
Abstract
The common vole (Microtus arvalis) and the field vole (Microtus agrestis) are morphologically similar species but are ecological distinctive and differ in the details of their evolutionary history as revealed by mitochondrial DNA (mtDNA). The aim of this study is to describe patterns of genetic variability using microsatellite markers in populations of the common and field vole in Poland using museum specimens, to assess the degree of congruence with mtDNA variation and thereby determine the factors that influence current patterns of gene flow. We genotyped 190 individuals of the common vole at 11 loci and 190 individuals of the field vole at 13 loci. Overall differentiation based on F ST was higher for the common vole than in the field vole. We detected a significant isolation by distance pattern for both species. Bayesian analysis in STRUCTURE identified Eastern and Western geographic groups in Poland based on microsatellites for both species. The location of river barriers is likely to be the main factor in these partitions. The eastern-western subdivision with microsatellites does not coincide with the distribution of mtDNA lineages for either species. Unlike previous studies in the common and field vole elsewhere in Europe, we found no evidence of reproductive isolation between the mtDNA lineages of these species at their contact zones in Poland. This study highlights the different roles of evolutionary history and landscape in shaping contemporary genetic structure in voles in Poland.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Last Glacial Maximum (LGM), which lasted from approximately 27.5–19 thousand years before present (ka bp; Clark et al. 2009), forced European species to survive the severe climatic conditions by retreating to refugia located in the south (in three peninsulas: Apennine, Balkan and Iberian; Taberlet et al. 1998; Hewitt 1999) and further north (Dordogne and the Carpathian Basin; Kotlík et al. 2006; Sommer and Nadachowski 2006). In Eastern Europe, post-LGM expansion has led to the formation of a phylogeographic ‘suture zone’ in present-day Poland in various mammalian species between multiple mitochondrial DNA (mtDNA) lineages originating from different refugia (Wójcik et al. 2010; McDevitt et al. 2012). In this region, three mtDNA lineages in the bank vole (Clethrionomys glareolus; Wójcik et al. 2010), four lineages in the weasel (Mustela nivalis; McDevitt et al. 2012), two lineages in the field vole (Microtus agrestis; Herman et al. 2014) and two lineages in the common vole (Microtus arvalis; Stojak et al. 2015) are present.
Contrasting population histories and ecological preferences of different species likely promoted the various patterns of re-colonization during the return of more favourable conditions after the LGM (Heckel et al. 2005). If we consider the two aforementioned Microtus vole species (the common and field vole), they are morphologically similar species but differ in their population histories in Europe (Herman et al. 2014; Stojak et al. 2015). For the common vole, two different chromosomal forms have been identified using cytogenetics, arvalis and obscurus. The hybrid zone between them occurs in central European Russia (Meyer et al. 1997; Haynes et al. 2003; Jaarola et al. 2004; Bulatova et al. 2010a, b, 2013). For the arvalis form, six main mtDNA lineages have been described in Europe: Western-South, Western-North, Central, Italian, Eastern and Balkan (Haynes et al. 2003; Tougard et al. 2008; Bužan et al. 2010; Martínková et al. 2013; Stojak et al. 2015). For the field vole, three main evolutionary units (which may represent cryptic species) have been described—Portuguese, Southern and Northern (Paupério et al. 2012). For the Northern evolutionary unit in the field vole, six main mtDNA lineages have been identified: Eastern, Scandinavian, Central European, French, North British and Western (Herman and Searle 2011; Herman et al. 2014). The differences in the distribution and origin of the mtDNA lineages between the common and field vole are evident in Poland (Herman et al. 2014; Stojak et al. 2015). The phylogeographic suture zone in Poland that was formed after the LGM in the common vole consists of two lineages, the Eastern lineage likely originating from the Carpathian refugium (based on fossil records and molecular data; Pazonyi 2004; Sommer and Nadachowski 2006; Stojak et al. 2015) and the Central lineage from a refugium originally suggested to be Italy (Haynes et al. 2003) but likely located further north (Heckel et al. 2005; based on molecular data). For the field vole, the two lineages that occur in Poland (Central European and Western; Fig. 1) are thought to represent expansion following the Younger Dryas cold period (12.9–11.7 ka bp; Herman and Searle 2011; Herman et al. 2014). There appears to have been a single LGM refugium for the Northern evolutionary unit of the field vole (Herman and Searle 2011), but the location of this is uncertain. The Carpathians is a possibility, following the suggestion of Jaarola and Searle (2002); however, a later fossil assessment (Pazonyi 2004) suggested that field voles were not part of the LGM community in the Carpathians.

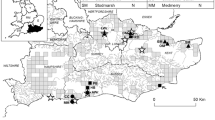
a Genetic structure of Microtus arvalis (top) and Microtus agrestis (bottom) populations in Poland based on a Bayesian analysis of microsatellite data in STRUCTURE, coloured according to their assignment to one of two genetic clusters at q ≥ 0.8 (yellow—the Western group; blue—the Eastern group). Individuals coloured in red represent admixed individuals between the two genetic clusters (0.2 < q < 0.8). Numbers on maps match sampling localities presented in Tables 1 and 2. The diameter of the circles represents the number of sampled individuals within the population. b Mitochondrial DNA lineage distributions described by Stojak et al. (2015) for M. arvalis and Herman et al. (2014) for M. agrestis. See the aforementioned publications for further information regarding population localities and the number of individuals used (Colour figure online)
In terms of the ecology of the common and field vole, both are grassland species, but the common vole inhabits open areas including farmlands (Amori et al. 2008), while the field vole prefers more damp habitats like meadows, marshes or river banks (Kryštufek et al. 2008). It is therefore apparent that different historical and contemporary processes may impact upon patterns of genetic structure in these two Microtus species.
Several studies have now described the phylogeography and genetic structure of multiple mammalian species using mtDNA in the vicinity of present-day Poland (Wójcik et al. 2010; McDevitt et al. 2012; Stojak et al. 2015). However, there has been little attempt to study the influence of these ancient processes on contemporary genetic structure in this region. The application of genetic markers such as microsatellites will allow an assessment of contemporary patterns of gene flow and differentiation in areas where different mtDNA lineages come into contact (Heckel et al. 2005; Godinho et al. 2008; Beysard et al. 2012). For the common vole, mtDNA and microsatellites have been used in other regions in Europe to find general congruence in patterns of genetic structure between the two types of marker (Heckel et al. 2005; Braaker and Heckel 2009; Beysard and Heckel 2014), including in an area of contact involving the same mtDNA lineages as found in Poland (Eastern and Central) but along the German-Czech Republic border (Beysard and Heckel 2014). Given that differences in genetic structure might be expected between mtDNA and microsatellite markers, because of differences in mutation rate, effective population sizes and inheritance, the congruent patterns for the two types of marker in the previous studies in the common vole indicate that a degree of contemporary reproductive isolation exists between mtDNA lineages in this species. Nuclear-mitochondrial congruence has also been found in a contact zone between the Southern and Northern evolutionary units (‘cryptic species’) of the field vole in the Swiss Jura mountains, revealing a degree of post-mating isolation (Beysard et al. 2012); the contact zone in Poland involves two mtDNA lineages within the Northern unit.
The purposes of our study were as follows: (i) to describe the patterns of genetic variability using microsatellite markers in populations of the common and field vole in Poland and (ii) to determine whether contemporary genetic structure demonstrates agreement with mtDNA lineage distribution found in these species across Poland (Herman et al. 2014, Stojak et al. 2015). Similar to these previous studies on mtDNA in both species, we are utilizing well-preserved museum specimens collected between 1960 and 1970 to allow for direct temporal comparisons of genetic structure between the marker types. Even though DNA degradation is an obvious concern for these types of specimens (Bi et al. 2013), a number of studies in different species have now successfully amplified microsatellite loci in museum specimens to infer genetic structure and diversity (e.g. Bourke et al. 2010; Rubidge et al. 2012).
Materials and methods
Samples
In this study, we used 380 museum specimens archived in the scientific zoological collection of the Mammal Research Institute of the Polish Academy of Sciences in Białowieża that were collected from different parts of Poland between 1960 and 1970. Specimens that yielded high DNA quantity and quality were chosen. We analysed 190 M. arvalis from 39 localities and 190 M. agrestis from 23 localities (Fig. 1). Sample sizes for the populations varied for both species, ranging from one to 11 collected individuals. A complete list of all specimens, localities and number of individuals per population is given in Tables 1 and 2.
Molecular methods
DNA was extracted from dried legs, using the Qiagen DNeasy Tissue Kit according to the manufacturer’s protocol. Based on their cross-species amplification in voles (Rudá et al. 2009; Czajkowska et al. 2010), an initial panel of 23 microsatellite markers from five species of vole (M. arvalis, M. agrestis, Microtus oeconomus, Microtus montebelli and Arvicola terrestris) was screened in a subset of individuals from both species to assess amplification success, ease of genotyping and variation (Table 3). Of these 23 loci, 11 were demonstrated to reliably genotype common vole individuals, and 13 loci were used for genotyping field vole individuals (Table 3). Microsatellites were amplified by polymerase chain reaction (PCR) in 10-μl reactions containing 5 μl of Multiplex PCR Master Mix (Qiagen), 1 μl template DNA, ddH2O and primer volumes ranging from 0.2 to 0.4 μl (from a stock concentration of 10 μM). Two or three primer pairs were combined in each multiplex reaction (see Tables S1 and S2 for details of each multiplex reaction). Negative PCR controls without template DNA for each set of samples and each multiplex were included. Amplification conditions were as follows: initial denaturation of 95 °C for 15 min, 35 cycles of 94 °C for 45 s, 54 °C for 90 s and 72 °C for 60 s, followed by a final elongation step of 72 °C for 45 min. Then, 1 μl of product was added to 0.2 μl of size standard GeneScan 500 Liz dye (Life Technologies) and 10 μl of Buffer HiDi Formamide (Applied Biosystems). After 3 min of incubation at 96 °C, the mixture was then analysed with an ABI Prism 3100 DNA sequencer and GeneMarker v1.7.0 was used to visualise the microsatellite alleles and determine the genotypes.
Following the procedure of Bourke et al. (2010), we repeated each PCR three times to avoid artefacts and mistakes caused by potential fragmentation of DNA in museum samples. All genotypes were independently scored twice for these three repeats. We accepted a homozygote if it was scored as such on all three occasions (or two out of three if it did not amplify in one of the repeats). A heterozygote was scored if it appeared on two out of three occasions. If these criteria were not met, alleles were scored as missing data (Table 4).
Genetic structure of populations
Only populations with five or more individuals were included in all population-level analyses. Observed and expected heterozygosity (H O and H E, respectively) and allelic richness (AR) were calculated in Arlequin v. 3.5 (Excoffier and Lischer 2010) and FSTAT v. 2.9.3 (Goudet 2001). We used FSTAT to estimate the inbreeding coefficient (F IS) for each locus and each population, with significance levels calculated by randomizing alleles among individuals within each population. We then compared this to the observed data to determine deviations from Hardy-Weinberg equilibrium (HWE), using 10,000 simulations. Tests for linkage disequilibrium between all pairs of loci were carried out in FSTAT. Pairwise F ST values (Weir and Cockerham 1984) between populations within each species were estimated in FSTAT, and significance was determined using 10,000 permutations. Pairwise comparisons were corrected for type I errors using sequential Bonferroni corrections (Rice 1989). Null alleles were identified in Micro-Checker v. 2.2.3 (van Oosterhout et al. 2004). The relationship between pairwise genetic (F ST) and geographic distances was tested using a Mantel test (Mantel 1967) using IBDWS v. 3.23 (Jensen et al. 2005).
We carried out a Bayesian analysis in STRUCTURE v. 2.3.3 (Pritchard et al. 2000) to establish the number of genetic clusters of both species in Poland using all available individuals. For each value of K (1-10), we performed ten independent runs of 500,000 generations following 100,000 generations of burn-in under the admixture model and with the assumption that allele frequencies among populations are correlated. We identified the optimal number of clusters for both datasets using the Evanno method (ΔK; Evanno et al. 2005) in STRUCTURE HARVESTER (Earl and vonHoldt 2012). Individuals were assigned as belonging to a particular cluster with an assignment probability (q) of ≥0.8, with individuals with an assignment probability of 0.2 < q < 0.8 classified as ‘admixed’ (Vähä and Primmer 2006; Bergl and Vigilant 2007; Shafer et al. 2011; McDevitt et al. 2013). We additionally carried out a discriminant analysis of principal component (DAPC; Jombart et al. 2010) in RStudio (2012). This is a two-step process, the first of which transforms the genotypes using principal component analysis (PCA) and then maximises the differentiation between populations, while minimizing within-population variation. Unlike the previous method, it does not assume HWE or linkage disequilibrium (Jombart et al. 2010). Twenty-two PCA components were retained in the discriminant analysis for the common vole and 24 for the field vole.
Results
The total dataset of 380 individuals (190 individuals of each species) were successfully genotyped from Poland. There were very few instances where the three genotyping repeats differed (recorded as missing data): 0.8 % in common voles and 2.6 % in field voles (Table 4).
No loci showed significant linkage disequilibrium tested over all populations. Results of H O and H E estimation and null allele identification per locus are presented in Tables S3 and S4. For the 11 loci in the common vole, diversity values ranged from 0.450 to 0.899 (H O) and 0.629 to 0.960 (H E). Null alleles were identified for Moe6 and AV12 in some populations of the common vole. As these loci had little influence on the overall results, they were not excluded from further analyses (see below). For the 13 loci in the field vole, diversity values ranged from 0.492 to 0.957 (H O) and 0.774 to 0.974 (H E). We identified no null alleles in this species.
Looking at variation at the population level in the common vole, values of allelic richness (AR), observed heterozygosity (H O) and expected heterozygosity (H E) varied from 4.35 to 6.10, 0.600 to 0.814 and 0.724 to 0.917, respectively, and inbreeding coefficients (F IS) from 0.049 to 0.356 (Table 1). In the field vole populations, they ranged from 4.02 to 4.59 (AR), 0.657 to 0.827 (H O), 0.816 to 0.884 (H E) and 0.012 to 0.229 (F IS; Table 2). No locus for any population within either species significantly differed from HWE expectations after Bonferroni correction.
Overall population differentiation was higher for the common vole (F ST = 0.058, 95 % confidence interval (CI) 0.042–0.077) than for the field vole (F ST = 0.047, 95 % CI 0.040–0.055). For M. arvalis, this value was almost the same after removing the Moe6 and AV12 loci (F ST = 0.056, 95 % CI 0.037–0.079). Most population pairs were significantly differentiated from each other within both species (Tables S5 and S6). Mantel tests of the relationships between genetic and geographic distances revealed a significant isolation by distance pattern for both species (r = 0.326 for M. arvalis and r = 0.397 for M. agrestis; P < 0.001). Bayesian analysis in STRUCTURE showed that two genetic groups of the common vole and field vole were present in Poland (ΔK = 2 was selected in both cases; Figs. 2 and S1). However, the distribution of these groups and the contact zone between them are not exactly the same (Fig. 1). For the common vole, the Western group occurs in western and northern Poland, whereas the Eastern group inhabits the eastern, central and southern parts of Poland. For the field vole, the Western group inhabits western, southern and northern Poland, whereas the Eastern group occurs mostly in the eastern part of Poland. The majority of individuals (176 out of 190 in the common vole and 167 out of 190 in the field vole) were assigned to one of the clusters at q ≥ 0.8. In the common vole, the admixed individuals (0.2 < q < 0.8) were as follows: four individuals from population 7; two individuals each from population 4 and 15; and one individual each from populations 20, 23, 33, 38 and 39 (Fig. 1). In the field vole, the admixed individuals were as follows: five individuals from population 7; two individuals each from populations 4, 5, 6 and 17; three individuals from population 13; and one individual each from populations 8, 9, 10, 15, 19 and 20 (Fig. 1).

Evanno et al. (2005) ΔK (dashed line, right Y axis) and the mean log probability LnP(K) (continuous line, left Y axis) result from STRUCTURE for Microtus arvalis (a) and Microtus agrestis (b) based on the microsatellite data
The two species of vole exhibited relatively little population structure detected by the DAPC (Fig. 3). Genetic structure is more pronounced between populations of the common vole than in the field vole. There are four populations of the common vole which are distinguished from the others: Pułkownikówka, Górowo Iławieckie and Pomorze from the northern part of Poland and Świerzawa from south-western Poland. In case of the field vole, populations largely overlapped (Fig. 3).

Discriminant Analysis of principal components (DAPC) of genetic variation based on the microsatellite datasets for Microtus arvalis (a) and Microtus agrestis (b). Only populations with ≥5 individuals sampled were included
Discussion
In this study, we utilised museum samples collected between 1960 and 1970 and found that these samples gave very reliable and consistent results for the microsatellite panels used. Only a few genotypes were rejected from further analyses (Table 4). Therefore, we contend that the data that we obtained from these well-preserved museum specimens are a valuable resource for population genetic studies of these two vole species.
Herein, we describe genetic structure and variability in populations of the common vole and field vole in Poland using microsatellites. We can also determine whether geographic structure revealed by microsatellites is in agreement with the mtDNA lineage distributions across Poland found in previous studies (Herman et al. 2014, Stojak et al. 2015). For one of these species, the common vole, studies from other regions in Europe have already found general congruence in patterns of genetic structure between mtDNA and microsatellites at contacts between lineages (Heckel et al. 2005; Braaker and Heckel 2009; Beysard and Heckel 2014). For the field vole, Beysard et al. (2012) demonstrated an overall pattern of congruence between mtDNA, microsatellites and Y chromosome markers at the contact of the Northern and Southern evolutionary units but also showed evidence of asymmetrical and differential gene introgression. Our study in Poland allows us to look for congruence between microsatellites and mtDNA in the contact zones between mtDNA lineages of the two species there (Herman et al. 2014; Stojak et al. 2015). These two vole species are morphologically similar but are characterised by different post-glacial evolutionary histories and different ecologies.
We found similar levels of genetic diversity across populations within each species and between them (Tables 1 and 2). The overall differentiation was higher for the common vole (F ST = 0.058) than for the field vole (F ST = 0.047). Almost all pairwise comparisons between populations were significant with the exception of those involving the field vole population from Kobiór (KOB; Tables S5 and S6) which may reflect the small sample size for this population. We detected a significant but moderate isolation by distance pattern for both species. The DAPC revealed a lack of obvious genetic structure for both species with individuals mostly overlapping between populations (Fig. 3). In agreement with overall F ST, the common vole showed evidence of higher levels of differentiation between populations in the DAPC.
Bayesian analysis in STRUCTURE revealed that there were two geographic groups for each species in Poland based on microsatellites (Figs. 1 and 2). Although strong isolation by distance can influence inferences of genetic structure based on STRUCTURE (Frantz et al. 2009), we did not observe this in our data (see above). The distribution patterns of the geographic groups differ slightly between the common and field voles, but they can both be roughly categorised into an Eastern and Western subdivision (Fig. 1). The Eastern group of the common vole has a wider distribution in Poland than the Eastern group of the field vole. For the common vole, the contact zone between the two microsatellite groups begins in the north, going through the western part of Poland, ending in the south, whereas for the field vole, the contact zone starts in the north-western part of Poland and goes straight through central Poland to the south-eastern border (Fig. 1). The Eastern group is located on the eastern side of the Vistula and San rivers and the Western group on the western side (Fig. 1). Additionally for the field vole, several mixed genetic groups are observed in northern Poland, separated from the Western group by the Noteć river (Fig. 1).
The distributions of mtDNA lineages of common and field voles in Poland do not match the Eastern and Western microsatellite groups. In Poland, there are two mtDNA lineages (Central and Eastern) of the common vole and the contact zone is located in the north-western part of Poland (Fig. 1; Stojak et al. 2015). Individuals from the Central lineage apparently recolonised Eastern Europe from a refugium in an ill-defined area north of Italy (Heckel et al. 2005), whereas the Eastern lineage most likely stemmed from the Carpathian refugium (Stojak et al. 2015). The contact zone of the two genetic groups based on microsatellites of the common vole is shifted considerably to the east (described above, see Fig. 1). For the field vole, the contact zone between the Western and Central European mtDNA lineages was in south-western Poland (Fig. 1) from unspecified Younger Dryas refugia (Herman et al. 2014). The proposed contact zone between the two genetic groups based on microsatellites of the field vole is located in the central part of Poland (Fig. 1).
Beysard and Heckel (2014) analysed the genetic structure and dynamics of three contact zones between mitochondrial lineages of the common vole located in Switzerland and Bavaria. They found very narrow zones of hybridization (using microsatellites) between the Central and Western lineages and a much wider hybrid zone between the Central and Eastern lineages (which are the lineages present in Poland; Stojak et al. 2015). The Eastern and Central lineages are grouped within the same clade and have an estimated time to most recent common ancestor (tMRCA) of approximately 28 ka bp, whereas the Central and Western lineages belong to separate clades, with a tMRCA at approximately 56 ka bp (Stojak et al. 2015). Given that the Central and Eastern lineages are less divergent than the Western and Central lineages, it is perhaps not surprising that Beysard and Heckel (2014) found stronger evidence for reproductive isolation for the Western and Central lineages. Our results are in general agreement with the aforementioned study because the microsatellites employed here demonstrate that individuals generally belong to a single genetic cluster (Western group) across the Central and Eastern mtDNA lineage contact zone in Poland (Fig. 1).
For the field vole, there is a similar lack of congruence between the distribution of mtDNA lineages and geographic groups based on microsatellites in Poland (Fig. 1). Beysard et al. (2012) revealed that there was a degree of reproductive isolation between the contact zone of what we now refer to as the Northern and Southern evolutionary units (Paupério et al. 2012) in Switzerland. These evolutionary units likely diverged at the LGM (Paupério et al. 2012). In Poland, the contact zone is between two more recently diverged mtDNA lineages within the Northern evolutionary unit, which apparently derived from two separate Younger Dryas refugia (Herman et al. 2014). Given that the field vole populations in Poland marked by mitochondrial differences were not isolated from each other for a long period, it is perhaps unsurprising that there is no evidence for reproductive isolation between those mtDNA lineages.
Given the similarities in the patterns of contemporary genetic structure in the common and field vole, it appears that major river valleys restrict or act as barriers to contemporary gene flow between populations of these taxa in Poland (Fig. 1), leading to differentiation between Eastern and Western geographical groups. Rivers have been identified as significant barriers to gene flow in many terrestrial mammals (e.g. Gerlach and Musolf 2000; Mullins et al. 2014). At the broad scale of this study, our results indicate that the end glacial population history has less of an impact on contemporary genetic structure in these two species than the landscape. Beysard and Heckel (2014) concluded that the divergence times between lineages play an important role in the level of gene introgression between them. A recent study by Beysard et al. (2015) demonstrated that female common voles from the Western mtDNA lineage preferentially selected males from their own lineage under lab conditions in preference to Central lineage males. At present, we do not know if there is such a preference between voles of the Central and Eastern lineages present in Poland (which diverged more recently than the Central and Western lineages; Stojak et al. 2015) or between lineages in the field vole (Beysard et al. 2012). Various studies in the European house mouse hybrid zone (between the subspecies Mus musculus musculus and Mus musculus domesticus) have demonstrated introgression between the subspecies, but reduced male fertility maintains a degree of reproductive isolation between the subspecies (Turner et al. 2012; Albrechtová et al. 2012). A similar mechanism was proposed to account for the partial reproductive isolation between the Northern and Southern evolutionary units of the field vole (Beysard et al. 2012) and the Central and Western mtDNA lineages of the common vole (Beysard and Heckel 2014). In Poland, however, where there are contact zones between mtDNA lineages that have diverged more recently in both species, there does not appear to be the same pattern of reproductive isolation between the mtDNA lineages based on the contemporary genetic structure inferred from microsatellite data. However, it is clear that further sampling would be beneficial to define more accurately the contact zones between the mtDNA lineages and the genetic clusters based on microsatellites in these two species in Poland (Beysard et al. 2012; Beysard and Heckel 2014). Newly collected specimens from Poland would also allow us (i) to investigate in more detail the influence of river barriers and other environmental and landscape features on gene flow in the common vole and field vole populations in Poland and (ii) to compare if the patterns of genetic structure based on microsatellites have changed or been retained over the last ca. 50 years (the samples that we analysed in the present study were of that age).
References
Albrechtová J, Albrecht T, Baird SJE, Macholán M, Rudolfsen G, Munclinger P, Tucker PK, Piálek J (2012) Sperm-related phenotypes implicated in both maintenance and breakdown of a natural species barrier in the house mouse. Proc R Soc B 279:4803–4810
Amori G, Hutterer R, Kryštufek B, Yigit N, Mitsain G, Palomo LJ (2008) Microtus arvalis. In: The IUCN red list of threatened species. Version 2014.3. <www.iucnredlist.org>.
Bergl RA, Vigilant L (2007) Genetic analysis reveals population structure and recent migration within the highly fragmented range of the Cross River gorilla (Gorilla gorilla diehli). Mol Ecol 16:501–516
Beysard M, Heckel G (2014) Structure and dynamics of hybrid zones at different stages of speciation in the common vole (Microtus arvalis). Mol Ecol 23:673–687
Beysard M, Perrin N, Jaarola M, Heckel G, Vogel P (2012) Assymetric and differential gene introgression at a contact zone between two highly divergent lineages of field voles (Microtus agrestis). J Evol Biol 25:400–408
Beysard M, Krebs-Wheaton R, Heckel G (2015) Tracing reinforcement through asymmetrical partner preference in the European common vole Microtus arvalis. BMC Evol Biol 15:170
Bi K, Linderoth T, Vanderpool D, Good JM, Nielsen R, Moritz C (2013) Unlocking the vault: next-generation museum population genomics. Mol Ecol 22:6018–6032
Bourke BP, Frantz AC, Lavers CP, Davison A, Dawson DA, Burke TA (2010) Genetic signatures of population change in the British golden eagle (Aquila chrysaetos). Conserv Gen 11:1837–1846
Braaker S, Heckel G (2009) Transalpine colonisation and partial phylogeographic erosion by dispersal in the common vole Microtus arvalis. Mol Ecol 18:2518–2531
Bulatova NS, Golenishchev FN, Koval’skaya YM, Emelyanova LG, Bystrakova NV, Pavlova SV, Nadzhafova RS, Lavrechenko LA (2010a) Cytogenetic study of the parapatric contact zone between two 46-chromosomal forms of the common vole in European Russia. Russ J Genet 46:443–448
Bulatova NS, Potapov SG, Lavrechenko LA (2010b) Genomic versus chromosomal polytypy in studies of mitochondrial and nuclear DNA markers in the Microtus arvalis group. Russ J Genet 46:586–594
Bulatova NS, Pavlova SV, Romanenko SA, Serdiukova NA, Golenishchev FN, Malygin VM, Lavrenchenko LA (2013) Molecular cytogenetic markers of cryptic species and hybrids of the common vole superspecies complex Microtus arvalis s. l. Tsitologiia 55:268–270
Bužan EV, Förster DW, Searle JB, Kryštufek B (2010) A new cytochrome b phylogroup of the common vole Microtus arvalis endemic to the Balkans and its implications for the evolutionary history of the species. Biol J Linn Soc 100:788–796
Clark PU, Dyke AS, Shakun JD, Carlson AE, Clark J, Wohlfarth B, Mitrovica JX, Hostetler SW, McCabe AM (2009) The last glacial maximum. Science 325:710–714
Czajkowska M, Borkowska A, Wieczorek M, Zub K (2010) Application of microsatellite markers developed for arvicoline species in a population genetic study of the root vole Microtus oeconomus. Acta Theriol 55:123–128
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Res 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetic analyses under Linux and Windows. Mol Ecol Res 10:564–567
Frantz AC, Celina S, Krier A, Schley L, Burke T (2009) Using spatial Bayesian methods to determine the genetic structure of a continuously distributed population: clusters or isolation by distance? J App Ecol 46:493–505
Gauffre B, Galan M, Bretagnolle V, Cosson JF (2007) Polymorphic microsatellite loci and PCR multiplexing in the common vole Microtus arvalis. Mol Ecol Notes 7:830–832
Gerlach G, Musolf K (2000) Fragmentation of landscape as a cause of genetic subdivision in bank voles. Conserv Biol 14:1066–1074
Godinho R, Crespo EG, Ferrand N (2008) The limits of mtDNA phylogeography: complex patterns of population history in a highly structured Iberian lizard are only revealed by the use of nuclear markers. Mol Ecol 17:4670–4683
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Department of Ecology and Evolution, University of Lausanne, Switzerland, Available at: http://www2.unil.ch/popgen/softwares/fstat.htm
Haynes S, Jaarola M, Searle JB (2003) Phylogeography of the common vole Microtus arvalis with particular emphasis on the colonization of the Orkney archipelago. Mol Ecol 12:951–956
Heckel G, Burri R, Fink S, Desmet JF, Excoffier L (2005) Genetic structure and colonization processes in European populations of the common vole Microtus arvalis. Evolution 59:2231–2242
Herman JS, Searle JB (2011) Post-glacial partitioning of mitochondrial genetic variation in the field vole. Proc R Soc B 278:3601–3607
Herman JS, McDevitt AD, Kawałko A, Jaarola M, Wójcik JM, Searle JB (2014) Land-bridge calibration of molecular clocks and the post-glacial colonization of Scandinavia by the Eurasian field vole Microtus agrestis. PLoS One 9:e103949
Hewitt GM (1999) Post-glacial re-colonization of European biota. Biol J Linn Soc 68:87–112
Ishibashi Y, Yoshinaga Y, Saitoh T, Abe S, Iida H, Yoshida MC (1999) Polymorphic microsatellite DNA markers in the field vole Microtus montebelii. Mol Ecol 8:163–164
Jaarola M, Searle JB (2002) Phylogeography of field voles (Microtus agrestis) in Eurasia inferred from mitochondrial DNA sequences. Mol Ecol 11:2613–2621
Jaarola M, Martínková N, Gündüz İ, Brunoff C, Zima J et al (2004) Molecular phylogeny of the speciose vole genus Microtus (Arvicolinae, Rodentia) inferred from mitochondrial DNA sequences. Mol Phylogenet Evol 33:647–663
Jaarola M, Ratkiewicz M, Ashford RT, Brunhoff C, Borkowska A (2007) Isolation and characterization of polymorphic microsatellite loci in the field vole, Microtus agrestis, and their cross-utility in the common vole, Microtus arvalis. Mol Ecol Notes 7:1029–1031
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6:13, v.3.23 http://ibdws.sdsu.edu/
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Kotlík P, Deffontaine V, Mascheretti S, Zima J, Michaux JR, Searle JB (2006) A northern glacial refugium for bank voles Clethrionomys glareolus. Proc Natl Acad Sci U S A 103:14860–14864
Kryštufek B, Vohralík V, Zima J, Zagorodnyuk I (2008) Microtus agrestis. In: The IUCN red list of threatened species. Version 2014.3. <www.iucnredlist.org>.
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Martínková N, Barnett R, Cucchi T, Struchen R, Pascal M, Pascal M, Fischer MC, Higham T, Brace S, Ho SYW, Quéré JP, O’Higgins P, Excoffier L, Heckel G, Hoelzel AR, Dobney KM, Searle JB (2013) Divergent evolutionary processes associated with colonization of offshore islands. Mol Ecol 22:5205–5220
McDevitt AD, Zub K, Kawałko A, Oliver MK, Herman JS, Wójcik JM (2012) Climate and refugial origin influence the mitochondrial lineage distribution of weasels Mustela nivalis in a phylogeographic suture zone. Biol J Linn Soc 106:57–69
McDevitt AD, Oliver MK, Piertney SB, Szafrańska PA, Konarzewski M, Zub K (2013) Individual variation in dispersal associated with phenotype influences fine-scale genetic structure in weasel. Conserv Genet 14:499–509
Meyer MN, Golenishchev FN, Bulatova NS, Artobolevsky GV (1997) On distribution of two Microtus arvalis chromosomal forms in European Russia. Zool Zh 76:487–493 (in Russian)
Mullins J, McDevitt AD, Kowalczyk R, Ruczynska I, Gorny M, Wójcik JM (2014) The influence of habitat structure on genetic differentiation in red fox populations in north-eastern Poland. Acta Theriol 59:367–376
Paupério J, Herman JS, Melo-Ferreira J, Jaarola M, Alves PC, Searle JB (2012) Cryptic speciation in the field vole: a multilocus approach confirms three highly divergent lineages in Eurasia. Mol Ecol 21:6015–6032
Pazonyi P (2004) Mammalian ecosystem dynamics in the Carpathian Basin during the last 27000 years. Palaeogeog Palaeoclim Palaeoecol 212:295–314
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
RStudio (2012) RStudio: integrated development environment for R (Version 0.96.122). Computer software, Boston, MA
Rubidge EM, Patton JL, Lim M, Burton AC, Brashares JS, Moritz C (2012) Climate-induced range contraction drives genetic erosion in an alpine mammal. Nature Clim Change 2:285–288
Rudá M, Žiak D, Gauffre B, Zima J, Martínková N (2009) Comprehensive cross-amplification of microsatellite multiplex sets across the rodent genus Microtus. Mol Ecol Res 9:974–978
Shafer ABA, Côté SD, Coltman DW (2011) Hot spots of genetic diversity descended from multiple Pleistocene refugia in an alpine ungulate. Evolution 65:125–138
Sommer RS, Nadachowski A (2006) Glacial refugia of mammals in Europe: evidence from fossil records. Mamm Rev 36:251–265
Stewart WA, Piertney SB, Dallas JF (1998) Isolation and characterization of highly polymorphic microsatellites in the water vole Arvicola terrestris. Mol Ecol 7:1258–1259
Stojak J, McDevitt AD, Herman JS, Searle JB, Wójcik JM (2015) Post-glacial colonization of eastern Europe from the Carpathian refugium: evidence from mitochondrial DNA of the common vole Microtus arvalis. Biol J Linn Soc 115:927–939
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464
Tougard C, Renvoisé E, Petitjean A, Quéré JP (2008) New insight into the colonization processes of common voles: inferences from molecular and fossil evidence. PLoS One 3:e3532
Turner LM, Schwahn DJ, Harr B (2012) Reduced male fertility is common but highly variable in form and severity in a natural house mouse hybrid zone. Evolution 66:443–458
Vähä JP, Primmer CR (2006) Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol Ecol 15:63–72
van de Zande L, van Apeldoorn RC, Blijdenstein AF, de Jong D, van Delden W, Bijlsma R (2000) Microsatellite analysis of population structure and genetic differentiation within and between populations of the root vole Microtus oeconomus in the Netherlands. Mol Ecol 9:1651–1656
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Res 4:535–538
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wójcik JM, Kawałko A, Marková S, Searle JB, Kotlík P (2010) Phylogeographic signatures of northward post-glacial colonization from high-latitude refugia: a case study of bank voles using museum specimens. J Zool 281:249–262
Acknowledgments
The authors thank Barbara Marczuk for her help in the laboratory. We also thank the two anonymous reviewers for their valuable comments. The study was financed by the National Science Centre, Poland, grant no. N N304 058340 to JMW and grant no. UMO-2013/09/N/NZ8/03205 to JS. This study forms part of the PhD thesis of JS, and she was awarded a PhD scholarship from the National Science Centre in Poland according to decision number DEC-2015/16/T/NZ8/00015. ADM was supported by a postdoctoral fellowship under the project BIOCONSUS (Research Potential in Conservation and Sustainable Management of Biodiversity), funded by the European Union 7th Framework Programme (FP7 2010–2013; Agreement No. 245737).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Mabel D. Giménez
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Table S1. PCR multiplex protocols for Microtus arvalis. (DOC 31 kb)
ESM 2
Table S2. PCR multiplex protocols for Microtus agrestis. (DOC 32 kb)
ESM 3
Table S3. Characteristics of loci used in the microsatellite analysis of Microtus arvalis including observed (HO) and expected (HE) heterozygosity and null alleles. (DOC 32 kb)
ESM 4
Table S4. Characteristics of loci used in microsatellite analysis of Microtus agrestis including observed (HO) and expected (HE) heterozygosity and null alleles. (DOC 33 kb)
ESM 5
Table S5. Unbiased estimates of multilocus FST between all population pairs of Microtus arvalis based on microsatellites with population labelling following Table 1. All significant values are after Bonferroni correction. Non-significant values are in italics. (DOC 30 kb)
ESM 6
Table S6. Unbiased estimates of multilocus FST between all population pairs of Microtus agrestis based on microsatellites with population labelling following Table 1. All significant values are after Bonferroni correction. Non-significant values are in italics. (DOC 32 kb)
ESM 7
Figure S1. Results of the Bayesian clustering analysis implemented in STRUCTURE under the admixture model for Microtus arvalis (A) and Microtus agrestis (B) based on the microsatellite dataset. Vertical columns represent the assignment probabilities to each of the inferred clusters identified for ΔK = 2 in both species. Numbers on horizontal axis match population localities presented on maps in Fig. 1. (DOC 185 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Stojak, J., Wójcik, J.M., Ruczyńska, I. et al. Contrasting and congruent patterns of genetic structuring in two Microtus vole species using museum specimens. Mamm Res 61, 141–152 (2016). https://doi.org/10.1007/s13364-015-0260-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13364-015-0260-y