterned AChR clusters and, at E18.5, the innervated clusters are enlarged, whereas the other primitive clusters disappear in synaptic and extrasynaptic regions [8, 9]. Conversely, other studies in cultured myotubes have indicated that motor neurons induce postsynaptic differentiation ignoring preexisting aneural clusters [11, 12]. It has also been recently hypothesized that some already formed AChR clusters are recognized by the nerve on muscle fibers, while others are induced by motor axons [13]. It is also possible that the myogenic and the neurogenic component role might depend on the species and on the developmental context, although the topic keeps being highly controversial [14].
3 NMJ maturation and AChR clusterization
The mechanisms that lead to the clusterization of ACh receptors within the postsynaptic membrane require a precise interaction of signals among motoneurons and skeletal muscle fibers and occur through different modalities. It has been described that AChRs move from the extrasynaptic region and are trapped to the synaptic pool through a rapid lateral diffusion [2]. Clustering also induces a higher stability of the AChRs; at early stages of development, the half-life of junctional non-clustered AChRs is about 1 day, whereas in adult, is increased to 8–14 days [15]. Moreover, transcriptional regulation has a pivotal role in AChR clusterization. Nuclei that are associated at the postsynaptic membrane (synaptic nuclei) actively transcribe the AChR subunit genes and other postsynaptic component genes at a higher rate than the non-synaptic nuclei. This leads to a localized synthesis and accumulation of AChRs [16–19]. The formation of AChR clusters depends not only on positive signals that enhance AChR concentration in the synaptic area but also on negative signals necessary to decrease AChR concentration outside the synaptic area, along the entire fiber.
3.1 Positive signaling
The best characterized signaling responsible for AChR accumulation at the NMJs is the agrin/lipoprotein receptor-related protein 4 (Lrp4)/muscle-specific tyrosine kinase receptor (MuSK) pathway. Agrin is a large heparan sulfate proteoglycan first isolated from the Torpedo electric organ [20] synthesized in the motoneurons, transported along axons, and released in the synaptic basal lamina, which surrounds the muscle fiber [21]. Agrin is necessary for clustering AChRs at synaptic sites (Fig. 1a). Agrin is sufficient to induce AChR ectopic clusters in adult muscle and postsynaptic specialization in denervated fibers [22, 23]. Although it is not essential for prepatterning, in agrin−/−mice, NMJs and AChRs are uniformly distributed in the muscle fiber and not clustered [8, 9, 24], and postsynaptic differentiation is inhibited.

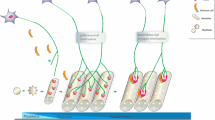
The agrin–MuSK–Lrp4 and neuregulin–ErbB pathways induce NMJ assembly positive signals. a Agrin is released by the motor axon terminal and induces AChR clustering, phosphorylation, and stabilization at the postsynaptic membrane. Lrp4 associates with MuSK in the absence of agrin. Agrin binds to the preformed MuSK–Lrp4 complex by interacting with Lrp4 and promotes MuSK transphosphorylation and activation. Once phosphorylated, MuSK recruits the adapter protein Dok-7 which binds Crk and CrkL and stimulates further MuSK phosphorylation and kinase activity. This induces phosphorylation and stabilization of nascent AChR clusters. Rapsyn is a coeffector in AChR assembly which anchorates AChRs at the muscle membrane. b Neuregulin (NGR-1) is released by the nerve and induces AChR transcription in synaptic nuclei. NRG-1 acts by binding tyrosine kinases receptors ErbBs. ErbB phosphorylation induced by NRG stimulates ERK and JNK kinase activity which phosphorylates GABP-α and GABP-β transcription factors. GABP-α heterodimerizes with GABP-β and binds DNA at the N-box thereby enhancing transcription of AChR genes
Agrin acts by activating another important player in the NMJ assembly pathway, MuSK. MuSK is a tyrosine kinase receptor expressed in the postsynaptic membrane of NMJs [25] where it co-localizes with AChRs [26]. The main role of agrin is to initiate MuSK autophosphorylation and activation [27] (Fig. 1a). However, MuSK can also be activated in an agrin-independent manner [28]. Indeed, in MuSK−/−fibers, no NMJs are formed, and also, prepatterning before innervation is absent [8, 9, 29]. Like AChR, MuSK gene is a target for the transcription factor myogenin (see below). Myogenin mediates MuSK upregulation during muscle fiber denervation and MuSK downregulation upon innervation [30]. However, upon innervation, in the synaptic nuclei, MuSK is upregulated by different transcriptional pathways.
Although agrin stimulates MuSK phosphorylation, the two proteins do not interact directly, but additional proteins (coreceptors) are engaged to allow agrin-mediated signaling in myotubes [31, 32]. A recently discovered agrin coreceptor is the transmembrane protein Lrp4 [33]. Lrp4 is concentrated at the NMJs and is necessary for agrin-stimulated MuSK phosphorylation and AChR clustering. According to this model, Lrp4 self-associates and interacts with MuSK in the absence of agrin. When agrin binds to this preformed complex, it triggers a reorganization of MuSK, so promoting its transphophorylation and kinase activity. Once phopshorylated, MuSK activates signaling pathways that lead to synaptic differentiation including clustering of AChRs [34, 35] (Fig. 1a). Agrin activation of MuSK also leads to concentration at synapses of other proteins such as acetylcholinesterase (AChE), rapsyn, and neuregulin (NRG) receptors (ErbBs; see below). Moreover, Lrp4 has a crucial role in muscle prepatterning since it promotes MuSK phosphorylation and activation in the absence of agrin [36].
Once phosphorylated, MuSK recruits docking protein-7 (Dok-7), a non-catalytic adapter protein, which stimulates further MuSK phosphorylation and increases MuSK kinase activity (Fig. 1a). To properly regulate NMJ formation, Dok-7 engages at two phosphorylation sites two adapter proteins, Crk and CrkL. In mouse models lacking Dok-7, Crk, and CrkL, neuromuscular synapse fails to form [37–40].
A coeffector in AChR assembly is rapsyn, a membrane protein associated with the cytoplasmic side of the plasma membrane (Fig. 1a). Rapsyn anchorates AChRs at the muscle membrane from the earliest stages of synaptogenesis, including prepatterning [41]. Rapsyn also interacts with cytoskeletal proteins, thus being essential for AChR clustering. In mice lacking rapsyn, AChRs fail to aggregate [42]. It has been proposed that MuSK induces phosphorylation of nascent AChR clusters and promotes also the binding of additional rapsyn to AChR aggregates, which contributes to AChR cluster growth and stability. Rapsyn may function as a scaffolding protein presenting a MuSK-activated Src-related kinase to the AChRs [43].
Upon phosphorylation and activation, MuSK interacts with a large number of effectors and pathways converging toward the regulation of AChR clusterization through different mechanisms. MuSK indeed controls (1) AChR scaffolding and redistribution via actin cytoskeleton reorganization, (2) AChR stabilization through AChR phosphorylation, (3) its proper endocytosis and turnover are also important for AChR clustering, and (4) enhancement of transcription of AChR genes selectively in subsynaptic nuclei (reviewed in [14]).
AChR subunits are selectively transcribed in the synaptic nuclei of myofibers. mRNAs encoding other synaptic proteins such as AChE, MuSK, and rapsyn are also concentrated at the synapses. The pathway activating AChR transcription in the subsynaptic regions still remains unclear. One candidate activator is neuregulin-1 (NGR-1), a glycoprotein which works as an extracellular signal that stimulates AChR and other synapse-specific components transcription. NRG-1 acts by binding to membrane-associated tyrosine kinase receptors related to the EGF receptor (erbB proteins 2-3-4) [44] (Fig. 1b). In vivo studies on NGR-1 role in NMJ differentiation are difficult because nrg-1−/−embryos die far before NMJs start forming. Heterozygous mice are viable and fertile and show a decreased number of AChRs at the NMJs [45, 46]. However, some recent in vivo studies, in which neuregulin-1 and erbB proteins were conditionally deleted, demonstrated that neuregulin signaling is dispensable [47, 48]. For this reason, the idea that NRG-1 is the main signal responsible for the increased transcription of AChRs is highly controversial. Recent evidences also indicate that neuregulin may have a role in AChR clustering and trafficking by modulating rather than determining AChR expression at the NMJs [49].
The pathways leading from NRG-1–ErbB binding to regulation of AChR gene transcription have been identified. Both in cultured muscle cells and in vivo, ErbB phosphorylation by NRG-1 concomitantly stimulates ERK [50, 51] and the c-JUN N-terminal kinase (JNK) activity [52, 53]. The activation of these pathways results in increased levels and phosphorylation of the E-twenty-six (ETS)-domain-binding transcription factor GA-binding protein (GABP)-α which binds DNA, heterodimerizes with GABP-β, and increases AChR transcription [54, 55] (Fig. 1b). ETS transcription factors bind DNA at the N-box, a DNA element located not only on the AChR promoter but also on the promotor of other NMJ components (utrophin and acelylcholinesterase) [56–58]. In vivo inhibition of GABP results in reduced expression of AChRs [59] indicating the GABP signaling pathway, activated directly or indirectly by neuregulin, is necessary for the formation of functional synapses. As stated before, the agrin–MuSK pathway also contributes to increase synaptic AChR gene transcription by a NRG/ErbB-independent but Rac-dependent JNK activation (Fig. 1b) and also by interacting with the NRG/ErbB pathway [43, 60, 61].
3.2 Negative signals
The positive signals we described enhance AChR accumulation only in the synaptic area. To achieve a correct formation of AChR clusters in the NMJs, the presence of some negative signals that decrease AChR concentration in the rest of the fiber, in non-synaptic areas, is necessary.
Muscle depolarization induced by released ACh is a negative signal for AChRs. It inhibits AChR localization, stability [62], and transcription along the muscle fiber by stimulation of cyclin-dependent kinase 5 [63, 64], protein kinase C (PKC), and Ca2+/calmodulin-dependent kinase II (CaMKII) [65]. Indeed, mutant mice lacking acetyltransferase, a biosynthetic enzyme for ACh, develop faster and larger AChR clusters; they also exhibit hyperinnervation due to a broader distribution of AChR clusters along the muscle fiber [66, 67].
In detail, through muscle action potential, ACh induces downregulation of AChR transcription along the entire fiber. However, since the previously described positive signals enhance AChR accumulation only in the synaptic area (Fig. 1), this results in a ACh-dependent suppression of AChR subunit gene transcription in extrasynaptic nuclei (Fig. 2) [68]. Suppression of AChR transcription occurs through myogenin inhibition. Myogenin is a basic helix-loop-helix myogenic transcription factor that activates AChR genes in the absence of innervation or by blockade of muscle electrical activity by denervation [69]. Myogenin regulates AChR and many other muscle gene transcription by binding the E-boxes located on their promoter/enhancers. Myogenin is downregulated as the muscle matures around the time when innervation is completed, and it is upregulated upon denervation [70]. Forced expression of myogenin in vivo is able to induce AChR expression along the entire muscle fiber [71]. Multiple mechanisms have been proposed to explain how neural activity controls myogenin in postnatal muscle. On the basis of one of these mechanisms, following muscle depolarization, extracellular calcium enters through voltage-activated channels and activates different signal transduction cascades mediated by PKC and CaMKII, leading to myogenin phosphorylation [63, 72]. This posttranslational modification inhibits myogenin binding to E-boxes of AChR promoters, resulting in reduced AChR expression (Fig. 2) [73, 74]. A second regulatory mechanism restraining myogenin activity involves transcriptional repression of myogenin (Fig. 2). The posttranslational and the transcriptional mechanisms of myogenin repression could act on a different timescale to ensure that myogenin expression remains blocked as long as the muscle is innervated [30]. Although it is very well known that myogenin is transcriptionally modulated as a consequence of muscle depolarization, the mechanisms that are upstream of this regulation have only recently started to be elucidated. Several studies reported that in myogenin repression during innervation, class II deacetylases are recruited at the myogenin promoter. The hystone deacetylase 9 (HDAC9) splice variant MITR is a transcriptional repressor induced in innervated muscle. It contributes to myogenin repression by chromatin acetylation and acts as a corepressor in a complex with the myocyte enhancer factor-2 (MEF2) transcription factor (Fig. 2) [75]. Another myogenin repressor is dachshund homolog 2 (DACH2) (Fig. 2). It has been shown that, during denervation, DACH2 is inhibited by HDAC4, another class II deacetylase, which induces myogenin expression thereby activating AChRs [76–78]. A third circuit of postnatal myogenin expression regulation is mediated by MSY3/CsdA, a Y-box factor that binds the myogenin promoter at a new binding motif (highly conserved element, HCE) and exerts a repression function (Fig. 2). MSY-3 is therefore partially responsible for the restricted spatiotemporal expression of AChR in the subsynaptical regions of muscle fibers [79]. The conserved binding motifs of HDAC9/MEF2, DACH2, and MSY-3 are adjacent sequences on the myogenin promoter, which suggests a strong interplay between the three cis–trans systems to ensure a proper myogenin and AChR repression in adult and innervated muscle.

The ACh–AChR interaction triggers NMJ assembly negative signals. Global AChR transcription is inhibited by ACh released by the nerve. AChR gene transcription suppression occurs through myogenin inhibition. The myogenic transcription factor myogenin activates muscle-specific genes including AChRs by binding the E-box element. When the action potential reaches the axon terminals, it induces the opening of the voltage-gated Ca2+ channels on the presynaptic nerve membrane. The Ca2+ influx into the neuron leads synaptic vesicles to fuse with the presynaptic membrane and to release the neurotransmitter acetylcholine (ACh) in the synaptic cleft. From here, ACh diffuses and binds to acetylcoline receptors (AChRs) localized on the postsynaptic membrane on the muscle fiber. This binding makes AChRs permeable to ions so that Na+ flows into the fiber while K+ flows out of the muscle cytosol. In this way, the muscle membrane locally depolarizes, and this local depolarization opens voltage-gated Na+ channels on the membrane, allowing a propagation of the depolarization (action potential) that spreads to involve the entire plasma membrane. The action potential induced by ACh release leads to the release of Ca2+ from the sarcoplasmic reticulum into the cytosol which causes muscle contraction. The high concentration of Ca2+ inside the cytosol also activates CaMKII and PKC kinases which mediate signaling pathways leading to myogenin phosphorylation and inactivation. When myogenin is inactive, AChR expression is suppressed. In addition to myogenin phosphorylation, a second mechanism, which represses myogenin, is activated by Ca2+ signaling upon innervation and leads to the activation of three transcription repressors (MSY-3, DACH2, and HDAC9). These repressors bind myogenin promoter and inhibit its transcriptional activation. The binding motifs for MSY-3, DACH2, and HDAC9 (respectively HCE, SIX, and MEF2) are located on adjacent sequences on the myogenin promoter
In conclusion, while during development the first events leading to AChR clusterization are MuSK- and rapsyn-dependent but nerve-independent (AChR prepatterning), the stabilization of some prepatterned AChR clusters requires the innervation. Indeed, once the muscle is contacted by the nerve, ACh released by the motor neuron induces a postsynaptic potential which stabilizes previous AChR clusters in the contacted area and prevents AChR clustering in non-contacted area. Moreover, agrin released by the neuron also stabilizes the AChR clusters and, as well as neuregulin, strongly increases AChR transcription in subsynaptic nuclei (Fig. 1). Concomitantly, Ca2+-dependent signals triggered by nerve-induced muscle electrical activity cause myogenin downregulation and inhibition of AChR transcription in extrasynaptic regions of the muscle fiber (Fig. 2). The integration of these two mechanisms makes possible the compartmentalization of AChR gene expression in subsynaptic nuclei and the stabilization of AChR clusters only at NMJs.
4 Neuromuscular disorders associated to defects in synaptogenesis
When NMJs are severely impaired, the muscles develop congenital myasthenic syndromes. In such disorders, defects in neuromuscular transmission cause a fatigable weakness in limb, ocular, bulbar, truncal, and respiratory muscles. Generally, weakness becomes more severe with exercise and improves with rest. CMS have been identified worldwide and occur at a frequency of <1/500,000 people, although this incidence is constantly growing [80]. Diagnosis can be difficult, often requiring a high risk of clinical suspicion. Most patients present symptoms during infancy, although in some syndromes, symptoms are not manifested until childhood or adult life. Depending on the neuromuscular transmission defect, CMS can be classified as presynaptic, synaptic, or postsynaptic, even though it is not always possible to identify accurately the cause of the pathology [81]. Over the past few years, many causative genes and mutations for CMS have been identified, although in many patients, no causative mutations can be found. In most cases, CMS causative mutations are missense, truncation, or splice-site mutations and result in structural changes of AChRs or in AChR low affinity for ACh. CMS-associated genetic mutations have been mapped in AChR subunits, choline acetyltransferase, the collagen tail subunit of acetylcholinesterase, rapsyn, MuSK, and skeletal muscle–sodium channel Na1.4 [82–84].
Dok-7 CMS is a newly identified synaptopathy characterized by a “limb girdle” phenotype that mainly affects proximal muscles rather than the distal ones with ptosis present from early age [85]. Mutations in the Dok-7 locus were identified in groups of patients who had small EPs, a reduced number of AChRs at the EPs, and a limb-girdle phenotype with no mutations in rapsyn or AChRs. Dok-7 mutants generally have abnormally small and simplified NMJs but show normal AChR and AChE functions, correct MuSK activation, and AChR clusterization, although Dok-7 acts in concert with MuSK in activating rapsyn to concentrate AChRs at the junctional folds. It has therefore been suggested that the altered size and integrity of NMJs observed were probably the consequence of a high and widespread degeneration and remodeling of the EPs [86, 87].
Together with genetic alterations in the coding region of components of the synaptic apparatus, also mutations in their regulative regions can account for the congenital myasthenic phenotype. A mutation in the N-box of the AChRε subunit promoter, resulting in a decreased GABP transcription factor binding activity [88], causes a reduction in the AChR number which is the cause of CMS clinical symptoms in human [89, 90].
Besides being congenital, myasthenia can also be caused by a complex autoimmune disorder of neuromuscular transmission (myasthenia gravis, MG). MG is characterized by a fluctuating weakness of the head, neck, and upper extremity muscles that worsens with activity and improves with rest [91, 92]. When the bulbar and the respiratory muscle deteriorate, the disease becomes life-threatening, and ventilation with medication is required. Approximately 80% of MG patients have autoantibodies against AChR and, consequently, they show a decreased number of AChRs at the postynaptic membrane [93]. In addition, the deposit of the complement causes a structural and functional damage of the postsynaptic membrane, resulting in a neuromuscular transmission failure. In some MG patients, autoantibodies against MuSK have also been found, but their pathogenic role remains unclear since the number of AChRs is not reduced and no deposits of complement are found at the postsynaptic membrane. It has been therefore suggested that, since MuSK can be required for retrograde signal, it is possible that MuSK autoantibodies interfere with the presynaptic apparatus organization [94, 95]. Some other MG patients have autoantibodies to Lrp4, which inhibit Lrp4 function [96].
5 NMJ degeneration in sarcopenia
The age-related muscle loss (sarcopenia) strongly impairs the quality of life in the elderly, and it is associated with increased risk of morbidity and mortality [97]. The etiology of sarcopenia is not clear yet. However, many studies suggest that NMJ structural and functional impairment plays a key role in muscle wasting during aging. NMJ degeneration is a feature of sarcopenia, and it has been documented both in animal models and in humans. Changes in NMJ morphology depend on the type of muscle and include variation of the nerve terminal area size, of the end plate size, of the number of synaptic vesicles, of mitochondrial content, and of smooth endoplasmic reticulum content (reviewed in [98]). By in vivo live animal imaging, it has been revealed that at 12–18 months, in mice, a few NMJs start to display loss of motor neuron terminal branches and AChRs. At this point, probably in order to compensate for loss of some synaptic sites, motor neuron sprouting increases and accounts for the formation of new NMJs. However, this compensatory effect strongly decreases after 18 months of age. Moreover, the newly formed synapses are more sensitive to degenerative changes. Therefore, by 24–36 months, most NMJs are completely denervated, and postsynaptic EPs are highly fragmented [98].
Many mechanisms have been evoked as cause of NMJ degeneration during aging. Age-related changes in the morphology of NMJs can be related to alteration of the axonal transport, which impairs the availability of trophic factors and organelles, such as mitochondria, essential for neuronal survival [99]. Age-related changes in myelinated Schwann cells have also been reported and might be associated with NMJ degeneration [100]. It has interestingly been shown that neurotrophic and myotrophic factors such as BDNF, NT-3, insulin-like growth factor (IGF)-I, and IGF-II, which are necessary for the maintenance of presynaptic and postsynaptic apparatus at the NMJ, also play a modulating role in aging [101, 102]. For example, IGF-1 injections into muscle inhibit motor neuron and NMJ degeneration and prevent age-related force decline in mice [103].
Recent studies suggest that motor neuron degeneration and consequent denervation of myofibers are a major cause of muscle mass loss [104, 105]. In aged people, muscle fibers undergo cycles of denervation and innervation. During these cycles, some myofibers are lost, and others, which were innervated by fast motor neurons (type II fibers), are reinnervated by slow ones (type I fibers). This results in an increased percentage of type I fibers and atrophy, which characterizes sarcopenia. It has also been demonstrated that chronic exercise training and higher neuromuscular activity counteract the alterations in motor neurons and delay the onset of denervation and sarcopenia in aged people [104–106].
Although the causes of age-associated NMJ degeneration remain unclear, other mechanisms might contribute to compromised muscle neurotransmission and sarcopenia in aging. For example, sarcopenia is also characterized by mitochondrial dysfunctions. Presynaptic terminals and postsynaptic EPs contain a high concentration of mitochondria, which are critical for NMJ function since they account for energy support, Ca2+ buffering, synaptic transmission, and apoptosis [107]. Mitochondrial dysfunctions might lead to altered calcium buffering, less ATP generation and more ROS production, and oxidative damage during aging, which impairs NMJs [108, 109]. In particular, the impaired buffering of Ca2+ determines an increased concentration of cytosolic Ca2+, which might activate some proteases called calpains. It has been shown that calpains interact with rapsyn thereby disrupting AChR clusters. Through this mechanism, mitochondrial impairment might partially account for the age-related dispersion of AChR clusters [110]. Moreover, a direct correlation between increased oxidative stress and neuromuscular function has been recently proposed. Mice lacking the antioxidant enzyme CuZnSOD (Cu/Zn superoxide dismutase, Sod 1) show high oxidative damage and rapid sarcopenia. The morphology of NMJs at 11 months in Sod1−/−mice is altered, and it is comparable to NMJ degeneration in 33-month-old, wild-type mice. Furthermore, denervated NMJs and fragmentated AChRs observed in young Sod1−/−mice notably reduce muscle contractile force [111, 112]. These observations suggest that NMJ degeneration during aging might be caused also by age-related oxidative stress, which usually is produced by mitochondrial dysfunctions. Oxidative stress also induces damage in myelinated peripheral nerves and consequent increased inflammatory cytokines during aging (Reviewed in [98]).
Interestingly, it has been recently reported that calorie restriction slows down the aging process and, in particular, decreases deterioration of peripheral nerve by upregulating autophagy [113]. It has also been shown that calorie restriction reduces oxidative damage, NMJ degeneration, and muscle atrophy in Sod1−/−mice ,and this further suggests that oxidative damage affects NMJs [111, 114].
6 Conclusions and prospects
The complexity of the neuromuscular junction apparatus and of the regulative systems responsible for its organization indicates that the functional modulation of the connection between neurons and muscle fibers is crucial. The right concentration of the acetylcholine receptors in the area where the axon terminals juxtaposes the postsynaptic membrane accounts for the NMJ’s ability to generate the action potential that provokes muscle contraction and consequently movements. For this reason, multiple levels of regulation are committed to accurately control this process. Animal models show that impairing only one of these regulative pathways can cause severe phenotypes characterized by compromised coordinate movements and breath capacity which usually prove fatal. We have also seen that transcriptional control plays an important role in muscle physiological response to nerve activity. CMS and myasthenia gravis are human pathologies characterized by an extreme fatigability and muscle weakness and in which insufficiency of a single component of NMJ frequently accounts for their pathophysiology. Studying NMJ transmission in aging and in those pathological conditions characterized by different levels of muscle weakness may reveal new mechanisms that contribute to the fine-tuning of the interplay between motor neurons and muscles. Understanding the mechanisms underlying age- and disease-associated atrophy and alteration of NMJs should open up new potential therapeutic avenues to these skeletal muscle dysfunctions.
References
Engel AG, Ohno K, Shen XM, Sine SM. Congenital myasthenic syndromes: multiple molecular targets at the neuromuscular junction. Ann N Y Acad Sci. 2003;998:138–60.
Hartzell HC, Fambrough DM. Acetycholine receptor production and incorporation into membranes of developing muscle fibers. Dev Biol. 1973;30:153–65.
Cohen SA, Fischbach GD. Clusters of acetylcholine receptors located at identified nerve-muscle synapses in vitro. Dev Biol. 1977;59:24–38.
Kummer TT, Misgeld T, Sanes JR. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr Opin Neurobiol. 2006;16:74–82.
Bevan S, Steinbach JH. The distribution of alpha-bungarotoxin binding sites of mammalian skeletal muscle developing in vivo. J Physiol. 1977;267:195–213.
Braithwaite AW, Harris AJ. Neural influence on acetylcholine receptor clusters in embryonic development of skeletal muscles. Nature. 1979;279:549–51.
Ziskind-Conhaim L, Bennett JI. The effects of electrical inactivity and denervation on the distribution of acetylcholine receptors in developing rat muscle. Dev Biol. 1982;90:185–97.
Yang X, Arber S, William C, Li L, Tanabe Y, Jessell TM, et al. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron. 2001;30:399–410.
Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–64.
Yang X, Li W, Prescott ED, Burden SJ, Wang JC. DNA topoisomerase II beta and neural development. Science. 2000;287:131–4.
Anderson MJ, Cohen MW. Nerve-induced and spontaneous redistribution of acetylcholine receptors on cultured muscle cells. J Physiol. 1977;268:757–73.
Frank E, Fischbach GD. Early events in neuromuscular junction formation in vitro: induction of acetylcholine receptor clusters in the postsynaptic membrane and morphology of newly formed synapses. J Cell Biol. 1979;83:143–58.
Burden SJ. SnapShot: neuromuscular junction. Cell. 2011;144:826–6.
Wu H, Xiong WC, Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;137:1017–33.
Young SH, Poo MM. Spontaneous release of transmitter from growth cones of embryonic neurones. Nature. 1983;305:634–7.
Burden S. Development of the neuromuscular junction in the chick embryo: the number, distribution, and stability of acetylcholine receptors. Dev Biol. 1977;57:317–29.
Merlie JP, Sanes JR. Concentration of acetylcholine receptor mRNA in synaptic regions of adult muscle fibres. Nature. 1985;317:66–8.
Fontaine B, Sassoon D, Buckingham M, Changeux JP. Detection of the nicotinic acetylcholine receptor alpha-subunit mRNA by in situ hybridization at neuromuscular junctions of 15-day-old chick striated muscles. EMBO J. 1988;7:603–9.
Simon AM, Hoppe P, Burden SJ. Spatial restriction of AChR gene expression to subsynaptic nuclei. Development. 1992;114:545–53.
McMahan UJ. The agrin hypothesis. Cold Spring Harb Symp Quant Biol. 1990;55:407–18.
Ruegg MA, Bixby JL. Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci. 1998;21:22–7.
Jones G, Meier T, Lichtsteiner M, Witzemann V, Sakmann B, Brenner HR. Induction by agrin of ectopic and functional postsynaptic-like membrane in innervated muscle. Proc Natl Acad Sci U S A. 1997;94:2654–9.
Herbst R, Burden SJ. The juxtamembrane region of MuSK has a critical role in agrin-mediated signaling. EMBO J. 2000;19:67–77. Erratum in: EMBO J 2000;19:1167.
Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, et al. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–35.
Jennings CG, Dyer SM, Burden SJ. Muscle-specific trk-related receptor with a kringle domain defines a distinct class of receptor tyrosine kinases. Proc Natl Acad Sci U S A. 1993;90(7):2895–9.
Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, et al. Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron. 1995;15:573–84.
DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–12.
Mittaud P, Camilleri AA, Willmann R, Erb-Vögtli S, Burden SJ, Fuhrer C. A single pulse of agrin triggers a pathway that acts to cluster acetylcholine receptors. Mol Cell Biol. 2004;24:7841–54.
Kim N, Burden SJ. MuSK controls where motor axons grow and form synapses. Nature Neurosci. 2008;11:19–27.
Thang H, Veldman MB, Goldman D. Characterization of a muscle-specific enhancer in human MuSK promoter reveals the essential role of myogenin in controlling activity-dependent gene regulation. Proc Natl Acad Sci U S A. 2006;103:16977–82.
Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, et al. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–23.
Glass DJ, Apel ED, Shah S, Bowen DC, DeChiara TM, Stitt TN, et al. Kinase domain of the muscle-specific receptor tyrosine kinase (MuSK) is sufficient for phosphorylation but not clustering of acetylcholine receptors: required role for the MuSK ectodomain? Proc Natl Acad Sci U S A. 1997;94:8848–53.
Weatherbee SD, Anderson KV, Niswander LA. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development. 2006;133:4993–5000.
Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, et al. Lrp4 is a receptor for agrin and forms a complex with MuSK. Cell. 2008;135:334–42.
Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–97.
Kummer TT, Misged T, Sanes JR. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr Opin Neurobiol. 2006;16:74–82.
Okada K, Inoue A, Okada M, Murata Y, Kakuta S, Jigami T, et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. 2006;312:1802–5.
Inoue A, Setoguchi K, Matsubara Y, Okada K, Sato N, Iwakura Y, et al. Dok-7 activates the muscle receptor kinase MuSK and shapes synapse formation. Sci Signal. 2009;2(59):ra7.
Bergamin E, Hallock PT, Burden SJ, Hubbard SR. The cytoplasmic adaptor protein Dok7 activates the receptor tyrosine kinase MuSK via dimerization. Mol Cell. 2010;39:100–9.
Hallock PT, Xu CF, Park TJ, Neubert TA, Curran T, Burden SJ. Dok-7 regulates neuromuscular synapse formation by recruiting Crk and Crk-L. Genes Dev. 2010;24:2451–61.
Burden SJ, DePalma RL, Hume RI, Akaaboune M. Identification of nicotinic acetylcholine receptor recycling and its role in maintaining receptor density at the neuromuscular junction in vivo. Cell. 1983;35:687–92.
Gautam M, Mudd J, Copeland NG, Gilbert DJ, Jenkins NA, Merlie JP. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 1995;377:232–6.
Strochlic L, Cartaud A, Cartaud J. The synaptic muscle-specific kinase (MuSK) complex: new partners, new functions. Bioassays. 2005;27:1129–35.
Falls DL. Neuregulins and the neuromuscular system: 10 years of answers and questions. J Neurocytol. 2003;32:619–47.
Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–90. Erratum in: Nature 1995;378:753.
Sandrock Jr AW, Dryer SE, Rosen KM, Gozani SN, Kramer R, Theill LE, et al. Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science. 1997;276:599–603.
Escher P, Lacazette E, Courtet M, Blindenbacher A, Landmann L, Bezakova G, et al. Synapses form in skeletal muscles lacking neuregulin receptors. Science. 2005;308:1920–3.
Jaworski A, Burden SJ. Neuromuscular synapse formation in mice lacking motor neuron- and skeletal muscle-derived neuregulin-1. J Neurosci. 2006;26:655–61.
Rimer M. Neuregulins at the neuromuscular synapse: past, present, and future. J Neurosci Res. 2007;85:1827–33.
Tansey MG, Chu GC, Merlie JP. ARIA/HRG regulates AChR epsilon subunit gene expression at the neuromuscular synapse via activation of phosphatidylinositol 3-kinase and Ras/MAPK pathway. J Cell Biol. 1996;134:465–76.
Altiok N, Altiok S, Changeux JP. Heregulin-stimulated acetylcholine receptor gene expression in muscle: requirement for MAP kinase and evidence for a parallel inhibitory pathway independent of electrical activity. EMBO J. 1997;16:717–25.
Si J, Luo Z, Mei L. Induction of acetylcholine receptor gene expression by ARIA requires activation of mitogen-activated protein kinase. J Biol Chem. 1996;271:19752–9.
Si J, Wang Q, Mei L. Essential roles of c-JUN and c-JUN N-terminal kinase (JNK) in neuregulin-increased expression of the acetylcholine receptor epsilon-subunit. J Neurosci. 1999;19:8498–508.
LaMarco K, Thompson CC, Byers BP, Walton EM, McKnight SL. Identification of Ets- and notch-related subunits in GA binding protein. Science. 1991;253:789–92.
Thompson CC, Brown TA, McKnight SL. Convergence of Ets- and notch-related structural motifs in a heteromeric DNA binding complex. Science. 1991;253:762–8.
Schaeffer L, Duclert N, Huchet-Dymanus M, Changeux JP. Implication of a multisubunit Ets-related transcription factor in synaptic expression of the nicotinic acetylcholine receptor. EMBO J. 1998;17:3078–90.
Fromm L, Burden SJ. Transcriptional pathways for synapse-specific, neuregulin-induced and electrical activity-dependent transcription. J Physiol Paris. 1998;92:173–6.
Sapru MK, Florance SK, Kirk C, Goldman D. Identification of a neuregulin and protein-tyrosine phosphatase response element in the nicotinic acetylcholine receptor epsilon subunit gene: regulatory role of an Rts transcription factor. Proc Natl Acad Sci U S A. 1998;95:1289–94.
Briguet A, Ruegg MA. The Ets transcription factor GABP is required for postsynaptic differentiation in vivo. J Neurosci. 2000;20:5989–96.
Lacazzette E, Le Calvez S, Gajendran N, Brenner HR. A novel pathway for MuSK to induce genes in neuromuscular synapse formation. J Cell Biol. 2003;161:727–36.
Strochlic L, Cartaud A, Mejat A, Graihe R, Shaeffer L, Changeux JP, et al. 14-3-3 gamma associates with muscle specific kinase and regulates synaptic gene transcription at vertebrate neuromuscular synapse. Proc Natl Acad Sci. 2004;101:18189–94.
Salpeter MM, Cooper DL, Levitt-Gilmour T. Degradation rates of acetylcholine receptors can be modified in the postjunctional plasma membrane of the vertebrate neuromuscular junction. J Cell Biol. 1986;103(4):1399–403.
Fu AK, Ip FC, Fu WY, Cheung J, Wang JH, Yung WH, et al. Aberrant motor axon projection, acetylcholine receptor clustering, and neurotransmission in cyclin-dependent kinase 5 null mice. Proc Natl Acad Sci U S A. 2005;102:15224–9.
Lin W, Dominguez B, Yang J, Aryal P, Brandon EP, Gage FH, et al. Neurotransmitter acetylcholine negatively regulates neuromuscular synapse formation by a Cdk-5-dependent mechanism. Neuron. 2005;46:569–79.
Tang H, Sun Z, Goldman D. CaM kinase II-dependent suppression of nicotinic acetylcholine receptor delta-subunit promoter activity. J Biol Chem. 2001;276:26057–65.
Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR. Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron. 2002;36:635–48.
Brandon EP, Lin W, D’Amour KA, Pizzo DP, Dominguez B, Sugiura Y, et al. Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci. 2003;23:539–49.
Laufer R, Changeux JP. Calcitonin gene-related peptide and cyclic AMP stimulate phosphoinositide turnover in skeletal muscle cells. Interaction between two second messenger systems. J Biol Chem. 1989;264:2683–9.
Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;11:791–805.
Eftimie R, Brenner HR, Buonanno A. Myogenin and MyoD join a family of skeletal muscle genes regulated by electrical activity. Proc Natl Acad Sci U S A. 1991;88:1349–53.
Gundersen K, Rabben I, Klocke BJ, Merlie JP. Overexpression of myogenin in muscles of transgenic mice: interaction with Id-1, negative crossregulation of myogenic factors, and induction of extrasynaptic acetylcholine receptor expression. Mol Cell Biol. 1995;15:7127–34.
Li L, Zhou J, James G, Heller-Harrison R, Czech MP, Olson EN. FGF inactivates myogenic helix-loop-helix proteins through phosphorylation of a conserved protein kinase C site in their DNA-binding domains. Cell. 1992;71:1181–94.
Mendelzon D, Changeux JP, Nghiêm HO. Phosphorylation of myogenin in chick myotubes: regulation by electrical activity and by protein kinase C. Implications for acetylcholine receptor gene expression. Biochemistry. 1994;33:2568–75.
Macpherson P, Kostrominova T, Tang H, Goldman D. Protein kinase C and calcium/calmodulin-activated protein kinase II (CaMK II) suppress nicotinic acetylcholine receptor gene expression in mammalian muscle. A specific role for CaMK II in activity-dependent gene expression. J Biol Chem. 2002;277:15638–46.
Méjat A, Ramond F, Bassel-Duby R, Khochbin S, Olson EN, Schaeffer L. Histone deacetylase 9 couples neuronal activity to muscle chromatin acetylation and gene expression. Nat Neurosci. 2005;8:313–21.
Tang H, Goldman D. Activity-dependent gene regulation in skeletal muscle is mediated by a histone deacetylase (HDAC)-Dach2-myogenin signal transduction cascade. Proc Natl Acad Sci U S A. 2006;103:16977–82.
Cohen TJ, Waddell DS, Barrientos T, Lu Z, Feng G, Cox GA, et al. The histone deacetylase HDAC4 connects neural activity to muscle transcriptional reprogramming. J Biol Chem. 2007;282:33752–9.
Tang H, Macpherson P, Marvin M, Meadows E, Klein WH, Yang XJ, et al. A histone deacetylase 4/myogenin positive feedback loop coordinates denervation-dependent gene induction and suppression. Mol Biol Cell. 2009;20:1120–31.
Berghella L, De Angelis L, De Buysscher T, Mortazavi A, Biressi S, Forcales SV, et al. A highly conserved molecular switch binds MSY-3 to regulate myogenin repression in postnatal muscle. Genes Dev. 2008;22:2125–38.
Vincent A, Newland C, Croxen R, Beeson D. Genes at the junction—candidates for congenital myasthenic syndromes. Trends Neurosci. 1997;20:15–22.
Engel AG, Sine SM. Current understanding of congenital myasthenic syndromes. Curr Opin Pharmacol. 2005;5:308–21.
Ohno K, Quiram PA, Milone M, Wang HL, Harper MC, Pruitt 2nd JN, et al. Congenital myasthenic syndromes due to heteroallelic nonsense/missense mutations in the acetylcholine receptor epsilon subunit gene: identification and functional characterization of six new mutations. Hum Mol Genet. 1997;6:753–66.
Ohno K, Engel AG, Shen XM, Selcen D, Brengman J, Harper CM, et al. Rapsyn mutations in humans cause end-plate acetylcholine-receptor deficiency and myasthenic syndrome. Am J Hum Genet. 2002;70:875–85.
Nogajski JH, Kiernan MC, Ouvrier RA, Andrews PI. Congenital myasthenic syndromes. J Clin Neurosci. 2009;16:1–11.
Beeson D, Higuchi O, Palace J, Cossins J, Spearman H, Maxwell S, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;29(313):1975–8.
Selcen D, Milone M, Shen XM, Harper CM, Stans AA, Wieben ED, et al. Dok-7 myasthenia: phenotypic and molecular genetic studies in 16 patients. Ann Neurol. 2008;64:71–87.
Yamanashi Y, Higuch O, Beeson D. Dok-7/MuSK signaling and a congenital myasthenic syndrome. Acta Myol. 2008;27:25–9.
Duclert A, Savatier N, Schaeffer L, Changeux JP. Identification of an element crucial for the sub-synaptic expression of the acetylcholine receptor epsilon-subunit gene. J Biol Chem. 1996;19(271):17433–8.
Ohno K, Anlar B, Engel AG. Congenital myasthenic syndrome caused by a mutation in the Ets-binding site of the promoter region of the acetylcholine receptor epsilon subunit gene. Neuromuscul Disord. 1999;9:131–5.
Nichols P, Croxen R, Vincent A, Rutter R, Hutchinson M, Newsom-Davis J, et al. Mutation of the acetylcholine receptor epsilon-subunit promoter in congenital myasthenic syndrome. Ann Neurol. 1999;45:439–43.
Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116:2843–54.
Vincent A, Lang B, Kleopa KA. Autoimmune channelopathies and related neurological disorders. Neuron. 2006;5(52):123–38.
Fambrough DM, Drachman DB, Satyamurti S. Neuromuscular junction in myasthenia gravis: decreased acetylcholine receptors. Science. 1973;182:293–5.
Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7:365–8.
Shiraishi H, Motomura M, Yoshimura T, Fukudome T, Fukuda T, Nakao Y, et al. Acetylcholine receptors loss and postsynaptic damage inMuSK antibody-positive myasthenia gravis. Ann Neurol. 2005;57:289–93.
Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol. 2011;69:418–22.
Janssen DS, Shepard PT, Katzmarzyk R, Roubenoff. The healthcare costs of sarcopenia in the United States. J Am Geriatr Soc. 2004;52:80–5.
Balice-Gordon RJ. Age-related changes in neuromuscular innervations. Muscle Nerve. 1997;5:S83–7.
Gutmann E, Hanzlikova V. Basic mechanisms of aging in the neuromuscular system. Mech Ageing Dev. 1973;1:327–49.
Ceballos D, Cuadras J, Verdu E, Navarro X. Morphometric and ultrastructural changes with ageing in mouse peripheral nerve. J Anat. 1999;195:563–76.
Gonzalez M, Ruggiero FP, Chang Q, Shi YJ, Rich MM, Kraner S, et al. Disruption of Trkb-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron. 1999;24:567–83.
Belluardo N, Westerblad H, Mudo G, Casabona A, Bruton J, Caniglia GO, et al. Neuromuscular junction disassembly and muscle fatigue in mice lacking neurotrophin-4. Mol Cell Neurosci. 2001;18:56–7.
Payne AM, Zheng Z, Messi ML, Milligan CE, Gonzalez E, Delbono O. Motor neurone targeting of IGF-1 prevents specific force decline in ageing mouse muscle. J Physiol. 2006;570:283–94.
Kim JH, Kwak HB, Leeuwenburgh C, Lawler JM. Lifelong exercise and mild (8%) caloric restriction attenuate age-induced alterations in plantaris muscle morphology, oxidative stress and IGF-1 in the Fischer-344 rat. Exp Gerontol. 2008;43:317–29.
Deschenes MR, Roby MA, Eason MK, Harris MB. Remodeling of the neuromuscular junction precedes sarcopenia related alterations in myofibers. Exp Gerontol. 2010;45:389–93.
Vandervoort AA. Aging of the human neuromuscular system. Muscle Nerve. 2002;25:17–25.
Hiona A, Leeuwenburgh C. The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Exp Gerontol. 2008;43:24–33.
Zhou J, Yi J, Fu R, Liu, Siddique ET, Rios E, et al. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J Biol Chem. 2010;285:705–12.
Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, et al. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 2010;24:1376–90.
Payne AM, Zheng Z, Gonzalez E, Wang ZM, Messi ML, Delbono O. External Ca(2+)-dependent excitation–contraction coupling in a population of ageing mouse skeletal muscle fibres. J Physiol. 2004;560:137–55.
Jang YC, Van Remmen H. Age-associated alterations of the neuromuscular junction. Exp Gerontol. 2011;46:193–8.
Muller FL, Song W, Liu Y, Chaudhuri A, Pieke-Dahl S, Strong R, et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic Biol Med. 2006;40:1993–2004.
Rangaraju S, Hankins D, Madorsky I, Madorsky E, Lee WH, Carter CS, et al. Molecular architecture of myelinated peripheral nerves is supported by calorie restriction with aging. Aging Cell. 2009;8:178–91.
Opalach K, Rangaraju S, Madorsky I, Leeuwenburgh C, Notterpek L. Lifelong calorie restriction alleviates age-related oxidative damage in peripheral nerves. Rejuvenation Res. 2010;13:65–74.
von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle. 2010;1:7–8.
Acknowledgments
The authors have been suppported in this work by the European Union Grant SICA-HF project. All authors of this manuscript comply with the guidelines of ethical authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle [115]. European Union Seventh Framework Program FP7/2007-2011 under grant agreement 241558 (SICA-HF).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Ferraro, E., Molinari, F. & Berghella, L. Molecular control of neuromuscular junction development. J Cachexia Sarcopenia Muscle 3, 13–23 (2012). https://doi.org/10.1007/s13539-011-0041-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13539-011-0041-7