
Abstract
A subset of patients with coronavirus 2 disease (COVID-19) experience neurological complications. These complications include loss of sense of taste and smell, stroke, delirium, and neuromuscular signs and symptoms. The etiological agent of COVID-19 is SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), an RNA virus with a glycoprotein-studded viral envelope that uses ACE2 (angiotensin-converting enzyme 2) as a functional receptor for infecting the host cells. Thus, the interaction of the envelope spike proteins with ACE2 on host cells determines the tropism and virulence of SARS-CoV-2. Loss of sense of taste and smell is an initial symptom of COVID-19 because the virus enters the nasal and oral cavities first and the epithelial cells are the receptors for these senses. Stroke in COVID-19 patients is likely a consequence of coagulopathy and injury to cerebral vascular endothelial cells that cause thrombo-embolism and stroke. Delirium and encephalopathy in acute and post COVID-19 patients are likely multifactorial and secondary to hypoxia, metabolic abnormalities, and immunological abnormalities. Thus far, there is no clear evidence that coronaviruses cause inflammatory neuromuscular diseases via direct invasion of peripheral nerves or muscles or via molecular mimicry. It appears that most of neurologic complications in COVID-19 patients are indirect and as a result of a bystander injury to neurons.
Similar content being viewed by others
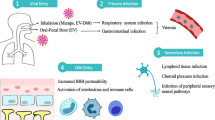

In 2019, a new coronavirus disease (COVID-19) emerged, sparking a worldwide pandemic that has led to significant morbidity and mortality and stressed the health systems globally. Since the beginning of the pandemic, a deluge of unfiltered information has streamed into the scientific journals and mainstream media about COVID-19 and its etiological agent, SARS-CoV-2. This “infodemic” has often been in equal parts valuable, redundant, confusing, and contradictory. Although SARS-CoV-2 initially injures the respiratory tract, it also affects other organs including the nervous system (Helms et al. 2020a, b; Guan et al. 2020; Mao et al. 2020). Here, we review the salient features of SARS-CoV-2 pathogenesis and highlight the mechanisms of neurologic complications of the SARS-CoV-2 infection.
Human coronavirus 2 (SARS-CoV-2)
The family of coronaviruses are a successful group of microbial pathogens that depend on the living cells for replication and infect many species of birds and mammals (Poutanen 2018). During the millions of years of coexistence, a complex system of cohabitation has evolved between viruses and their host cells. At different stages of this evolution, flare-ups of destructive infection in the host population can occur that eventually reach a homeostasis. COVID-19 can be considered such a destructive infection phase. SARS-CoV-2 is an RNA virus that belongs to the beta coronavirus genus and similar to SARS-CoV-1 and the Middle Eastern respiratory syndrome (MERS) is host- and cell-specific (Lu et al. 2020; Luis et al. 2015). The virus is a zoonotic pathogen native to bats and is likely transmitted to humans via an as yet unknown intermediary (Lu et al. 2020; Luis et al. 2013). Bats and rodents are rich sources of zoonotic viruses because different species of these animals share the same viruses allowing the virus to adapt to different hosts (Luis et al. 2013). Once the coronavirus achieved a hold in the human host, it has made further adaptations. New clades of virus have accelerated human-to-human transmission, contributing to the severity of the pandemic (Li et al. 2020; Korber et al. 2020; Andersen et al. 2020). SARS-CoV-2 is primarily transmitted from human to human by inhalation of virus containing airborne droplets in crowded closed quarters and also via a possible fecal to oral route (Lamers et al. 2020; Xu et al. 2020a). Although SARS-CoV-2 carries “respiratory” in its moniker, it exploits the broadly expressed angiotensin-converting enzyme 2 (ACE2) for entry into the target endothelial and epithelial cells (Li et al. 2003; Shang et al. 2020a, b). This contributes to both the specificity and breadth of infection, as other organs and organ systems that contain epithelial cells, including the nervous system, are affected (Guan et al. 2020; Mao et al. 2020).
SARS-CoV-2 is a large positive-sense, single-stranded, non-segmented RNA that is enclosed in a nuclear capsid. The RNA genome is over 29,000 bases. Another noteworthy feature of coronaviruses in general, SARS-CoV-2 included, is their glycoprotein-studded viral envelope. The lipid envelope, a remnant of the previous host cell membrane, dictates viral stability and its infectivity. The proteins imbedded in this envelope mediate viral entry into the host cells (Figs. 1 and 2) and are important in allowing the virus to evade host immune surveillance (Zhang et al. 2020a, b, c; Shang et al. 2020b; Alcami and Koszinowski 2000). Each of the spike proteins in the viral ectodomain is comprised of a trimer of glycoproteins (S1, Figs. 1 and 2) that are mounted on a stalk (S2) like an ice cream cone (Fig. 1b) (Walls et al. 2020; Wrapp et al. 2020; Shang et al. 2020a, b). Spike proteins are translated from the viral RNA and determine the virulence and tropism of the virus (Lu et al. 2020; Luis et al. 2015). Therefore, attention has quickly focused on the structure and function of the SARS-CoV-2 spike proteins as therapeutic targets and for vaccine development (Lan et al. 2020; Shang et al. 2020a, b; Corbett et al. 2020; Ju et al. 2020).

Images of SARS-CoV-2 and its spike protein. a Electron microscope images of crown-shaped SARS-CoV-2 with protein spikes (red arrows). b Structure of protein spikes showing the receptor-binding domain (green) that attaches to ACE2 and the site of enzymatic cleavage (yellow) that allow conformational changes in the spike protein to facilitate fusing of viral envelope with host cell membrane. c Another view of spike protein with sticky binding sites (red). (From Wrapp et al. 2020, with permission)

Diagram depicting a compilation of data on attachment and fusion of coronavirus with the host cell. Attachment occurs by fastening of the receptor binding domain (RBD, Fig. 1c) to the ACE2 as a functional receptor. RBD is a variable segment and a target of treatment (see text). The spike proteins undergo conformational changes to fuse virus and cell membranes in order to allow delivery of the viral genome into the target cells. The structural rearrangements are accomplished by enzyme assisted proteolysis involving the transmembrane serine protease 2 (TMPRSS2), Furin, and ACE2. The substrate for these proteolytic enzymes is the polybasic cleavage site (yellow) at the S1-S2 junction of the spike protein. ACE2 plays an indispensable role as it provides a docking port for SARS-CoV2 and facilitates fusion via enzymatic modification of the spike proteins
Infection of host cells by SARS-CoV-2
Identifying the steps involved in the mode of infection of host cells by SARS-CoV-2 will help us understand the phenotype of neurological disease in COVID-19. SARS-CoV-2 spike proteins mediate viral entry into the host cells through a complex molecular dance with the target host cells. This interaction occurs in two steps, attachment of viral envelope glycoproteins (S1) to ACE2, and fusion of viral envelope to the host cell membrane (Tortorici and Veesler 2019; Lan et al. 2020; Walls et al. 2016, 2020). Attachment occurs when the receptor-binding domain (RBD) of spike protein fastens to ACE2 (Figs. 1, 2). RBD is a variable domain located on the outer side of the spike protein (Fig. 1c) and has been the target for treatment of COVID-19 by neutralizing antibodies (Ju et al. 2020; Shi et al. 2020; Wrapp et al. 2020; Walls et al. 2020; Shang et al. 2020b). ACE2 is a cognate cell surface receptor for SARS-CoV-2 and determines its tropism. The next step in target cell infection is fusion of the coronavirus envelope with the host cell membrane that is driven by changes in the conformation of the viral spike proteins (Belouzard et al. 2012; Heald-Sargent and Gallagher 2012). The conformational changes are accomplished by the host enzymes including transmembrane serine protease 2 (TMPRSS2); Furin and ACE2 are epithelial specific proteolytic enzymes (Stopsack et al. 2020; Braun and Sauter 2019; Walls et al. 2020; Shang et al. 2020a). They cleave a polybasic site at the S1-S2 junction of the spike protein (Fig. 2) and rearrange its structure (Belouzard et al. 2012; Heald-Sargent and Gallagher 2012) allowing the viral envelope to fuse with the target cell membrane.
Since cells that harbor ACE2 are the main targets of SARS-CoV-2 infection, ACE2 determines viral tropism and by extension disease phenotype and virulence. Thus, it is important to appreciate its localization in the body. Studies of the protein distribution demonstrate that ACE2 is concentrated primarily on the surface of endothelial cells and the epithelium of the exposed mucosa, such as the linings of the nasal and oral cavities, the lungs, and the gut ((Ackermann et al. 2020; Hamming et al. 2004; Sungnak et al. 2020; Xu et al. 2020a, b). Since lungs are mostly comprised of epithelial and endothelial cells and are exposed to the aerosol transmission, they become infected early and severely. Also, the initial clinical manifestation of SARS-CoV-2 infections has been reported in conjunctiva (Chen et al. 2020), nasal cavity lining (Sungnak et al. 2020), oral cavity lining (Xu et al. 2020a, b), and gastrointestinal lining (Zhang et al. 2020a, b, c). All these areas are lined with epithelial cells that harbor ACE2 on their surface accounting for early clinical findings in COVID-19.
While ACE2 serendipitously facilitates coronavirus entry into the host cells (Li et al. 2003; Shang et al. 2020a, b), it also has significant physiological roles in homeostatic regulation of blood circulation as a vasoprotective and cardioprotective agent (Kuba et al. 2013). ACE2 serves as a counterbalance to ACE (angiotensin-converting enzyme and deactivates angiotensin II) by reducing the blood pressure (Kuba et al. 2013). In the gut mucosa, ACE2 regulates the intestinal amino acid transporters (Zhang et al. 2020a, b, c). There has been much speculation as to whether a commonly used class of antihypertensive drugs, the ACE inhibitors, has any influence on COVID-19. The available data (Reynolds et al. 2020; Mancia et al. 2020) indicate that these drugs have no effect on prevalence or infectivity of SARS-CoV-2. Once the viral RNA enters the target cells by endocytosis, it starts to translate and efficiently replicate (Sawicki 2009) and injure the host cells. Enveloped viruses such as SARS-CoV-2 generally form a cytopathic infection in the host cells and these cells can become viral reservoirs. Each of these steps represent a possible target for therapeutic intervention in COVID-19 (Corbett et al. 2020).
Neurological manifestations of COVID-19
The above discussion lays the ground work for understanding neuropsychiatric manifestations COVID-19. While a number of viruses infect the non-neural cells in the nervous system, only a few viruses such as herpes (Kleinschmidt-DeMasters and Gilden 2006) and rabies (Wang et al. 2018; Lafon 2008) directly infect neurons via specific receptors. This is because evolutionarily most pathogenic viruses require a complex chemical dance with the target cells before entering the cell, as discussed above. The phenotypes of the virally mediated neurological diseases depend on the type of infected cells and the affected neural networks. For example, human immunodeficiency virus (HIV) establishes a cytopathic infection in microglia and astrocytes (Castellano et al. 2017) and these cells can serve as reservoirs of viral particles (Ghafouri et al. 2006) and indirectly affecting neurons. Common systemic viral infections, including SARS-CoV-2, are known to indirectly involve the nervous system triggering acute and or chronic neuropsychiatric syndromes (Helms et al. 2020a; Mao et al. 2020; De Felice et al. 2020). The nervous system is vulnerable to immune dysregulations because of its special immune status (Billingham et al. 1953); thus, an immune reaction that is triggered by a systemic viral infection can injure the nervous system (Ghafouri et al. 2006). A number of nervous system injuries have been associated with COVID-19 including stroke (Yaghi et al. 2020; Oxley et al. 2020), loss of sense of taste and smell (Menni et al. 2020; Cooper et al. 2020), Guillain-Barre syndrome (Toscano et al. 2020), and upper motor neuron signs with abnormal MRI and EEGs (Helms et al. 2020a, b) as well as neuropsychiatric disorders (Helms et al. 2020a, b; Rogers et al. 2020). The mechanisms of neurological manifestations of COVID-19 and the sequence of pathogenesis of these conditions are not fully understood yet.
Stroke
Recent reports (Yaghi et al. 2020; Oxley et al. 2020) associated COVID-19 with large vessel strokes in a group of relatively young patients without significant stroke risk factors. Also, many patients with stroke have tested positive for SARS-CoV-2 and vice versa (see Table 1). These studies suggest a potential increased risk for stroke and stroke as a contributor of increased morbidity and mortality in patients with COVID-19 (Yaghi et al. 2020; Qin et al. 2020; Mao et al. 2020). However, the total number of patients admitted to the hospitals including ours for evaluation of acute stroke during COVID-19 crisis declined (Kansagra et al. 2020). This is similar to the rate of admissions for evaluation of acute myocardial infarction (Solomon et al. 2020a). The decline in hospitalizations during the peak of outbreak was likely due to the fear of COVID-19, where patients with signs and symptoms of stroke avoid or postpone seeking help and also aggressive triage by the ED personnel to avoid unnecessary hospital admissions because of paucity of hospital beds. The question that remains unanswered is whether COVID-19 is an independent or a contributory risk factor for stroke? If so, what is the mechanism of stroke in COVID-19 patients? Stroke is an acute and focal loss of neurological function brought on by an abrupt cessation of blood supply to a portion of the brain. Pathogenesis of stroke involves the heart, large and small blood vessels, and blood itself, harking back to the Virchow’s triad of thrombosis: irritation of vessels, blood coagulation, and interruption of blood flow (Bagot and Arya 2008). It appears that in COVID-19 patients the proximate causes of stroke are coagulopathy and injury to cerebral vascular endothelial cells (Fig. 3).

A box-and-arrow diagram depicting the pathogenesis of stroke in COVID-19 patients. The two proximate causes of stroke are cerebral vascular endothelialitis and coagulopathy; these can result in embolic and thrombotic strokes
Once the nosocomial infection with SARS-CoV-2 is established, spilling of viral particles into the blood stream causes viremia that can then infect cerebral blood vessels. The endothelial lining of the cerebral blood vessels is rich in ACE2 (discussed above; Fig. 4 in Hamming et al. 2004), which makes them targets for SARS-CoV-2 infection. Viral infection and proliferation of viral particles in the vascular endothelial cells injure the lining of blood vessels causing endothelialitis (Pugin et al. 2020; Varga et al. 2020). The injury can initiate thrombosis leading to strokes (Fig. 3). Also, a significant SARS-CoV-2 viremia can disrupt function of ACE2 in the cerebral vessels and interrupt its vasoprotective function (Kuba et al. 2013). Local immune response can be another cause of injury to the cerebral vessels. Viral antigens can activate the complement system, macrophages, and neutrophils that then damage the endothelial cells. The complement system forms the membrane attack complex (MAC) that if left unregulated, MAC injures endothelial cells (Magro et al. 2020; Song and Fitzgerald 2020). Even a small injury to the lining of the cerebral vessels can initiate in situ thrombosis (Dubois et al. 2007; Atkinson et al. 2010) and subsequently large or small strokes (Fig. 3).

A box-and-arrow diagram showing the clinical genesis and multifactoriality of encephalopathy and delirium in COVID-19 patients
The second major reason for stroke is coagulopathy, which is associated with COVID-19 (Klok et al. 2020a, b; Helms et al. 2020b). Autopsy of COVID-19 decedents and the pathological analysis revealed that both veins and arteries in multiple areas of body contained blood clots (Varga et al. 2020). Another study revealed that 31% of patients admitted to ICUs with COVID-19 showed complications of thrombosis such as pulmonary embolism and deep venous thrombosis and stroke (Klok et al. 2020a, b). The high rate of coagulopathy is attributed to a robust immune response triggered by SARS-CoV-2 infection. Humoral immune response is known to contribute to thrombosis by forming antibodies such as anti-phospholipid antibodies. In patients with systemic lupus erythematosus, anti-phospholipid antibodies in circulation are used as clinical markers of thrombosis and stroke (Janardhan et al. 2004; Kitagawa 2005). Increased levels of anti-phospholipid antibodies have been reported in COVID-19 patients with coagulopathy (Zhang et al. 2020c). It is also known that shortly after the onset of COVID-19, an antibody response to SARS-CoV-2 develops (Ni et al. 2020; Haveri et al. 2020; Shi et al. 2020). Some of the antibodies are neutralizing antibodies against the virus and preliminary clinical observations suggest that they may mitigate SARS-CoV-2 infection (Kreer et al. 2020; Shi et al. 2020; Bloch et al. 2020). However, this protective antibody reaction is not a likely risk factor for stroke (Joyner et al. 2020).
In addition to antibody response, another reason for the coagulopathy is cytokine and metabolic dysregulation triggered by SARS-CoV-2 (Shen et al. 2020). Activation of neutrophils by the complement system promotes formation of neutrophil external traps (NETs) that can trigger clot formation (Barnes et al. 2020; Becker 2020). Release of cytokines, particularly IL-6, activate cell-mediated immune response against the viral antigens. However, reports of basic laboratory values and our own observations indicate only mild to moderate leukocytosis and minimal elevation of neutrophils in majority of COVID-19 patients (Zhang et al. 2020a, b, c; Huang et al. 2020). On the other hand, spilling of viral particles, antigenic proteins, and debris from injured infected cells can represent risk factors for thrombosis. For example, it is known that free RNA, released after cell injury into blood stream, can initiate thrombosis (Kannemeier et al. 2007). In patients with COVID-19, viral proteins and RNA as well as cellular debris after cytolytic destruction of endothelial cells can present a significant risk for coagulopathy and stroke. Neuroimaging studies of COVID-19 patients with neurological symptoms reveal that hypoxia and stroke are the most common findings in these patients (Jain et al. 2020). In summary, SARS-CoV-2 infection can contribute to stroke by initially causing vasculitis and coagulopathy that can lead to both thrombotic and embolic strokes (Fig. 3).
Loss of sense of taste and smell
Loss of sense of taste and smell has been reported as the initial clinical manifestations of COVID-19. They are the most common self-reported initial symptoms among patients who tested positive for SARS-CoV-2 infection (Menni et al. 2020; Cooper et al. 2020). The clinical validity of these symptoms is clear and follows our discussion about the mode of contagion and tropism of the SARS-CoV-2 infection. SARS-CoV-2 containing airborne droplets first come in contact with the ACE2-rich epithelium of the nasal and oral mucosa (Sungnak et al. 2020; Xu et al. 2020a, b) infecting these cells. Since epithelial cells are the chemical receptors for the sense of taste and smell, injury to these cells result in alteration of sense of taste and smell. Transient olfactory bulb edema in a COVID-19 anosmic patient has been reported; this indicates involvement of neurovascular complex (Laurendon et al. 2020). Though a neuropathological study and RT-PCR of human olfactory bulb tissues infected with SARS-CoV2 did not reveal SARS-CoV-2 infection or injury to the neural tissues (Solomon et al. 2020b). Thus, loss of senses of taste and smell is likely due to epithelial injury and can be an initial clinical indicator of SARS-CoV-2 infection, and can point to a population that should be tested for SARS-CoV-2 (Menni et al. 2020) and triaged as such.
COVID-19 and neuromuscular system
A subset of patients with COVID-19 present with neuromuscular disorders including weakness, sensory loss, and acute neuropathy or Guillain-Barre syndrome (Alberti et al. 2020; Sedaghat and Karimi 2020; Toscano et al. 2020). It is conceivable that the observed neuropathy in COVID-19 patients is inflammatory through delayed immune response and molecular mimicry. This is similar to other viral and bacterial infections that are associated with Guillain-Barre syndrome including Campylobacter jejuni (Godschalk et al. 2004; Nachamkin et al. 1998), HIV (Goldstein et al. 1993; Shepherd et al. 2017), cytomegalovirus (Visser et al. 1996) and Zika virus (de Siqueira et al. 2016), where an immune response that is directed against an antigenic epitope of the pathogen also attacks a similar glycoprotein on the surface of myelin and or axon, causing immune-mediated injury. However, it is curious that there are only a few reported cases of inflammatory motor neuropathy associated with other similar coronaviruses such as SARS-CoV-1 and MERS virus (Kim et al. 2017).
A portion of neuromuscular disorders reported in gravely ill COVID-19 patients with prolonged convalescence could be due to critical illness neuropathy and myopathy (Latronico et al. 1996). As further reports of neuromuscular abnormalities are revealed, we will be able to determine the specific incidence and mechanism of these conditions in COVID-19 patients.
COVID-19 and neuropsychiatric issues
Neuropsychiatric issues including impaired consciousness, lethargy, disorientation, and headache have been reported in a group of COVID-19 patients (Kotfis et al. 2020; Rogers et al. 2020). In the acute phase of COVID-19 illness, critically ill patients exhibit encephalopathy in a range of intensities, from almost no effect to severe delirium. The intensity of condition is related to duration and severity of illness. During the acute and convalescent stages of COVID-19, vulnerable patients manifest impaired consciousness, disorientation, and altered mentation (Kotfis et al. 2020; Helms et al. 2020a, b; Larvie et al. 2020). Clinically, these effects are manifested as delirium and later as cognitive deficits (Helms et al. 2020a, b; Rogers et al. 2020; Fong et al. 2011). Delirium is an acute disruption of information processing by the brain sensory networks and the ensuing aberrant behavior. It is clinically manifested as alteration in both perceptual and behavioral domains and is often observed as agitation in the patients confined to the intensive care units. Delirium is caused by hypoxia, metabolic derangements, and inflammation; thus, it can be fluctuating and reversible (Fig. 4). The term encephalitis has been loosely used to refer to neurologic manifestations of systemic viral infections, though neurons are highly differentiated cells and remain resistant to viral entry and infection. No specific neuropsychiatric illness has been attributed to SARS-CoV-2 and as yet there is no evidence that SARS-CoV-2 infects neurons directly. It has been speculated that SARS-CoV-2 can spread to the temporal lobes via the olfactory tract infection, causing encephalopathy. However, a recent neuropathology study from an autopsy series of COVID-19 decedents demonstrate mainly hypoxic injury to the brain (Solomon et al. 2020b). A survey of COVID-19 hospitalized patients with neuropsychiatric symptoms did not reveal infected CSF (Helms et al. 2020a). On the other hand, the cerebral vessels are in direct contact with and are affected by the viral infection and inflammatory changes in the blood (Pugin et al. 2020). Patients with underlying brain disorders and metabolic derangements are at higher risk for delirium during hospitalization, because in addition to critical illness, there are environmental factors such as unfamiliar surroundings, lack of visitation, uncertainty, and anxiety. These “mental status changes” are likely due to effects of systemic chemical and inflammatory alterations, disruption of neurovascular unit, and the effects of general medical illness on the brain (Fig. 4). It is not clear whether long-term effects of COVID-19 on the nervous system are different from neuropsychiatric manifestation of other severe illnesses.
Neuroimaging studies of COVID-19 patients with neuropsychiatric symptoms reveal non-specific and variable changes in the brain (Kandemirli et al. 2020; Mahammedi et al. 2020). Magnetic resonance imaging data demonstrate lesions in the cortical white matter that are consistent with disruption of neurovascular unit and vasogenic edema (Mahammedi et al. 2020) and endothelialitis of cerebral vessels (Pugin et al. 2020).
Summary
At this juncture, enough molecular and organizational knowledge is available from different disciplines for us to infer some of the mechanisms of injury to human nervous systems by SARS-CoV-2 that can help in diagnosis and treatment of neurological manifestation of this disease. As more data become available, we will gain a more nuanced clarity regarding neuro-patho-physiology and genesis of COVID-19. SARS-CoV-2 is a highly adaptable zoonotic virus that uses ACE2 as an entry receptor into the host cells. Thus, presence of a functioning ACE2 protein on the exposed epithelial cells is responsible for the site of initial infection and the phenotype of COVID-19 clinical manifestation. The glycoprotein-studded viral envelope renders the virus agile and adaptable (Li et al. 2020; Korber et al. 2020). As such, SARS-CoV-2 will likely persist beyond the current pandemic requiring worldwide augmentation of clinical capacity for treatment. A detailed understanding of its mode of infection will assist us in devising appropriate therapies to mitigate neurological effects of the disease.
Loss of sense of taste and smell is an initial symptom of COVID-19 because the virus enters the nasal and oral cavities first and the epithelial cells are the receptors for these senses. COVID-19-associated stroke occurs because of likely injury to vascular endothelial cells and coagulopathy that then cause thrombo-embolism. Encephalopathy in acute and convalescent COVID-19 patients is multifactorial and likely secondary to hypoxia as well as metabolic and immunological abnormalities. Thus far, there is no clear evidence that coronaviruses cause inflammatory neuromuscular diseases via direct invasion of peripheral nerves or muscles or via molecular mimicry. It appears that neurologic injuries in COVID-19 patients are likely indirect and a bystander injury, since SARS-CoV-2 does not have tropism for the neural cells and tissues.
References
Ackermann M, Verlenden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstaplel A, Werlein C, Stark H, Zakov A, Li WW, Li VW, Mentzger SJ, Jonigk D (2020) Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. NEJM. 383:120–128. https://doi.org/10.1056/NEJMoa2015432
Alberti P, Beretta S, Piatti M, Karantzoulis A, Piatti ML, Santoro P, Viganò M, Giovannelli G, Pirro F, Montisano DA, Appollonio I, Ferrarese C (2020) Guillain-Barré syndrome related to COVID-19 infection. Neurol Neuroimmunol Neuroinflammation 7(4):e741
Alcami A, Koszinowski UH (2000) Viral mechanisms of immune evasion. Trends Microbiol 8(9):410–418. https://doi.org/10.1016/s0966-842x(00)01830-8
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF (2020) The proximal origin of SARS-CoV-2. Nat Med 26:450–452. https://doi.org/10.1038/s41591-020-0820-9
Atkinson BT, Jasuja R, Chen VM, Nandivada P, Furie B, Furie BC (2010) Laser-induced endothelial cell activation supports fibrin formation. Blood 116(22):4675–4683. https://doi.org/10.1182/blood-2010-05-283986
Avula A, Nalleballe K, Narula N, Sapozhnikov S, Dandu V, Toom S, Glaser A, Elsayegh D (2020) COVID-19 presenting as stroke. Brain Behav Immun 28:S0889–1591(20)30685–1. https://doi.org/10.1016/j.bbi.2020.04.077
Bagot CN, Arya R (2008) Virchow and his triad: a question of attribution. Br J Haematol 143(2):180–190. https://doi.org/10.1111/j.1365-2141.2008.07323.x
Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, Daßler-Plenker J, Guerci P, Huynh C, Knight JS, Loda M, Looney MR, McAllister F, Rayes R, Renaud S, Rousseau S, Salvatore S, Schwartz RE, Spicer JD, Yost CC, Weber A, Zuo Y, Egeblad M (2020) Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med 217(6):e20200652. https://doi.org/10.1084/jem.20200652
Becker RC (2020) Toward understanding the 2019 Coronavirus and its impact on the heart. J Thromb Thrombolysis 50(1):33–42. https://doi.org/10.1007/s11239-020-02107-6
Belouzard S, Millet JK, Licitra BN, Whittaker GR (2012) Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4:1011–1033. https://doi.org/10.3390/v4061011
Billingham RE, Brent L, Medawar PB (1953) ‘Actively acquired tolerance’ of foreign cells. 1953. J Immunol 184(1):5–8. https://doi.org/10.4049/jimmunol.0990109
Bloch EM, Shoham S, Casadevall A, Sachais BS, Shaz B, Winters JL, van Buskirk C, Grossman BJ, Joyner M, Henderson JP, Pekosz A, Lau B, Wesolowski A, Katz L, Shan H, Auwaerter PG, Thomas D, Sullivan DJ, Paneth N, Gehrie E, Spitalnik S, Hod EA, Pollack L, Nicholson WT, Pirofski LA, Bailey JA, Tobian AA (2020) Deployment of convalescent plasma for the prevention and treatment of COVID-19. J Clin Invest 130(6):2757–2765. https://doi.org/10.1172/JCI138745
Braun E, Sauter D (2019) Furin-mediated protein processing in infectious diseases and cancer. Clin Transl Immunol 8(8):e1073. https://doi.org/10.1002/cti2.1073
Castellano P, Prevedel L, Eugenin EA (2017) HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Sci Rep 7(1):12866. https://doi.org/10.1038/s41598-017-12758-w
Chen L, Liu M, Zhang Z, Qiao K, Huang T, Chen M, Xin N, Huang Z, Liu L, Zhang G, Wang J (2020) Ocular manifestations of a hospitalised patient with confirmed 2019 novel coronavirus disease. Br J Ophthalmol 104(6):748–751. https://doi.org/10.1136/bjophthalmol-2020-316304
Cooper KW, Brann DH, Farruggia MC, Bhutani S, Pellegrino R, Tsukahara T, Weinreb C, Joseph PV, Larson ED, Parma V, Albers MW, Barlow LA, Datta SR, Di Pizio A (2020) COVID-19 and the chemical senses: supporting players take center stage. Neuron. S0896–6273(20)30486–4. https://doi.org/10.1016/j.neuron.2020.06.032
Corbett KS, Edwards DK, Leist SR, Abiona OM, Boyoglu-Barnum S, Gillespie RA, Himansu S, Schäfer A, Ziwawo CT, DiPiazza AT, Dinnon KH, Elbashir SM, Shaw CA, Woods A, Fritch EJ, Martinez DR, Bock KW, Minai M, Nagata BM, Hutchinson GB, Wu K, Henry C, Bahi K, Garcia-Dominguez D, Ma L, Renzi I, Kong WP, Schmidt SD, Wang L, Zhang Y, Phung E, Chang LA, Loomis RJ, Altaras NE, Narayanan E, Metkar M, Presnyak V, Liu C, Louder MK, Shi W, Leung K, Yang ES, West A, Gully KL, Stevens LJ, Wang N, Wrapp D, Doria-Rose NA, Stewart-Jones G, Bennett H, Alvarado GS, Nason MC, Ruckwardt TJ, McLellan JS, Denison MR, Chappell JD, Moore IN, Morabito KM, Mascola JR, Baric RS, Carfi A, Graham BS (2020, 2020) SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature. https://doi.org/10.1038/s41586-020-2622-0
De Felice FG, Tovar-Moll F, Moll J, Munoz DP, Ferreira ST (2020) Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the central nervous system. TINS 43(6):355–357
de Siqueira IC, Guerreiro Rodrigues S, Caricio Martins L, Vasilakis N, Caires Novaes MA, Junior Alcântara LC, et al (2016) Guillain–Barré syndrome after zika virus infection in Brazil. Am J Trop Med Hyg 95(5):1157–1160
Dubois C, Laurence PD, Gainor JF, Furie B (2007) Thrombin-initiated platelet activation in vivo is vWF independent during thrombus formation in a laser injury model. J Clin Invest 117(4):953–960
Fong TG, Tulebaey SR, Inouye SK (2011) Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol 5(4):210–220. https://doi.org/10.1038/nrneurol.2009.24
Ghafouri M, Amini S, Khalili K, Sawaya B (2006) HIV-1 associated dementia: symptoms and causes. Retrovirology 3:28. https://doi.org/10.1186/1742-4690-3-28
Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, Raizada MK, Grant MB, Oudit GY (2020) Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res 126(10):1456–1474. https://doi.org/10.1161/CIRCRESAHA.120.317015
Gibb R, Redding DW, Chin KQ, Donnelly CA, Blackburn TM, Newbold T, Jones KE (2020) Zoonotic host diversity increases in human-dominated ecosystems. Nature. https://doi.org/10.1038/s41586-020-2562-8
Godschalk PCR, Heikema AP, Gilbert M, Komagamine T, Wim Ang C, Glerum J, Brochu D, Li J, Yuki N, Jacobs BC, van Belkum A, Endtz HP (2004) The crucial role of Campylobacter jejuni genes in anti-ganglioside antibody induction in Guillain-Barré syndrome. J Clin Investig 114(11):1659–1665
Goldstein JM, Azizi SA, Booss J, Vollmer TL. (1993) Human immunodeficiency virus-associated motor axonal polyradiculoneuropathy. Arch Neurol 50(12):1316–1319. https://doi.org/10.1001/archneur.00540120031009
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, Liu L, Shan H, Lei CL, DSC H, Du B, Li LJ, Zeng G, Yuen KY, Chen RC, Tang CL, Wang T, Chen PY, Xiang J, Li SY, Wang JL, Liang ZJ, Peng YX, Wei L, Liu Y, Hu YH, Peng P, Wang JM, Liu JY, Chen Z, Li G, Zheng ZJ, Qiu SQ, Luo J, Ye CJ, Zhu SY, Zhong NS, China Medical Treatment Expert Group for Covid-19 (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382(18):1708–1720. https://doi.org/10.1056/NEJMoa2002032
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203(2):631–637. https://doi.org/10.1002/path.1570
Haveri A, Smura T, Kuivanen S, Österlund P, Hepojoki J, Ikonen N, Pitkäpaasi M, Blomqvist S, Rönkkö E, Kantele A, Strandin T, Kallio-Kokko H, Mannonen L, Lappalainen M, Broas M, Jiang M, Siira L, Salminen M, Puumalainen T, Sane J, Melin M, Vapalahti O, Savolainen-Kopra C (2020) Serological and molecular findings during SARS-CoV-2 infection: the first case study in Finland. Euro Surveill 25(11):2000266. https://doi.org/10.2807/1560-7917.ES.2020.25.11.2000266
Heald-Sargent T, Gallagher T (2012) Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 4:557–580. https://doi.org/10.3390/v40400557
Helms J, Kremer S, Merdiji H, Clere-Jehl R, Schenck M, Kummerlen C, Collange O, Boulay C, Fafi-Kremer S, Ohana M, Anheim M (2020a) Neurologic Features in Severe SARS-CoV-2 Infection. N Engl J Med 382:2268–2270. https://doi.org/10.1056/NEJMc2008597
Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X et al (2020b) High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 46:1089–1098. https://doi.org/10.1007/s00134-020-06062-x
Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH 3rd, Kato T, Lee RE, Yount BL, Mascenik TM, Chen G, Olivier KN, Ghio A, Tse LV, Leist SR, Gralinski LE, Schäfer A, Dang H, Gilmore R, Nakano S, Sun L, Fulcher ML, Livraghi-Butrico A, Nicely NI, Cameron M, Cameron C, Kelvin DJ, de Silva A, Margolis DM, Markmann A, Bartelt L, Zumwalt R, Martinez FJ, Salvatore SP, Borczuk A, Tata PR, Sontake V, Kimple A, Jaspers I, O'Neal WK, Randell SH, Boucher RC, Baric RS (2020) SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 27:S0092–8674(20)30675–9. https://doi.org/10.1016/j.cell.2020.05.042
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395(10223):497–506. https://doi.org/10.1016/S0140-6736(20)30183-5
Jain R, Young M, Dogra S, Kennedy H, Nguyen V, Jones S, Bilaloglu S, Hochman K, Raz E, Galetta S, Horwtiz L (2020) COVID-19 related neuroimaging findings: a signal of thromboembolic complications and a strong prognostic marker of poor patient outcome. J Neurol Sci 414:116923. https://doi.org/10.1016/j.jns.2020.116923
Janardhan V, Wolf PA, Kase CS, Massaro JM, D'Agostino RB, Franzblau C, Wilson PW (2004) Anticardiolipin antibodies and risk of ischemic stroke and transient ischemic attack: the Framingham cohort and offspring study. Stroke 35(3):736–741. https://doi.org/10.1161/01.STR.0000117575.48205.2D
Joyner MJ, Wright RS, Fairweather D, Senefeld JW, Bruno KA, Klassen SA, Carter RA, Klompas M, Wiggins C, JRA S, Rea RF, Whelan ER, Clayburn AJ, Spiegel MR, Johnson PW, Lesser ER, Baker SE, Larson KF, Ripoll JG, Andersen KJ, Hodge DO, Kunze KL, Buras MR, MNP V, Herasevich V, Dennis JD, Regimbal RJ, Bauer PR, Blair JE, van Buskirk CM, Winters JL, Stubbs JR, Paneth NS, Verdun NC, Marks P, Casadevall A (2020) Early safety indicators of COVID-19 convalescent plasma in 5,000 patients. J Clin Invest. https://doi.org/10.1172/JCI140200
Ju B, Zhang Q, Ge J, Wang R, Sun J, Ge X, Yu J, Shan S, Zhou B, Song S, Tang X, Yu J, Lan J, Yuan J, Wang H, Zhao J, Zhang S, Wang Y, Shi X, Liu L, Zhao J, Wang X, Zhang Z, Zhang L (2020) Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 584(7819):115–119. https://doi.org/10.1038/s41586-020-2380-z
Kam YW, Okumura Y, Kido H, Ng LFP, Bruzzone R, altmeyer R (2009) Cleavage of the SARS Coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PLoS One 4(11):e7870. https://doi.org/10.1371/journal.pone.0007870
Kandemirli SG, Dogan L, Sarikaya ZT, Kara S, Akinci C, Kaya D, Kaya Y, Yildirim D, Tuzuner F, Yildirim MS, Ozluk E, Gucyetmez B, Karaarslan E, Koyluoglu I, Demirel Kaya HS, Mammadov O, Kisa Ozdemir I, Afsar N, Citci Yalcinkaya B, Rasimoglu S, Guduk DE, Kedir Jima A, Ilksoz A, Ersoz V, Yonca Eren M, Celtik N, Arslan S, Korkmazer B, Dincer SS, Gulek E, Dikmen I, Yazici M, Unsal S, Ljama T, Demirel I, Ayyıldız A, Kesimci I, Bolsoy Deveci S, Tutuncu M, Kizilkilic O, Telci L, Zengin R, Dincer A, Akinci IO, Kocer N (2020) Brain MRI findings in patients in the intensive care unit with COVID-19 infection. Radiology 8:201697. https://doi.org/10.1148/radiol.2020201697
Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, Song Y, Tzima E, Kennerknecht E, Niepmann M, von Bruehl ML, Sedding D, Massberg S, Günther A, Engelmann B, Preissner KT (2007) Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A 104(15):6388–6393. https://doi.org/10.1073/pnas.0608647104
Kansagra A, Goyal M, Hamilton S, Albers G (2020) Collateral effects of COVID-19 on stroke evaluation in the United States. NEJM 383:400–401. https://doi.org/10.1056/NEJMc2014816
Khailany R, Safdar M, Ozaslan M (2020) Genomic characterization of a novel SARS-CoV2. Gene Rep 19:100682. https://doi.org/10.1016/j.genrep.2020.100682
Kim JE, Heo JH, Kim HO, Song SH, Park SS, Park TH, Ahn JY, Kim MK, Choi JP (2017) Neurological complications during treatment of Middle East respiratory syndrome. J Clin Neurol 13(3):227–233. https://doi.org/10.3988/jcn.2017.13.3.227
Kitagawa Y (2005) Antiphospholipid syndrome and stroke. Rinsho Shinkeigaku. 45(11):852–855
Kleinschmidt-DeMasters BK, Gilden D (2006) The expanding spectrum of herpesvirus infections of the nervous systems. Brain Pathol 11(4):440–451. https://doi.org/10.1111/j.1750-3639.2001.tb00413.x
Klok FA, Kruip MJ, van der Meer NJ, Arbous MS, Gommers DA, Kant KM, Aptein FH, van Paassan J, Stals MA, Huisman MV, Endeman H (2020a) Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res 191:145–147. https://doi.org/10.1016/j.thromres.2020.04.013
Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers D, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, Endeman H (2020b) Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb Res 191:148–150. https://doi.org/10.1016/j.thromres.2020.04.041
Konig MF, Powell M, Staedtke V, Bai RY, Thomas DL, Fischer N, Huq S, Khallafallah A et al (2020) Preventing cytokine storm syndrome in COVID-19 using α-1 adrenergic receptor antagonists. J Clin Invest 130:3345–3347. https://doi.org/10.1172/JCI139642
Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, Hastie KM, Parker MD, Partridge DG, Evans CM, Freeman TM, de Silva TI, McDanal C, Perez LG, Tang H, Moon-Walker A, Whelan SP, LaBranche CC, Saphire EO, Montefiori DC, Angyal A, Brown RL, Carrilero L, Green LR, Groves DC, Johnson KJ, Keeley AJ, Lindsey BB, Parsons PJ, Raza M, Rowland-Jones S, Smith N, Tucker RM, Wang D, Wyles MD (2020) Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182(4):812–827.e19
Kotfis K, Roberson SW, Wilson JE, Dabrowski W, Pun BT, Ely EW (2020) COVID-19: ICU delirium management during SARS-CoV-2 pandemic. Crit Care 24:176. https://doi.org/10.1186/s13054-020-02882-x
Kreer C, Zehner M, Weber T, Ercanoglu MS, Gieselmann L, Rohde C, Halwe S, Korenkov M, Schommers P, Vanshylla K, Di Cristanziano V, Janicki H, Brinker R, Ashurov A, Krähling V, Kupke A, Cohen-Dvashi H, Koch M, Eckert JM, Lederer S, Pfeifer N, Wolf T, Vehreschild MJGT, Wendtner C, Diskin R, Gruell H, Becker S, Klein F (2020) Longitudinal isolation of potent near-germline SARS-CoV-2-neutralizing antibodies from COVID-19 patients. Cell. S0092–8674(20)30821–7. https://doi.org/10.1016/j.cell.2020.06.044
Kuba K, Imai Y, Penninger JM (2013) Multiple functions of angiotensin-converting enzyme 2 and its relevance in cardiovascular diseases. Circ J 77(2):301–308. https://doi.org/10.1253/circj.cj-12-1544
Lafon M (2008) Rabies virus receptors. BMC Proc 2(Suppl 1):S26
Lamers MM, Beumer J, van der Vaart J, Knoops K, Puschhof J, Breugem TI, Ravelli RBG, Paul van Schayck J, Mykytyn AZ, Duimel HQ, van Donselaar E, Riesebosch S, HJH K, Schipper D, van de Wetering WJ, de Graaf M, Koopmans M, Cuppen E, Peters PJ, Haagmans BL, Clevers H (2020) SARS-CoV-2 productively infects human gut enterocytes. Science 369(6499):50–54. https://doi.org/10.1126/science.abc1669
Lan J, Ge J, Yu J, Yu J et al (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581(7807):215–220. https://doi.org/10.1038/s41586-020-2180-5
Larvie M, Lev MH, Hess CP (2020) More on neurologic features on severe SARS-CoV2 infection. N Engl J Med 382:e110. https://doi.org/10.1056/NEJMc2015132
Latronico N, Recupero D, Candiani A, Guarneri B, De Maria G, Antonini L, Fenzi F, Tomelleri G, Tonin P, Rizzuto N (1996) Critical illness myopathy and neuropathy. Lancet 347(9015):1579–1582
Laurendon T, Radulesco T, Mugnier J, Gérault M, Chagnaud C, El Ahmadi AA, Varoquaux A (2020) Bilateral transient olfactory bulb edema during COVID-19-related anosmia. Neurology 95(5):224–225. https://doi.org/10.1212/WNL.0000000000009850
Li W, Moore M, Vasilieva N, Sui J, Wong SK, Berne MA, Somasondara M, Sullivan JL, Luzuriaga K, Greenough T, Choe H, Farzan M (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454. https://doi.org/10.1038/nature02145
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, Tan W (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395(10224):565–574. https://doi.org/10.1016/S0140-6736(20)30251-8
Luis AD, Hayman DTS, O’Shea TJ, Cryan PM, Gilbert AT, Pulliam JRC, Mills JN, Timonin ME, Willis CKR, Cunningham AA, Fooks AR, Rupprecht CE, Wood JLN, Webb CT (2013) A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc R Soc B 280:20122753. https://doi.org/10.1098/rspb.2012.2753
Luis AD, O'Shea TJ, Hayman DTS, Wood JLN, Cunningham AA, Gilbert AT, Mills JN, Webb CT, Gomez JM (2015) Network analysis of host-virus communities in bats and rodents reveals determinants of cross-species transmission. Ecol Lett 18(11):1153–1162
Luo JJ, Dun N (2020) Could SARSCoV-2 invade the brain? J Neurol Exp Neurosci 6(S1):S4–S8. https://doi.org/10.17756/jnen.2020-S1-002
Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, Baxter-Stoltzfus, Laurence J (2020) Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases Transl Res S1931-5244(20)30070–0. DOI:https://doi.org/10.1016/j.trsl.2020.04.007, 30013
Mahammedi A, Saba L, Vagal A, Leali M, Rossi A, Gaskill M, Sengupta S, Zhang B, Carriero A, Bachir S, Crivelli P, Paschè A, Premi E, Padovani A, Gasparotti R (2020) Imaging in neurological disease of hospitalized COVID-19 patients: an Italian multicenter retrospective observational study. Radiology 21:201933. https://doi.org/10.1148/radiol.2020201933
Mancia G, Rea F, Ludergnani M, Apolone G, Corrao G (2020) Renin-angiotensin-aldosterone system blockers and the risk of Covid-19. N Engl J Med:NEJMoa2006923. https://doi.org/10.1056/NEJMoa2006923
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, Chang J, Hong C, Zhou Y, Wang D, Miao X, Li Y, Hu B (2020) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol 77:683. https://doi.org/10.1001/jamaneurol.2020.1127
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, HLH Across Speciality Collaboration, UK (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395(10229):1033–1034. https://doi.org/10.1016/S0140-6736(20)30628-0
Menni C, Valdes AM, Freidin MB, Sudre CH, Nguyen LH, Drew DA, Ganesh S, Varsavsky T, Jorje Cardoso M, El-Sayed Mustafa JS, Visconti A, Pirro H, Bowyer RC, Mangino M, Falchi M, Wolfe J, Ourselin S, Chan AT, Steves CJ, Spector TD (2020) Real-time tracking of self-reported symptoms to predict potential COVID-19. Nat Med 26:1037–1040. https://doi.org/10.1038/s41591-020-0916-2
Merkler AE, Parikh NS, Mir S, Gupta A, Kamel H, Lin E, Lantos J, Schenck EJ, Goyal P, Bruce SS, Kahan J, Lansdale KN, LeMoss NM, Murthy SB, Stieg PE, Fink ME, Iadecola C, Segal AZ, Cusick M, Campion TR Jr, Diaz I, Zhang C, Navi BB (2020) Risk of ischemic stroke in patients with coronavirus disease 2019 (COVID-19) vs patients with influenza. JAMA Neurol 2:e202730. https://doi.org/10.1001/jamaneurol.2020.2730
Nachamkin I, Allos BM, Ho T (1998) Campylobacter species and Guillain-Barre syndrome. Clin Microbiol Rev 11(3):555–567
Ni L, Ye F, Cheng ML, Feng Y, Deng YQ, Zhao H, Wei P, Ge J, Gou M, Li X, Sun L, Cao T, Wang P, Zhou C, Zhang R, Liang P, Guo H, Wang X, Qin CF, Chen F, Dong C (2020) Detection of SARS-CoV-2-specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity 52(6):971–977.e3. https://doi.org/10.1016/j.immuni.2020.04.023
Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, De Leacy RA, Shigematsu T, Ladner TR, Yaeger KA, Skliut M, Weinberger J, Dangayach NS, Bederson JB, Tuhrim S, Fifi JT (2020) Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med 382(20):e60. https://doi.org/10.1056/NEJMc2009787
Poutanen SM (2018) Human coronaviruses. In: Long S (ed) Principle and practice of pediatric infectious diseases. 5th edn. Elsevier, New York, pp 1148–1152. https://doi.org/10.1016/B978-0-323-40181-4.00222-X
Pugin D, Vargas MI, Thieffry C, Schibler M, Grosgurin O, Pugin J, Lalive PH (2020) COVID-19-related encephalopathy responsive to high doses glucocorticoids. Neurology. https://doi.org/10.1212/WNL.0000000000010354
Qin C, Zhou L, Hu Z, Yang S, Zhang S, Chen M, Yu H, Tian DS, Wang W (2020) Clinical characteristics and outcomes of COVID-19 patients with a history of stroke in Wuhan, China. Stroke 51(7):2219–2223. https://doi.org/10.1161/STROKEAHA.120.030365
Reynolds HR, Adhikari S, Pulgarin C, Troxel AB, Iturrate E, Johnson SB, Hausvater A, Newman JD, Berger JS, Bangalore S, Katz SD, Fishman GI, Kunichoff D, Chen Y, Ogedegbe G, Hochman JS (2020) Renin-angiotensin-aldosterone system inhibitors and risk of Covid-19. N Engl J Med. https://doi.org/10.1056/NEJMoa2008975
Rogers JP, Chesney E, Oliver D, Pollak T, McGuire P, Fusar-Poli P, Zandi MS, Lewis G, David AS (2020) Psychiatric and neuropsychiatric presentations associated with severe coronavirus infections: a systematic review and meta-analysis with comparison to the COVID-19 pandemic. Lancet Psychiatry. https://doi.org/10.1016/S2215-0366(20)30203-0
Sawicki SG (2009) Coronavirus genome replication. In: Cameron CE (ed) Viral genome replication. Springer, Boston, pp. 25–39. https://doi.org/10.1007/b135974_2
Sedaghat Z, Karimi N (2020) Guillain Barre syndrome associated with COVID-19 infection: A case report. J Clin Neurosci. 76:233–235. https://doi.org/10.1016/j.jocn.04.062
Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, Li F (2020a) Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci 117(21):11727–11734. https://doi.org/10.1073/pnas.2003138117
Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A, Li F (2020b) Structural basis of receptor recognition by SARS-CoV-2. Nature 581(7807):221–224. https://doi.org/10.1038/s41586-020-2179-y
Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, Quan S, Zhang F, Sun R, Qian L, Ge W, Liu W, Liang S, Chen H, Zhang Y, Li J, Xu J, He Z, Chen B, Wang J, Yan H, Zheng Y, Wang D, Zhu J, Kong Z, Kang Z, Liang X, Ding X, Ruan G, Xiang N, Cai X, Gao H, Li L, Li S, Xiao Q, Lu T, Zhu Y, Liu H, Chen H, Guo T (2020) Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 182(1):59–72.e15. https://doi.org/10.1016/j.cell.2020.05.032
Shepherd SJ, Black H, Thomson EC, Gunson RN (2017) HIV positive patient with GBS-like syndrome. JMM Case Rep 2017:4. https://doi.org/10.1099/jmmcr.0.005107
Shi R, Shan C, Duan X, Chen Z, Liu P, Song J, Song T, Bi X, Han C, Wu L, Gao G, Hu X, Zhang Y, Tong Z, Huang W, Liu WJ, Wu G, Zhang B, Wang L, Qi J, Feng H, Wang F-S, Wang Q, Fu Gao G, Yuan Z, Yan J (2020) A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 584(7819):120–124
Solomon MD, McNulty EJ, Rana JS, Leong TK, Lee C, Sung SH, Ambrosy AP, Sidney S, Go AS (2020a) The Covid-19 pandemic and the incidence of acute myocardial infarction. N Engl J Med 383:691–693. https://doi.org/10.1056/NEJMc2015630
Solomon IH, Normandin E, Mukerji SS, Keller K, Ali A, Adams G, Hornick J, Sabeti P (2020b) Neuropathological features of Covid-19. NEJM. https://doi.org/10.1056/NEJMc2019373
Song WC, Fitzgerald GA (2020) COVID-19, microangiopathy, hemostatic activation and complement. J Clin Invest. https://doi.org/10.1172/JCI140183
Stopsack KH, Mucci LA, Antonarakis ES, Nelson PS, Kantoff PW (2020) TMPRSS2 and COVID-19: serendipity or opportunity for intervention? Cancer Discov 10(6):779–782. https://doi.org/10.1158/2159-8290.CD-20-0451
Sungnak W, Huang N, Becavin C, Berg M, Queen R, Litvinukova M, Talavera-Lopez C, Worlock K, Yoshida M, Barnes JL (2020) SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med 26(5):681–687
Thomas G (2002) Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol 3(10):753–766. https://doi.org/10.1038/nrm934
Tortorici MA, Veesler D (2019) Structural insights into coronavirus entry. Adv In Virus Res 105:93. https://doi.org/10.1016/bs.aivir.019.08.002
Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Guzzoni MG, Franciotta D, Baldanti F, Dturi R, Pastorino P, Cavallini A, Micieli G (2020) Guillain-Barre syndrome associated with SARS-CoV-2. N Engl J Med 382:2574–2576. https://doi.org/10.1056/NEJMc2009191
Varga S, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel S, Mehra M, Schuepback RA, Ruschitzka F, Moch H (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395:1417–1418. https://doi.org/10.1016/S0140-6736(20)30937-5
Visser LH, van der Meche FGA, Meulstee J, Rothbarth P, Jacobs BC, Schmitz PIM, van Doorn PA (1996) Cytomegalovirus infection and Guillain-Barre syndrome: The clinical, electrophysiologic, and prognostic features. Neurology 47(3):668–673
Walls AC, Tortorici MA, Frenz B, Snijder J, Li W, Rey FA, DiMaio F, Bosch B-J, Veesler D (2016) Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat Struct Mol Biol 23(10):899–905
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181:281–292. https://doi.org/10.1016/j.cell.2020.02.058
Wang J, Wang Z, Liu R, Shuai L, Wang X, Luo J, Wang C, Chen W, Wang X, Ge J, He X, Wen Z, Bu Z (2018) Metabotropic glutamate receptor subtype 2 is a cellular receptor for rabies virus. PLoS Pathog 14(7):e1007189. https://doi.org/10.1371/journal.ppat.1007189
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367:1260–1263
Xiong W, Mu J, Guo J, Lu L, Liu D, Luo J, Li N, Liu J, Yang D, Gao H, Zhang Y, Lin M, Shen S, Zhang H, Chen L, Wang G, Luo F, Li W, Chen S, He L, Sander JW, Zhou D (2020) New onset neurologic events in people with COVID-19 infection in three regions in China. Neurology. https://doi.org/10.1212/WNL.0000000000010034
Xu H, Zhong L, Deng J, Peng J, Dan H, Zeng X, Li T, Chen Q (2020a) High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci 12:8. https://doi.org/10.1038/s41368-020-0074-x
Xu Y, Li X, Zhu B, Liang H, Fang C, Gong Y, Guo Q, Sun X, Zhao D, Shen J, Zhang H, Liu H, Xia H, Tang J, Zhang K, Gong S (2020b) Characteristics of pediatric SARS-CoV-2 infection and potential evidence for persistent fecal viral shedding. Nat Med 26:502–505. https://doi.org/10.1038/s41591-020-0817-4
Yaghi S, Ishida K, Torres J, Mac Grory B, Raz E, Humbert K, Henninger N, Trivedi T, Lillemoe K, Alam S, Sanger M, Kim S, Scher E, Dehkharghani S, Wachs M, Tanweer O, Volpicelli F, Bosworth B, Lord A, Frontera J (2020) SARS-CoV-2 and stroke in a New York Healthcare System. Stroke 51(7):2002–2011. https://doi.org/10.1161/STROKEAHA.120.030335 Erratum in: Stroke. 2020 Aug;51(8):e179
Zhang H, Li HB, Lyu JR, Lei XM, Li W, Wu G, Lyu J, Dai ZM (2020a) Specific ACE2 expression in small intestinal enterocytes may cause gastrointestinal symptoms and injury after 2019-nCoV infection. Int J Infect Dis 18(96):19–24. https://doi.org/10.1016/j.ijid.2020.04.027
Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, Chen H, Ding X, Zhao H, Zhang H, Wang C, Zhao J, Sun X, Tian R, Wu W, Wu D, Ma J, Chen Y, Zhang D, Xie J, Yan X, Zhou X, Liu Z, Wang J, Du B, Qin Y, Gao P, Qin X, Xu Y, Zhang W, Li T, Zhang F, Zhao Y, Li Y, Zhang S (2020b) Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med 382(17):e38. https://doi.org/10.1056/NEJMc2007575
Zhang X, Tan Y, Ling Y, Lu G, Liu F, Yi Z, Jia X, Wu M, Shi B, Xu S, Chen J, Wang W, Chen B, Jiang L, Yu S, Lu J, Wang J, Xu M, Yuan Z, Zhang Q, Zhang X, Zhao G, Wang S, Chen S, Lu H (2020c) Viral and host factors related to the clinical outcome of COVID-19. Nature. 583:437–440. https://doi.org/10.1038/s41586-020-2355-0
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Azizi, S.A., Azizi, SA. Neurological injuries in COVID-19 patients: direct viral invasion or a bystander injury after infection of epithelial/endothelial cells. J. Neurovirol. 26, 631–641 (2020). https://doi.org/10.1007/s13365-020-00903-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-020-00903-7