
Abstract
Gas-phase hydrogen deuterium exchange (HDX), collision cross section (CCS) measurement, and molecular dynamics simulation (MDS) techniques were utilized to develop and compare three methods for estimating the relative surface area exposure of separate peptide chains within bovine insulin ions. Electrosprayed [M – 3H]3− and [M – 5H]5− insulin ions produced a single conformer type with respective collision cross sections of 528 ± 5 Å2 and 808 ± 2 Å2. [M – 4H]4− ions were comprised of more compact (Ω = 676 ± 3 Å2) and diffuse (i.e., more elongated, Ω = 779 ± 3 Å2) ion conformer types. Ions were subjected to HDX in the drift tube using D2O as the reagent gas. Collision-induced dissociation was used to fragment mobility-selected, isotopically labeled [M – 4H]4− and [M – 5H]5− ions into the protein subchains. Deuterium uptake levels of each chain can be explained by limited inter-chain isotopic scrambling upon collisional activation. Using nominal ion structures from MDS and a hydrogen accessibility model, the deuterium uptake for each chain was correlated to its exposed surface area. In separate experiments, the per-residue deuterium content for the protonated and deprotonated ions of the synthetic peptide KKDDDDDIIKIIK were compared. The differences in deuterium content indicated the regional HDX accessibility for cations versus anions. Using ions of similar conformational type, this comparison highlights the complementary nature of HDX data obtained from positive- and negative-ion analysis.

ᅟ
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Over the last 20 years, the elucidation of protein structures has proceeded at an accelerating rate [1]. Because of the importance of protein complexes in performing specific biological functions, over recent years increased attention has been given to the development of techniques for quaternary structure determination. Ideally, X-ray crystallography would be used to provide high-resolution structural information for protein complexes; however, structural studies require the in vitro reconstitution of purified protein subunits as well as the ability to crystallize the protein complex [2]. Although ground breaking studies in the determination of protein quaternary structure occurred in 2000 [3–5], there are yet limitations in the elucidation of such structures by X-ray crystallography [2]. Nuclear magnetic resonance spectroscopy (NMR) also suffers from a limitation associated with molecular size; however, recent advances such as magic angle spinning NMR have enabled its limited application to the study of protein subunit architecture [6]. More recently, impressive advances in cryoelectron microscopy have allowed the study of large protein complexes although challenges remain with regard to resolution, throughput, and instrumental parameter optimization (e.g., electron energies) [7, 8].
To overcome some of the challenges associated with traditional methods of structure characterization, mass spectrometry (MS) techniques have been developed. A major area of research involves chemical modification of the proteins of interest followed by MS characterization of the intact protein (top-down approach) or proteolytic fragments (bottom-up approach). One strategy that is used extensively is solution hydrogen-deuterium exchange (HDX). For typical experiments, a protein is temporarily subjected to exchange (hydrogens for deuteriums – exchange in, or deuteriums for hydrogens – exchange out) followed by reaction quenching, enzymatic digestion, and liquid chromatography (LC)-tandem mass spectrometry (MS/MS) [9, 10]. Several challenges arise from this approach, including digestion efficiency and peptide identification (especially for samples producing complex peptide mixtures) as well as comparing the data with in-silico structures [9–12]. Another approach involves the use of chemical cross-linking of amino acid residues that are in three-dimensional proximity followed by enzymatic digestion and LC-MS/MS to identify neighboring sites [13, 14]. Similar to HDX studies, peptide ion identification presents a challenge to such an approach. Finally, more recently, a number of studies have sought to introduce other chemical modifications into proteins in solution followed by MS analysis [15–17]. Although promising, such techniques experience some of the same challenges of the HDX LC-MS/MS approaches and have largely been limited to smaller protein analysis.
During the mid1990s, researchers presented the idea of native MS whereby the composition of ions of noncovalent protein complexes could be analyzed by electrospray ionization (ESI)-MS [18]. Studies in this area have proceeded rapidly over the last two decades, allowing the determination of protein complex masses and stoichiometries [18, 19]. It is recognized that the size of ions that can be interrogated by the approach is nearly limitless, extending to the tens of megadaltons [20–22]. Additionally, collisional activation can be used to provide some information regarding individual protein subunits [23]. One breakthrough approach has been the use of surface-induced dissociation (SID), which allows the preservation of the charge distribution within the product ions [24]. Although native MS provides advantages in terms of sensitivity, throughput, and characterization breadth, challenges associated with sample purity as well as the requirement of unique MS instrumentation and operational conditions exist.
As an expansion of native MS studies, seminal experiments have demonstrated the utility of incorporating ion mobility spectrometry (IMS) measurements. This addition presents shape information for protein complex and aggregate ions in the form of collision cross section (CCS) measurements [25–27]. Although the concept of comparisons of CCS values for biomolecular ions was established nearly a decade earlier [28–31], theoretical approaches for the generation of trial structures for extremely large ions have proceeded at a rapid pace [32]. One notable advantage of the IMS-MS approach for protein complex studies is that collisional activation can be used to study protein complex, subcomplex, and subunit ions[23, 33, 34]. A limitation of IMS-MS studies is that only general information regarding ion shape is obtained. The extraction of higher-resolution structural information requires the ability to obtain relevant comparisons to accurate ion structures as may be obtained by molecular dynamics simulations (MDS). This not only presents challenges with obtaining accurate structures themselves but also with the ability to calculate reliable CCS for many trial structures [32].
One approach intended to provide additional structural information is the incorporation of gas-phase HDX with IMS-MS analysis. The relative degree of exchange site accessibility has been assessed for different protein ion conformations [35, 36]. Additionally, HDX site accessibility models have been presented for ion structures obtained from MDS [36, 37]. A limitation of the approach is that a detailed description of accessible exchange sites was not possible because of the potential for hydrogen-deuterium (HD) scrambling upon precursor ion fragmentation by collision-induced dissociation (CID) [38]. The introduction of nonergodic ion fragmentation techniques has provided a means for preserving the location of the deuterium uptake label [39]. Indeed, recent studies have demonstrated that per-residue deuterium uptake can be determined for specific ion conformers as distinguished by IMS [40, 41].
In the accompanying manuscript with this report, IMS-HDX-MS/MS has been performed on peptide anions whereupon precursor ion CID, the locations of charge sites can be determined. Additionally, the study accompanying this work shows some ability to use CID as an ion activation approach for the study of deuterium incorporation within large peptide anions. In this study, bovine insulin is used as a model system to examine the degree of label retention for a multi-chain protein system. In these proof-of-principle studies we find limited HD scrambling up to the separation of the peptide chains. Finally, models to estimate the relative exposed surface area of different insulin subchains are presented. The results are discussed with respect to the applicability of such approaches for protein complex structure characterization.
Experimental
Sample Preparation
Much of the experimental details are presented in the study accompanying this work. Here, only a brief description of the experimental parameters is presented. The synthetic peptide having the sequence of KKDDDDDIIKIIK and bovine insulin (each >90% purity) were purchased (Genscript, Piscataway, NJ, USA and Sigma Aldrich, St. Louis, MO, USA, respectively) and used without further purification. From stock solutions (1.0 mg∙mL−1), ESI solutions consisting of 10 mM ammonium hydroxide:acetonitrile (1:1) were prepared such that the final analyte concentration was 0.1 mg∙mL−1. Infused samples (300 nL∙min−1) were electrosprayed into the hybrid IMS-MS instrument [41–43] using a bias of −2200 V.
IMS-MS Measurements
Comprehensive descriptions of IMS instrumentation and applications exist in the literature [25, 44–50].The new IMS-MS instrument has also been described previously [41, 42] as well as in the accompanying manuscript. Using a dual ion gate drift tube configuration, electrosprayed anions were periodically pulsed into a drift tube with the first ion gate and mobility selected with the second ion gate. For a given mobility selection, MS and MS/MS spectra were recorded with the linear ion trap (LIT) mass spectrometer (LTQ Velos; ThermoScientific, San Jose, CA, USA). By scanning the delay times between the front and back ion gate, mass spectral characterization of the entire drift time (t D) distribution was accomplished. For all mobility-resolved MS datasets, the LIT scan parameters employed a m/z range of 80 to 2000. Sample injection times of 200 ms (5 microscans) were utilized; t D -selected precursor ion mass spectra were collected for 0.5 min each. Two-dimensional IMS-MS datasets were generated using an algorithm that created a three-column array file associating m/z and intensity (i) information with each t D selection.
HDX Measurements
D2O (>99%, Sigma Aldrich, St. Louis, MO, USA) was introduced into the He buffer gas using a separate leak valve (Granville Philips, Longmont, CO, USA); a D2O partial pressure setting of 0.04 Torr was used for these studies. Here we note that in previous studies of insulin anions, higher partial pressures of D2O were used to obtain maximum HDX levels [42]. A lower pressure is used here to allow the consideration of only the fast exchanging hydrogens [43]. These sites are the most amenable to modeling (exhibiting the same HDX rate) by the HDX accessibility method (see below); t D , m/z distributions were recorded in pure He and subsequently in the He:D2O mixture. For specific ion conformers, the number of incorporated deuteriums was determined from the difference in precursor ion average mass before and after exposure to D2O. The deuterium content of chain A and chain B fragment ions was also obtained by comparing the m/z values of these ions formed upon CID of precursor ions before and after undergoing HDX.
Because previous experiments have shown that multiple ion conformers can exist for given mobility selections [43], it is useful to consider the effect of the width of the mobility selection. We have increased the gate width up to 1 ms for the ion conformer types studied and observed no significant difference in the deuterium content of the chain A and chain B fragment ions. For these experiments, we have utilized selection widths of 200 μs to be consistent with the narrowest range of ion conformers possible for the current IMS-MS instrumentation.
Molecular Dynamics Simulations (MDS)
MDS experiments required the creation of the [M – 4H]4− and [M – 5H]5− insulin ion starting structures. Charge site location considerations were the same as those described in the accompanying manuscript. Indeed, the ion structures for the KKDDDDDIIKIIK anions are the same as reported therein. For bovine insulin, there are six acidic residues with one being adjacent to an arginine residue (E21 and R22 on chain B). This site was assumed to form a salt bridge and so, for the [M – 5H]5− insulin ions, all remaining acidic sites were deprotonated. For [M – 4H]−4 insulin ions, Coulomb energy considerations suggest that two negative charges be placed on chain A with the other two being placed on chain B, which has three acidic sites (E4, E17, C-terminus). The intrinsic gas-phase acidity of the E residue is much larger than the C-terminus [51]; therefore, the E4 and E17 residues were used as charge sites on chain B. Additionally, alternative charge placements for MDS resulted in structures that were too large for comparison with the IMS data and the HDX prediction data matched the above-mentioned charge site configuration.
Starting ion structures for simulated annealing were obtained as described here. Chain A and chain B ions were created as linear species. Each ion was heated separately to 1000 K and subsequently cooled and energy minimized. Low-energy structures for each peptide ion that exhibited similar distances between cysteine residues (as assessed by intra-chain disulfide bonds) were selected and joined with disulfide bonds to form the insulin precursor ion starting structures. Simulated annealing was performed as described previously [41, 43] and low-energy structures with matching CCSs for the [M – 4H]4− and [M – 5H]5− ions were used for the hydrogen accessibility scoring and surface area assessment. The CCS values for 1000 conformers from simulated annealing were calculated using the Mobcal [52] software employing the trajectory method (TM) [53]. Here, the MSMS program was used to calculate the solvent accessible (SAS) and solvent excluded (SES) surface areas [54]. To calculate the SES and SAS values, a probe sphere radius of 1.4 Å and a triangulation density of 1.0 vertex/Å2 were used.
Estimating Peptide Chain Deuterium Content Under 100% HD Scrambling Conditions
To estimate the expected deuterium content for 100% HD scrambling, the total number of labile hydrogens was first calculated. Per-site 100% HD scrambling values were calculated by dividing the total uptake by the number of labile hydrogens. The per-site values were then summed for each chain to estimate the expected deuterium content. As an example, consider a dimer peptide ion that contains 30 labile hydrogens and experiences, on average, an increase in mass of ~15 Da. Under 100% scrambling conditions, the per-site deuterium content would be ~0.5. These values can then be summed for each peptide chain according to its distinctive number of labile hydrogens to determine expected uptake values for individual peptide chain under 100% HD scrambling conditions. This method is the same as the generally accepted method presented by Jorgensen and coworkers [39].
Hydrogen Accessibility Scoring (HAS)
The model for scoring relative hydrogen accessibility was based on the generally accepted “relay” mechanism for HDX as presented by Beauchamp and coworkers and Chan and Enke [55, 56].The current model is similar to other models that considered threshold distances between charge sites and exchange sites; the current model also considers distances between exchange sites and the site of initial deuterium incorporation. Briefly, deuterium uptake predictions were performed for the subchains of bovine insulin using a modified method described previously [41, 57]. As before, the distances between charge sites and exchange sites were determined using nominal ion structures obtained from MDS. Heteroatom exchange sites were then scored (S HDXS ) based on a summation of the inverse distances according to Equation 1.
In Equation 1, d O-H represents the distance between the exchange site and a deprotonated carboxylic acid site. Although, the accessibility treatment presented by Equation 1 describes the relative accessibility of exchange sites to charge sites, the effects of protein fold protection are not taken into account. To investigate exchange site protection from the protein fold, two different methods of chain scoring were used, which are discussed comprehensively below. Insulin subchain scores (representing relative uptake capability) obtained from the S HDXS values modified by the protein fold effects (see below) were then normalized to the total deuterium uptake of the precursor ions and compared with the experimental values.
Results and Discussion
Peptide and Protein Ion Collision Cross Sections
Figure 1 shows the three-dimensional raised relief plot for insulin ions indicating t D and m/z values for different conformer types. A dominant feature comprising [M – 5H]5− insulin ions is observed at a t D of 9.5 ms corresponding to a relatively diffuse conformer type having a CCS of 808 ± 5 Å2. Here, the term diffuse refers to larger ion structures within a given charge state. Also observed for these ions are some unresolved lower mobility features. At m/z 1432 [M – 4H]4− insulin ions exhibit two major conformer types; one appears at a t D of 9.6 ms (Ω = 676 ± 3 Å2) and the other is observed at a t D of 10.3 ms (Ω = 779 ± 3 Å2). [M – 3H]3− insulin ions show the lowest intensities and the most compact structures. The most compact [M – 3H]3− insulin ions appear at a t D of 9.8 ms (Ω = 528 ± 2 Å2) and are observed to be more intense than the more diffuse triply charged ions appearing at a t D of 10.8 ms (Ω = 595 ± 2 Å2 ) as shown in Supplementary Figure 1. It is instructive to consider the charge state dependence of collision cross sections for the insulin system. For the [M – 5H]5− ions only one diffuse conformer type exists, which is very similar to the more diffuse [M – 4H]4− ions (808 Å2 versus 779 Å2, respectively). This small increase in cross section—from the latter to the former ions—can be attributed to the minor structural change of ion elongation resulting from greater Coulomb repulsion. The more compact [M – 4H]4− ions are intermediate in size compared with the compact [M – 3H]3− ions and the diffuse conformer types, which shows a structural transition effected by Coulomb repulsion similar to that described previously for protein ions [58].

Three-dimensional (t D , m/z, i) raised-relief plot produced upon electrospraying a solution of bovine insulin. Dominant ions in the distribution are labeled
Collision cross sections for the dominant features in Figure 1 have been calculated using Equation 2 [59].
In Equation 2, ze and k B correspond to the charge of the ion and Boltzmann’s constant, respectively, and m I and m B correspond to the mass of the ion and the mass of the buffer gas, respectively. The variables E, L, T, and P correspond to the electric field, length of the drift tube, and the temperature and pressure of the buffer gas, respectively. N is neutral number density under standard temperature and pressure (STP) conditions.
Insulin as a Model System for Determining Subunit HD Scrambling
In the study of protein complex structure, a knowledge of the relative amount of exposed surface area of each subunit is desirable. Such information can provide insight about the overall morphology and the arrangement of subunits in a protein complex. Therefore, measurements of total deuterium uptake by each subunit may provide a method for estimating the relative exposed surface area of subunits. For such experiments, HDX-MS/MS data are useful only if no extensive inter-subunit HD scrambling occurs upon dissociation of the precursor ions. Dissociation must occur at an earlier time point than that of extensive proton mobilization between subunits. Insulin was chosen for this study because the single polypeptide chain A is strongly tethered to the separate peptide chain B by two disulfide bonds. The insulin system, therefore, can be considered a worst-case scenario for small protein complex ions. For comparison purposes, Supplementary Table 1 shows the full dissociation normalized collision energies for a series of related peptide dimers of similar size to the insulin system studied here. Notably, the values are nearly 2-fold smaller than those required to dissociate the insulin ions. The small protein was also selected because of the limited m/z range of the LIT mass spectrometer.
Admittedly, the ESI solution used in these studies does not resemble native MS conditions. The home-built IMS-MS instrument is significantly less sensitive in negative ion mode. Therefore, the ESI solvent composition for protein and peptide anions was selected based on its ability to provide the largest ion signal levels. Under such conditions, appreciable amounts of multimer ions are not observed for larger peptides. For this reason, disulfide bonded insulin was selected for the study. MDS shows that ion structures matching the IMS and HDX data are sufficiently compact to indicate comparatively close proximity of subchains [60].
CID of Insulin Anions to Assess Hydrogen-Deuterium Scrambling
Supplementary Figure 2 shows the CID spectra for the more diffuse [M – 4H]4− and [M – 5H]5− insulin ions. In Supplementary Figure 2a, an increase in the ion activation energy to a normalized collision energy (instrument parameter) of 30 results in three separate features in the mass spectrum. The remaining precursor ions are observed at m/z 1432. More intense features are observed at m/z 1167 and 1698. These features are assigned to doubly charged chain A and doubly charged chain B fragment ions, respectively. Here, we note that multiple studies have indicated different types of ions formed by dissociation of disulfide-bonded peptide and protein anions. Some experiments have shown retention or loss of both sulfur atoms by one peptide ion fragment [61–63]. Our data shows disulfide bond cleavage similar to one cleavage type (of several) observed in previous protein anion studies [63].
After exposing the diffuse [M – 4H]4− ions to D2O (partial pressure of ~0.04 Torr) and subsequently fragmenting the precursor ions using the same activation parameters, a nearly identical mass spectrum is produced. The relative intensities of the precursor ions and doubly charged chain A and chain B ions are similar to those observed prior to the introduction of D2O into the drift tube. A difference is that the ions observed after HDX are shifted to higher m/z values, indicating deuterium incorporation by the precursor ions. The [M – 5H]5− insulin ions exhibit similar behavior under CID conditions with the exception that another unassigned fragment ion peak near the precursor ion is observed as shown in Supplementary Figure 2b. The deuterium content for each peptide chain is calculated by subtracting the m/z of the fragment ion produced from precursor ions not subjected to HDX from the m/z of the same fragment ion produced after HDX.
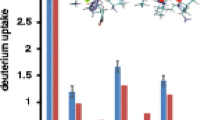
McLafferty and coworkers first presented the concern of deuterium scrambling within collisionally-activated organic compound ions [64]. Since that time, a number of researchers have demonstrated HD scrambling for collisionally activated biomolecular ions [38, 39, 65, 66]. Although some studies have demonstrated 100% scrambling, including studies of peptide anions [66, 67], others have suggested that scrambling is less significant and indeed some structural information can be preserved from HDX experiments [38, 65]. The peptide chain deuterium content for the insulin fragment ions has been compared with that expected from 100% HD scrambling (see the Experimental section). The results of this comparison are shown in Figure 2. Notably, there is a significant difference between the deuterium content values expected for 100% HD scrambling versus the experimentally determined values. Therefore, the data are consistent with the occurrence of insulin chain separation prior to significant proton mobilization leading to 100% HD scrambling. Studies have shown that MS instrument parameters such as collisional activation time can affect the degree of HD scrambling [68]. To support the claim of limited (or no) HD scrambling under these conditions, we have subjected the [M – 4H]4− insulin ions to increased CID activation times. Under these conditions, dissociation of the chain A and chain B peptides shows a shift toward increased inter-chain HD scrambling as presented in Supplementary Figure 3. It is also instructive to note that the HDX values were determined using average m/z values of the isotopic envelopes.

Bar graph showing the comparisons of deuterium uptake for the insulin chain ions. The blue and red bars represent the experimentally measured deuterium uptake and the deuterium uptake that would be observed upon 100% HD scrambling, respectively. The yellow bars show the expected deuterium uptake values using the hydrogen accessibility scoring for all of labile hydrogens (see the Experimental section). Panels (a) and (b) represent data for the more diffuse [M – 4H]4− and [M – 5H]5− insulin ions, respectively. The error bars represent one standard deviation obtained from three separate measurements of mobility-selected ions
To compare the behavior of deprotonated insulin and protonated insulin identical (MS instrument parameters), experiments have been repeated for positively charged insulin ions. Supplementary Figure 4 and Supplementary Figure 5 show the results for diffuse [M + 4H]4+ (Ω = 720 Å2) and [M + 5H]5+ (Ω = 818 Å2) insulin ions, respectively. A normalized collision energy increase from 20 to 25 results in backbone cleavage, and no intact chain A and chain B fragment ions are observed at higher CID energies. Such results are indicative of more extensive proton mobilization for these insulin ions compared with the deprotonated species. Indeed, for the deprotonated insulin ions, the separation of chain A and chain B requires an even larger normalized collision energy of 30. Here, we present a note of caution. The energy distribution of the ion population during the CID process is broad and these comparisons are, thus, only presented as semiquantitative assessments; more precise comparisons can be obtained by utilizing other ion dissociation techniques such as SID [69].
It is well established that the various fragments produced by CID of peptide ions result from the mobility of available protons. During collisional activation of isotopically labeled peptide ions, this mobility results in the scrambling of the deuterium label. Therefore, the relatively limited HD scrambling observed between chain A and chain B fragment ions may result from what is here termed as limited proton mobilization for these deprotonated species. For such ions, the source of protons is more limited than for protonated ions. For example, there is only one proton source for the [M – 5H]5− insulin ions (a neutral carboxylic acid group). In comparison, [M + 5H]5+ ions would have all protonated basic residues as well as six neutral carboxylic acid residues. Additionally, it should be noted that the proton from the carboxylic acid side chains has been termed “locally” mobile because of the observation of limited fragment ion types resulting from its translocation [70–72]. Therefore, the conditions of fewer available protons overall and the limited degree of mobility of some may account for the observed limited inter-chain scrambling (as well as the limited scrambling observed in the manuscript accompanying this work).
Hydrogen Accessibility Scoring Combined with Protein Fold Considerations
Figure 2 reveals the disparity in deuterium uptake by the different insulin peptide chains. For the more diffuse [M – 4H]4− insulin ions, chain B uptakes a considerably larger number (2.5-fold) of deuterium compared with chain A. Although chain B has a greater number of labile hydrogens than chain A (52 versus 42, red bars in Figure 2), this difference alone cannot account for the observed deuterium within the different peptide chains. Using a nominal ion structure (conformer type) obtained from MDS (shown in Figure 3) along with a hydrogen accessibility scoring method (see the Experimental section), the deuterium content ratio (chain B:chain A) should not be greater than 1.7-fold (yellow bars Figure 2). Therefore, it becomes necessary to consider other factors that may result in the greater protection of exchange sites on chain A.

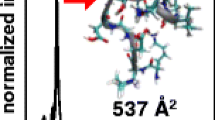
Panels (a) and (b) contain nominal structures obtained by MDS for the more diffuse [M – 4H]4− and [M – 5H]5− insulin ions, respectively. The upper portions of the panels show detailed structures and the lower portions show the overall surface accessible morphology. In these schematic depictions, chain A and chain B are colored blue and red, respectively. Collision cross sections for the nominal structures are shown above each panel
Supplementary Figure 6 shows a near linear relationship between the maximum deuterium uptake level and CCS of different ubiquitin and cytochrome c anions as reported in a recent study [42]. A question that arises is what accounts for the increased uptake levels with increased CCS. For the data shown in Supplementary Figure 6, ions with larger collision cross sections typically have a greater number of deprotonation sites. Additionally, the more diffuse species should have a greater number of exchange sites exposed to D2O. These two effects could account for the large increases in deuterium uptake with CCS for the ubiquitin and cytochrome c ions. The largest ubiquitin ions exhibit a greater charge density than the smallest cytochrome c ions of similar size and yet they exhibit lower levels of deuterium uptake. Previously it has been suggested that this relative level of protection for the ubiquitin ions results from the fact that a disproportionate number of hydrogens are buried compared with the more exposed sites on cytochrome c ions.[42] It is also noted here that increased ion temperature can enhance the accessibility of exchange sites for species of higher charge density due to more “effective” (i.e., the formation of a critical reaction complex) conformational fluctuations [37].
The effects of protein fold protection are expected to decrease dramatically with decreased ion size. For example, for small peptides typically the more diffuse species experience less deuterium uptake [41]. Considering that no residue is completely buried within the peptide, the maximum deuterium uptake levels have been shown to depend primarily on distance from charge sites (e.g., S HDXS values) [41]. In contrast, protein ions may have a considerable number of residues buried in the interior. For example, cytochrome c has more than 200 labile hydrogens, of which less than 40% on average are accessible at buffer gas temperatures of 300 K [37, 42]. Because the charge density does not change significantly with CCS, it may be argued that surface exposure has an increasing impact on hydrogen accessibility for these larger protein ions (even for elongated species). An extension of this concept is that quaternary structure may have a more pronounced effect upon relative deuterium uptake by subunits allowing the extraction of structural information in the form of assigning relative (periphery versus interior) positions.
It is, therefore, instructive to consider the effect of surface exposure for the insulin ions studied here. The ion structures exhibiting the lowest energies and having matching collision cross sections with the measured values for the more diffuse [M – 4H]4− and [M – 5H]5− insulin ions are shown in Figure 3a and b, respectively. Exposed regions of chain A and chain B are also shown in Figure 3. To model the expected deuterium content in chain A and chain B fragment ions while accounting for the effects of protein fold protection, two different approaches have been considered. In the first approach, the S HDXS value (Equation 1) of each hetero atom site is scaled by that site’s surface accessibility (H surface ) as estimated from SES values. The chain accessibility scores (S chain ) are then computed using Equation 3.
By scaling the sum of the S chain values to the total deuterium uptake of the precursor ions, comparisons with experimental deuterium content can be made as shown in Figure 4. Overall, the agreement between experiment and theory is quite good for both the [M – 4H]4− and the [M – 5H]5− ions. The effect of incorporating the surface accessibility parameter is illustrated in Figure 2 where the usage of S HDXS alone does not accurately model the experimental data. Supplementary Figures 7 and 8 show the effect of collision cross section of MDS structures on the ability to match deuterium uptake levels. In the example, a larger collision cross section for [M – 4H]4− ions results in deuterium uptake values that are significantly larger and smaller for peptide chain A and chain B, respectively.

Bar graph showing the comparisons of deuterium uptake for the insulin chain ions. The blue bars represent the experimentally measured deuterium uptake. The green bars show the expected deuterium uptake values using hydrogen accessibility scoring for all labile hydrogens scaled by surface exposure values (see Discussion section for details). The black bars demonstrate the expected hydrogen uptake values using the product of exposed area for each chain and their hydrogen accessibility score. Panels (a) and (b) contain data for the more diffuse [M – 4H]4−and [M – 5H]5− insulin ions, respectively. The error bars represent one standard deviation obtained from three separate measurements of mobility-selected ions
Although the first approach provides a good estimate of the deuterium uptake level for each chain, it does not take into account the total exposed surface area of each chain directly. It is instructive to evaluate a method that incorporates the complete exposed area of each peptide chain. One motivation for using the exposed area of the entire chain is to consider an approach where an estimated exposed surface area can be calculated directly from experimental uptake data. Here the chain surface area exposure (surface chain ) is computed from the sum of the SES values for the individual amino acid residues comprising a given chain. Thus, the new S chain values are determined from
Again, the S chain values can be scaled to the experimental deuterium uptake values and comparisons can be made to the experimental data as shown in Figure 4. The values are in good agreement with the experimental deuterium content. However, the agreement is not as good as that obtained when using the individual H surface values. Here we note that the good agreement of the former surface accessibility model with the experimental results further supports the idea that only limited HD scrambling occurs up to peptide chain dissociation for these experimental conditions. Although we cannot rule out the possibility of greater HD scrambling that is not accounted for because of the hypothetical structures obtained from MDS, previous experiments have shown that conformational types obtained from MDS do accurately account for deuterium uptake using accessibility modeling [41, 43]. Additionally, we note that surface accessibility and charge site accessibility have been combined in a previous model to account for deuterium uptake by peptide ions [36]. Having incorporated surface chain into the expression for theoretical deuterium uptake, it is useful to consider whether or not experimental values can be related to surface chain without using S HDXS. More interestingly, the question is whether or not the relative surface exposure for subunits in ions of protein complexes can be determined from deuterium uptake values alone. To accomplish such a comparison, another assumption is necessary. Here, we consider the average S HDXS values (\( {\overline{S}}_{HDXS}\Big) \) for both chains as given by:
In Equation 5, the variable n represents the total number of exchangeable hydrogens for a given peptide chain. The assumption to be made is that \( {\overline{S}}_{HDXS} \) (chain A) is equal to \( {\overline{S}}_{HDXS} \) (chain B). Using this relationship and rearranging Equation 5 followed by substitution into Equation 4, it is possible to derive the following ratio:
Here, the values of n a and n b correspond to the total number of exchangeable hydrogens on chain A and chain B, respectively.
Estimating Subchain Surface Area from Deuterium Uptake Measurements
From Equation 6, it is possible to express the fraction of surface area of each chain according to the manner shown in Equation 7.
For the purposes of this comparison, in Equation 7, S chainA and S insulin would correspond to the deuterium uptake values of chain A and precursor ions, respectively. Knowing values for n A and n B (and by extension n total ), allows the solving of the fractional representation of surface chainA and surface chainB . These can be compared with the fractions obtained from the model structures. Notably, the model structures are here considered as relatively accurate representations of only ion conformation type based on the analysis described above and depicted in Figure 4. Figure 5 shows the results of the calculation using Equation 7. Although the relative surface areas of chain A and chain B are in good agreement with those for the matching structure for the [M – 5H]5− ions, the model does not agree with the structure for the more diffuse [M – 4H]4− ions. In Figure 2, the greater difference between the 100% HD scrambling and the S HDXS models for the [M – 4H]4− ions suggests that the assumption that the accessibility of all hydrogens is the same (represented by \( {\overline{S}}_{HDXS}\Big) \) is less supported for these ions. The [M – 5H]5− ions may possess a more even distribution of charges than the [M – 4H]4− ions and the assumption that is made to generate Equation 6 is less valid for the latter ions. Notably, for protein complexes, the assumption could be more valid in that the hydrogens of the same subunit within a multiple-copy complex may be the same.

Bar graph showing the ratio of the exposed surface area for insulin subchains to the total exposed surface area of insulin. Red and blue bars compare results for the solvent excluded surface area (MDS) and the calculation using the experimental deuterium uptake values (Equation 6), respectively. Panels (a) and (b) represent values for the more diffuse [M – 4H]4− and [M – 5H]5− insulin ions, respectively
A limitation in the comparison in Figure 5 is the inability to estimate an error for the assumption regarding hydrogen accessibility that is necessary for formulating Equation 6. Therefore, the comparison only provides an estimation of the degree of dissimilarity in surface area assessment. It should be noted that the disagreement observed for the [M – 4H]4− ions may result from the fact that the average accessibility of hydrogens is more different between chain A and chain B compared with the [M – 5H]5− ions. Thus, the latter ions would represent a more uniform charge distribution. We cannot rule out the possibility that a more accurate structure (or subset of structures to reflect conformer dynamics) from MDS could provide better agreement for the former ions. Having noted the limitation, it is instructive to consider the potential applicability of the approach. Consider the possibility that \( {\overline{S}}_{HDXS} \) could be scaled according to a particular protein system (e.g., different exchange site accessibilities because of differences in charge site density). Supplementary Figure 6 suggests that it may be possible to estimate surface area using characteristic \( {\overline{S}}_{HDXS} \) for the different subunits within a protein complex.
Comparison of Protonated and Deprotonated KKDDDDDIIKIIK Deuterium Uptake Patterns
One reason for the selection of the KKDDDDDIIKIIK peptide for this study is the opportunity to study sequence effects on hydrogen accessibility. One comparison that can be performed is that of hydrogen accessibility for anions versus cations. The latter species are expected to have charges located on the terminal K residues; however, the anions should have deprotonation sites in the middle of the peptide. Comparing the deuterium uptake patterns of the protonated and deprotonated peptide ions is interesting because hydrogen accessibility in the respective polarity modes should be reversed. Unfortunately, ETD data for [M + 2H]2+ peptide ions is inefficient and the signal level for the compact [M – 3H]3− ions is insufficient for CID analysis. For this reason, comparisons of species of similar size (CCS) but not necessarily of the same charge state have been performed. To some degree, the hydrogen accessibility model results in a data normalization that allows the comparison of species of different charge state and opposite polarity.
Figure 6a shows the per-residue deuterium uptake comparison of compact [M + 3H]3+ and diffuse [M – 2H]2− KKDDDDDIIKIIK ions. It is also noted that these ions have relatively similar collision cross sections (340 Å2 versus 315 Å2, respectively) allowing a rough assessment of conformer type. A cursory comparison of deuterium uptake shows that the overall trend is as expected according to charge site locations. On the protonated peptide, the deuterium uptake at terminal K residues is more pronounced; on the deprotonated peptide, the deuterium uptake at interior D residues is increased. Exceptions are the D4 and D6 residues for which the protonated peptide ion has two labile hydrogens whereas the deprotonated peptide ions have only one; additionally, the possibility for partial HD scrambling may exacerbate this difference in deuterium uptake for the D4 and D6 residues (see the manuscript accompanying this work). This same trend is observed in the comparison of the more diffuse [M – 3H]3− and the more diffuse [M + 3H]3+ KKDDDDDIIKIIK ions shown in Figure 6b.

Bar graph showing the comparison of deuterium uptake for positively and negatively charged KKDDDDDIIKIIK peptide ions. Panel (a) compares deuterium uptake for the more compact (Ω = 340 Å2) [M + 3H]3+ KKDDDDDIIKIIK peptide ions (red bars) and the more diffuse (Ω = 315 Å2) [M – 2H]2− KKDDDDDIIKIIK peptide ions (blue bars); panel (b) shows the same comparison for the diffuse (Ω = 367 Å2) [M + 3H]3+ KKDDDDDIIKIIK ions (red bars) with the diffuse (Ω = 362 Å2) [M – 3H]3− KKDDDDDIIKIIK ions (blue bars). The error bars represent one standard deviation obtained from three separate measurements of mobility-selected ions
The Complementary Nature of Positive and Negative Polarity HDX
As mentioned above, a purpose of this work is to consider the utility of gas-phase HDX as a chemical probe to characterize the surfaces of subunits in protein complexes providing information about complex morphology. One advantage in using D2O as the deuterating reagent compared with other compounds (e.g., ND3) is the possibility to also determine charge site locations for gas-phase ions based upon deuterium uptake “patches” as described above and depicted in Supplementary Figure 9. Residues in closer proximity to charge sites will generally exhibit higher deuterium uptake levels and faster HDX kinetics [37]. Although deuterium uptake may occur on residues that are fairly distant to charge sites because of conformational fluctuations (“breathing”), residues within ~7 Å of a charge site exhibit more pronounced HDX activity [41]. Therefore, for considerations of exposed surface area, the surface residues that are within 7 Å of a charge site can be considered accessible and the summation of the surface area presented by such residues can represent the total surface accessible by HDX.
To obtain a better understanding of sites that are protected in subunit interface regions, it would be necessary to map the HDX accessible surface as completely as possible. Supplementary Figure 9 shows the HDX accessible surface for a nominal structure for [M – 5H]5− insulin ions according to the type of HDX experiment to be conducted. Notably, a large portion of the total surface area is not accessible (within 7 Å of a positive or negative site). The combination of negative ion analysis does provide a significant enhancement with regard to HDX accessible surface area. For example, for the structure shown in Supplementary Figure 9, each polarity alone can probe less than 40% of the total surface area; together, both polarities can probe about 60% of the surface area. There is an increase in surface area coverage of ~50% by performing HDX in positive- and negative-ion modes. A note of caution is presented. The relatively large inaccessible area should be considered cautiously. A single MDS structure does not provide information about which areas become accessible as a result of conformational fluctuations over the relatively long timescale of the mobility measurement.
These experiments that utilize D2O could be supplemented with those using ND3 as the gas-phase reagent. Since ND3 does not require the formation of a long-lived reaction intermediate bridging two ion locations as with D2O [37], it is suggested to be a more direct probe of hydrogen accessibility [73, 74]. Recently, foundational experiments have demonstrated the use of ND3 in HDX experiments in which ETD is used to locate sites of deuterium incorporation [40, 75]. Here, and in the manuscript accompanying the work, D2O has been used because of location constraints in the HDX process. The requirement that exchange occurs near charge sites provides evidence for the location of charge sites (an important consideration in MDS analysis) as well as the ability to check for inter-chain scrambling using a hydrogen accessibility scoring model described here. Future experiments would benefit from combined HDX experiments using ND3 and D2O. Here, it will be possible to gain information about potential charge site locations as well as to increase the surface area that can be characterized.
Conclusions
Studies with bovine insulin ions demonstrated one advantage of HDX analysis in negative ion mode. The lack of peptide bond cleavage and HD scrambling between chain A and chain B peptides upon dissociation of insulin ions suggests that the analysis of anions may, to some degree, allow the determination of deuterium incorporation for subunit ions within protein complexes. Such a result may be more suitable for an ion dissociation technique with rapid energy deposition such as SID [76]. Additionally, the potential for decreased proton mobilization upon CID of negatively charged ions may allow the generation of the intact peptide chain ions as opposed to the induction of backbone fragmentation, which challenges spectral interpretation as well as the ability to conduct multi-stage tandem mass spectrometry (MSn) of the individual peptide chain ions.
This work also shows that the deuterium uptake for each peptide chain correlates with its exposed surface area as shown by two different approaches. Although somewhat limited in the precision of the estimation of exposed surface area for some ions, values for each chain can be estimated without using nominal structures from MDS for some ions. This preliminary work still is of benefit for combined IMS-MS experiments. As an example, consider that MDS can determine numerous morphologies with matching collision cross sections. The approach described here can be used to increase the overall selectivity of structure comparisons in order to obtain relevant MDS structures.
Finally, although the HDX accessibility model analysis shows that relatively large portions of protein surfaces can be probed by HDX for select anion conformers, the fraction of the surface area that can be studied by HDX is increased significantly if complementary cation studies are conducted. Tthe different accessibility regions (with D2O as the deuterating reagent) for protonated and deprotonated peptides provides an opportunity for mapping a much larger portion of the ion’s surface. Such an enhancement may be valuable in the future for elucidating protein–protein interaction regions for protein complex ions as well as charge site locations.
References
Yearly Growth of Protein Structures. Available at: http://www.rcsb.org/pdb/statistics/contentGrowthChart.do?content=molType-protein&seqid=100. Accessed 20 March 2014
Robinson, C.V., Sali, A., Baumeister, W.: The molecular sociology of the cell. Nature 450, 973–982 (2007)
Wimberly, B.T., Brodersen, D.E., Clemons, W.M., Morgan-Warren, R.J., Carter, A.P., Vonrhein, C., Hartsch, T., Ramakrishnan, V.: Structure of the 30S ribosomal subunit. Nature 407, 327–339 (2000)
Ban, N., Nissen, P., Hansen, J., Moore, P.B., Steitz, T.A.: The complete atomic structure of the large ribosomal subunit at 2.4 angstrom resolution. Science 289, 905–920 (2000)
Schluenzen, F., Tocilj, A., Zarivach, R., Harms, J., Gluehmann, M., Janell, D., Bashan, A., Bartels, H., Agmon, I., Franceschi, F., Yonath, A.: Structure of functionally activated small ribosomal subunit at 3.3 angstrom resolution. Cell 102, 615–623 (2000)
Mainz, A., Religa, T.L., Sprangers, R., Linser, R., Kay, L.E., Reif, B.: NMR spectroscopy of soluble protein complexes at one mega-dalton and beyond. Angew. Chem. Int. Ed. 52, 8746–8751 (2013)
Bartesaghi, A., Matthies, D., Banerjee, S., Merk, A., Subramaniam, S.: Structure of beta-galactosidase at 3.2-angstrom resolution obtained by cryo-electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 111, 11709–11714 (2014)
Frank, J.: Story in a samplethe potential (and limitations) of cryo-electron microscopy applied to molecular machines. Biopolymers 99, 832–836 (2013)
Konermann, L., Pan, J., Liu, Y.-H.: Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 40, 1224–1234 (2011)
Pirrone, G.F., Iacob, R.E., Engen, J.R.: Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Anal. Chem. 87, 99–118 (2015)
Sowole, M.A., Konermann, L.: Effects of protein-ligand interactions on hydrogen/deuterium exchange kinetics: canonical and noncanonical scenarios. Anal. Chem. 86, 6715–6722 (2014)
McAllister, R.G., Konermann, L.: Challenges in the interpretation of protein H/D exchange data: a molecular dynamics simulation perspective. Biochemistry 54, 2683–2692 (2015)
Maiolica, A., Cittaro, D., Borsotti, D., Sennels, L., Ciferri, C., Tarricone, C., Musacchio, A., Rappsilber, J.: Structural analysis of multiprotein complexes by cross-linking, mass spectrometry, and database searching. Mol. Cell Proteomics 6, 2200–2211 (2007)
Sinz, A.: Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein–protein interactions. Mass Spectrom. Rev. 25, 663–682 (2006)
Vahidi, S., Stocks, B.B., Liaghati-Mobarhan, Y., Konermann, L.: Submillisecond protein folding events monitored by rapid mixing and mass spectrometry-based oxidative labeling. Anal. Chem. 85, 8618–8625 (2013)
Mendoza, V.L., Vachet, R.W.: Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom. Rev. 28, 785–815 (2009)
Kirk, B.B., Trevitt, A.J., Blanksby, S.J., Tao, Y.Q., Moore, B.N., Julian, R.R.: Ultraviolet action spectroscopy of iodine labeled peptides and proteins in the gas phase. J. Phys. Chem. A 117, 1228–1232 (2013)
Lightwahl, K.J., Schwartz, B.L., Smith, R.D.: Observation of the noncovalent quaternary associations of proteins by electrospray-ionization mass-spectrometry. J. Am. Chem. Soc. 116, 5271–5278 (1994)
Ruotolo, B.T., Robinson, C.V.: Aspects of native proteins are retained in vacuum. Curr. Opin. Chem. Biol. 10, 402–408 (2006)
Van Berkel, W.J.H., Van Den Heuvel, R.H.H., Versluis, C., Heck, A.J.R.: Detection of intact megaDalton protein assemblies of vanillyl-alcohol oxidase by mass spectrometry. Protein Sci. 9, 435–439 (2000)
Sharon, M., Robinson, C.V.: The role of mass spectrometry in structure elucidation of dynamic protein complexes. Annu Rev Biochem. 76, 167–193 (2007)
Keifer, D.Z., Pierson, E.E., Hogan, J.A., Bedwell, G.J., Prevelige, P.E., Jarrold, M.F.: Charge detection mass spectrometry of bacteriophage P22 procapsid distributions above 20 MDa. Rapid Commun. Mass Spectrom. 28, 483–488 (2014)
Benesch, J.L.P., Aquilina, J.A., Ruotolo, B.T., Sobott, F., Robinson, C.V.: Tandem mass spectrometry reveals the quaternary organization of macromolecular assemblies. Chem. Biol. 13, 597–605 (2006)
Blackwell, A.E., Dodds, E.D., Bandarian, V., Wysocki, V.H.: Revealing the quaternary structure of a heterogeneous noncovalent protein complex through surface-induced dissociation. Anal. Chem. 83, 2862–2865 (2011)
Woods, L.A., Radford, S.E., Ashcroft, A.E.: Advances in ion mobility spectrometry-mass spectrometry reveal key insights into amyloid assembly. Biochim. Biophys. Acta, Proteins Proteomics 1834, 1257–1268 (2013)
Bernstein, S.L., Dupuis, N.F., Lazo, N.D., Wyttenbach, T., Condron, M.M., Bitan, G., Teplow, D., Shea, J., Ruotolo, B., Robinson, C., Bowers, M.: Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nat. Chem. 1, 326–331 (2009)
Uetrecht, C., Rose, R.J., van Duijn, E., Lorenzen, K., Heck, A.J.R.: Ion mobility mass spectrometry of proteins and protein assemblies. Chem. Soc. Rev. 39, 1633–1655 (2010)
Mesleh, M.F., Hunter, J.M., Shvartsburg, A.A., Schatz, G.C., Jarrold, M.F.: Structural information from ion mobility measurements: effects of the long-range potential. J. Phys. Chem. A 100, 16082–16086 (1996)
Wyttenbach, T., Bleiholder, C., Bowers, M.T.: Factors contributing to the collision cross section of polyatomic ions in the kilodalton to gigadalton range: application to ion mobility measurements. Anal. Chem. 85, 2191–2199 (2013)
Wyttenbach, T., vonHelden, G., Batka, J.J., Carlat, D., Bowers, M.T.: Effect of the long-range potential on ion mobility measurements. J. Am. Soc. Mass Spectrom. 8, 275–282 (1997)
Shvartsburg, A.A., Jarrold, M.F.: An exact hard-spheres scattering model for the mobilities of polyatomic ions. Chem. Phys. Lett. 261, 86–91 (1996)
Hall, Z., Politis, A., Robinson, C.V.: Structural modeling of heteromeric protein complexes from disassembly pathways and ion mobility-mass spectrometry. Structure 20, 1596–1609 (2012)
Benesch, J.L., Robinson, C.V.: Mass spectrometry of macromolecular assemblies: preservation and dissociation. Curr. Opin. Struct. Biol. 16, 245–251 (2006)
Quintyn, R.S., Yan, J., Wysocki, V.H.: Surface-induced dissociation of homotetramers with D-2 symmetry yields their assembly pathways and characterizes the effect of ligand binding. Chem. Biol. 22, 583–592 (2015)
Valentine, S.J., Clemmer, D.E.: H/D exchange levels of shape-resolved cytochrome c conformers in the gas phase. J. Am. Chem. Soc. 119, 3558–3566 (1997)
Wyttenbach, T., Bowers, M.T.: Gas phase conformations of biological molecules: the hydrogen/deuterium exchange mechanism. J. Am. Soc. Mass Spectrom. 10, 9–14 (1999)
Valentine, S.J., Clemmer, D.E.: Temperature-dependent H/D exchange of compact and elongated cytochrome c ions in the gas phase. J. Am. Soc. Mass Spectrom. 13, 506–517 (2002)
Smith, D.L., Zhang, Z.Q., Liu, Y.Q.: Amide hydrogen-exchange and mass-spectrometry—a probe of high-order structure in proteins. Pure Appl. Chem. 66, 89–94 (1994)
Rand, K.D., Adams, C.M., Zubarev, R.A., Jorgensen, T.J.D.: Electron capture dissociation proceeds with a low degree of intramolecular migration of peptide amide hydrogens. J. Am. Chem. Soc. 130, 1341–1349 (2008)
Mistarz, U.H., Brown, J.M., Haselmann, K.F., Rand, K.D.: Simple setup for gas-phase H/D exchange mass spectrometry coupled to electron transfer dissociation and ion mobility for analysis of polypeptide structure on a liquid chromatographic time scale. Anal. Chem. 86, 11868–11876 (2014)
Khakinejad, M., Kondalaji, S.G., Maleki, H., Arndt, J.R., Donohoe, G.C., Valentine, S.J.: Combining ion mobility spectrometry with hydrogen-deuterium exchange and top-down MS for peptide ion structure analysis. J. Am. Soc. Mass Spectrom. 25, 2103–2115 (2014)
Donohoe, G.C., Khakinejad, M., Valentine, S.J.: Ion mobility spectrometry-hydrogen deuterium exchange mass spectrometry of anions: Part 1. peptides to proteins. J. Am. Soc. Mass Spectrom. 26, 564–576 (2015)
Khakinejad, M., Kondalaji, S., Tafreshian, A., Valentine, S.: Gas-phase hydrogen-deuterium exchange labeling of select peptide ion conformer types: a per-residue kinetics analysis. J. Am. Soc. Mass Spectrom. 26(7), 1115–1127 (2015)
Stlouis, R.H., Hill, H.H.: Ion mobility spectrometry in analytical-chemistry. Crit. Rev. Anal. Chem. 21, 321–355 (1990)
Wyttenbach, T., Pierson, N.A., Clemmer, D.E., Bowers, M.T.: Ion mobility analysis of molecular dynamics. Annu. Rev. Phys. Chem. 65, 175–196 (2014)
Clemmer, D.E., Jarrold, M.F.: Ion mobility measurements and their applications to clusters and biomolecules. J. Mass Spectrom. 32, 577–592 (1997)
Maurer, M.M., Donohoe, G.C., Valentine, S.J.: Advances in ion mobility-mass spectrometry instrumentation and techniques for characterizing structural heterogeneity. Analyst. 140(20), 6782–6798 (2015)
Hyung, S.-J., Ruotolo, B.T.: Integrating mass spectrometry of intact protein complexes into structural proteomics. Proteomics 12, 1547–1564 (2012)
May, J.C., McLean, J.A.: Ion mobility-mass spectrometry: time-dispersive instrumentation. Anal. Chem. 87, 1422–1436 (2015)
Cumeras, R., Figueras, E., Davis, C.E., Baumbach, J.I., Gracia, I.: Review on ion mobility spectrometry. Part 1. Current instrumentation. Analyst 140, 1376–1390 (2015)
Jones, C.M., Bernier, M., Carson, E., Colyer, K.E., Metz, R., Pawlow, A., Wischow, E., Webb, I., Andriole, E., Poutsma, J.: Gas-phase acidities of the 20 protein amino acids. Int. J. Mass Spectrom. 267, 54–62 (2007)
Mobcal Software. Available from: http://www.indiana.edu/~nano/software.html (1997). Accessed 4 May 2015
Mesleh, M.F., Hunter, J.M., Shvartsburg, A.A., Schatz, G.C., Jarrold, M.F.: Structural information from ion mobility measurements: effects of the long-range potential. J. Phys. Chem. 100, 16082–16086 (1996)
Sanner, M.F., Olson, A.J., Spehner, J.C.: Reduced surface: an efficient way to compute molecular surfaces. Biopolymers 38, 305–320 (1996)
Campbell, S., Rodgers, M.T., Marzluff, E.M., Beauchamp, J.L.: Deuterium exchange reactions as a probe of biomolecule structure. Fundamental studies of cas phase H/D exchange reactions of protonated glycine oligomers with D2O, CD3OD, CD3CO2D, and ND3. J. Am. Chem. Soc. 117, 12840–12854 (1995)
Chan, S., Enke, C.G.: Mechanistic study of hydrogen-deuterium exchange between M-1- ions of chlorinated benzenes and D2O or ND3. J. Am. Soc. Mass Spectrom. 5, 282–291 (1994)
Khakinejad, M., Kondalaji, S., Tafreshian, A., Valentine, S.: Gas-phase hydrogen-deuterium exchange labeling of select peptide ion conformer types: a per-residue kinetics analysis. J. Am. Soc. Mass Spectrom. 26(7), 1115–1127 (2015)
Shelimov, K.B., Clemmer, D.E., Hudgins, R.R., Jarrold, M.F.: Protein structure in vacuo: gas-phase confirmations of BPTI and cytochrome c. J. Am. Chem. Soc. 119, 2240–2248 (1997)
Mason, E.A., McDaniel, E.W.: Transport properties of ions in gaxses. Wiley, New York (1988)
Arndt, J.R., Kondalaji, S.G., Maurer, M.M., Parker, A., Legleiter, J., Valentine, S.J.: Huntingtin N-terminal monomeric and multimeric structures destabilized by covalent modification of heteroatomic residues. Biochemistry 54, 4285–4296 (2015)
Bilusich, D., Bowie, J.H.: Fragmentations of (M-H)(−) anions of underivatised peptides. PART 2. Characteristic cleavages of ser and cys and of disulfides and other post-translational modifications, together with some unusual internal processes. Mass Spectrometry Reviews 28, 20–34 (2009)
Mentinova, M., Han, H., McLuckey, S.A.: Dissociation of disulfide-intact somatostatin ions: the roles of ion type and dissociation method. Rapid Commun. Mass Spectrom. 23, 2647–2655 (2009)
Zhang, M., Kaltashov, I.A.: Mapping of protein disulfide bonds using negative ion fragmentation with a broadband precursor selection. Anal. Chem. 78, 4820–4829 (2006)
Howe, I., McLafferty, F.W.: Hydrogen scrambling in organic ions as a function of internal energy–extension of energy range. J. Am. Chem. Soc. 92, 3797 (1970)
Xiao, H., Kaltashov, I.A.: Transient structural disorder as a facilitator of protein-ligand binding: native H/D exchange-mass spectrometry study of cellular retinoic acid binding protein I. J. Am. Soc. Mass Spectrom. 16, 869–879 (2005)
Hamuro, Y., Tomasso, J.C., Coales, S.J.: A simple test to detect hydrogen/deuterium scrambling during gas-phase peptide fragmentation. Anal. Chem. 80, 6785–6790 (2008)
Bache, N., Rand, K.D., Roepstorff, P., Ploug, M., Jorgensen, T.J.D.: Hydrogen atom scrambling in selectively labeled anionic peptides upon collisional activation by maldi tandem time-of-flight mass spectrometry. J. Am. Soc. Mass Spectrom. 19, 1719–1725 (2008)
Hoerner, J.K., Xiao, H., Dobo, A., Kaltashov, I.A.: Is there hydrogen scrambling in the gas phase? Energetic and structural determinants of proton mobility within protein ions. J. Am. Chem. Soc. 126, 7709–7717 (2004)
Paizs, B., Suhai, S.: Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 24, 508–548 (2005)
Wysocki, V.H., Tsaprailis, G., Smith, L.L., Breci, L.A.: Special feature: commentary – mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom. 35, 1399–1406 (2000)
Gu, C.G., Tsaprailis, G., Breci, L., Wysocki, V.H.: Selective gas-phase cleavage at the peptide bond terminal to aspartic acid in fixed-charge derivatives of asp-containing peptides. Anal. Chem. 72, 5804–5813 (2000)
Herrmann, K.A., Wysocki, V.H., Vorpagel, E.R.: Computational investigation and hydrogen/deuterium exchange of the fixed charge derivative tris(2,4,6-trimethoxyphenyl) phosphonium: implications for the aspartic acid cleavage mechanism. J. Am. Soc. Mass Spectrom. 16, 1067–1080 (2005)
Rand, K.D., Pringle, S.D., Murphy, J.P., Fadgen, K.E., Brown, J., Engen, J.R.: Gas-phase hydrogen/deuterium exchange in a traveling wave ion guide for the examination of protein conformations. Anal. Chem. 81, 10019–10028 (2009)
Geller, O., Lifshitz, C.: A fast flow tube study of gas phase H/D exchange of multiply protonated ubiquitin. J. Phys. Chem. A 109, 2217–2222 (2005)
Rand, K.D., Pringle, S.D., Morris, M., Brown, J.M.: Site-specific analysis of gas-phase hydrogen/deuterium exchange of peptides and proteins by electron transfer dissociation. Anal. Chem. 84, 1931–1940 (2012)
Ma, X., Zhou, M., Wysocki, V.H.: Surface induced dissociation yields quaternary substructure of refractory noncovalent phosphorylase B and glutamate dehydrogenase complexes. J. Am. Soc. Mass Spectrom. 25, 368–379 (2014)
Acknowledgments
The authors gratefully acknowledge financial support from the Eberly College of Arts and Sciences at West Virginia University for providing laboratory research startup funds. Additionally, partial support for this work was provided by the National Institutes of Health (1R15NS090380-01 and 1R01GM114494-01).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 4020 kb)
Rights and permissions
About this article

Cite this article
Khakinejad, M., Ghassabi Kondalaji, S., Donohoe, G.C. et al. Ion Mobility Spectrometry-Hydrogen Deuterium Exchange Mass Spectrometry of Anions: Part 3. Estimating Surface Area Exposure by Deuterium Uptake. J. Am. Soc. Mass Spectrom. 27, 462–473 (2016). https://doi.org/10.1007/s13361-015-1305-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-015-1305-9