
Abstract
Chemical-ionization techniques that use metastable species to ionize analytes traditionally use a flat pin or a sharp solid needle onto which the high potential needed to generate the discharge plasma is applied. We report here that direct analysis of samples containing volatile and semivolatile compounds, including saturated and unsaturated aliphatic hydrocarbons, can be achieved on any electrospray-ionization mass spectrometer by passing helium though the sample delivery metal capillary held at a high potential. In the helium plasma ionization source (HPIS) described here, the typical helium flow required (about 20–30 mL/min), was significantly lower than that needed for other helium-ionization sources. By this procedure, positive ions were generated by nominal hydride ion removal from molecules emanating from heated saturated hydrocarbons as large as tetratetracontane (C44H90), at capillary voltages ranging from 2.0 to 4.0 kV. Unsaturated hydrocarbons, on the other hand, underwent facile protonation under much lower capillary voltages (0.9 to 2.0 kV). Although saturated and monounsaturated hydrocarbons bearing the same number of carbon atoms generate ions of the same m/z ratio, a gas-phase deuterium exchange method is described to ascertain the identity of these isomeric ions originating from either protonation or hydride abstraction mechanisms. Moreover, mass spectrometric results obtained by exposing unsaturated hydrocarbons to D2O vapor in an HPIS-MS instrument confirmed that the proton donor for ionization of unsaturated hydrocarbons is protonated water.
Similar content being viewed by others

1 Introduction
The introduction of ambient ionization techniques has revolutionized mass spectrometry. Direct-analysis-in-real-time (DART) [1] and desorption electrospray ionization (DESI) [2] are two of the most widely used ambient pressure ion sources for mass spectrometry [3]. The DESI procedure, which is more suited for polar compounds, is less efficient for compounds of low polarity such as aliphatic hydrocarbons [4, 5]. For less polar compounds, a more complicated procedure called reactive DESI has been shown to be more practical [5]. Recently, a more efficient ionization method using a nebulized spray of ionized toluene for compounds of low proton affinity and low acidity, such as cholesterol, has been described [6].
In contrast to electrospray related liquid-spray techniques, the atmospheric pressure chemical ionization techniques employ direct-current corona discharges [7, 8]. For example, samples can be heated and ionized in an atmospheric-pressure chemical ionization (APCI) source and then transferred to a mass analyzer to obtain chemical ionization spectra [9, 10]. Sometimes, ionization can also be achieved under lower voltages than voltages used for APCI when a gas such as helium is employed to obtain a glow discharge instead of a corona discharge [1, 11, 12]. In glow-discharge techniques such as DART, an atmospheric pressure gas is passed over a needle that is maintained at a lower potential against a grounded counter electrode than those used for corona discharges. The glow discharge generated in this way, produces metastable species of the gas used, which subsequently transfers its energy to analyte molecules by Penning ionization [13].
Although the use of the afterglow of a helium discharge had been proposed decades ago as an alternative for gas chromatographic flame-ionization detection [14], it has not become a practical method for generating gaseous ions for mass spectrometry. In fact, methods for generating ions under atmospheric conditions from high-molecular weight materials are highly sought [15]. The petroleum industry is particularly interested in novel methods of obtaining spectra of hydrocarbons present in petroleum distillates [16–18]. Generally, atmospheric pressure ionization methods are not well suited for hydrocarbon analysis. In our pursuit for novel methods of ionization [6, 19], we investigated the possibility of producing gaseous ions by passing helium though the charged metal capillary of a commercial electrospray ionization source. Here, we report that DART-like spectra can be produced from hydrocarbons by this simple modification.
2 Experimental
2.1 Chemicals
High-purity helium (99.999%; Airgas, Radnor, PA, USA) was used for all experiments. Heneicosane, (Z)-9-tricosene, tricosane, (Z)-3-pentacosene, (Z)-7-pentacosene, and pentacosane were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). All other compounds used in this study were from our collection of research samples. Candle wax was purchased from RiteAid (Hoboken, NJ, USA).
2.2 Insects
Baited with rotten apples, many fruit flies were trapped near an open window of our laboratory (Hoboken, New Jersey). Ten flies (males and females mixed) were anesthetized by placing them in a refrigerator for about 1 min. The flies were then extracted with hexane (50 μL). After 1 min, the supernatant was withdrawn and concentrated to 3–4 μL and used for chemical analysis.
2.3 GC/MS
GC/MS analysis of the insect extract (1.0 μL of the concentrated extract) was conducted on a Hewlett Packard 5971 Series Mass Selective Detector linked to a Hewlett Packard 5890 Series II gas chromatograph (Hewlett Packard, Palo Alto, CA, USA). Positive-ion electron-ionization mass spectra (70 eV) were acquired from m/z 35 to 450. Samples were introduced by splitless injection on a 25 m × 0.20 mm fused-silica column coated with DB-5 MS (0.33 μm) (Agilent JW Scientific, Santa Clara, CA, USA). The oven temperature was maintained at 100°C for 3 min and increased at 5°C/min to a final temperature of 285°C. After reaching 285°C, the GC oven was maintained isothermally for 15 min. The injector temperature was set at 270°C, and the transfer line temperature was 280°C.
2.4 Helium Plasma Ionization Source (HPIS) and Mass Spectrometry
The Z-Spray ion source of a Waters Micromass Q-TOF API-US mass spectrometer was modified as shown in Figure 1. Briefly, a slit (1 in. × 1 in.) was cut in the glass cylinder that surrounds the ionization region of the Z-Spray ion source, and a direct insertion probe (GCQ solids probe; Finnigan Co., San Jose, CA, USA) was inserted through the hole. The analytes were placed in the bottom of a glass sample tube (10 mm × 1.3 mm o.d. × 1.1 mm i.d.) and inserted into the port of the solids probe. Typically, the probe temperature was held at 140°C for 20 s and increased at 10°C/min to 200°C, and held for 2 min. To clean the probe, after each analysis, the temperature was increased at 50°C/min to 350°C and held for 2 min. The probe was then cooled to room temperature with a stream of N2.

A schematic that illustrates the modifications to an electrospray ionization source to function as a helium plasma ionization source (HPIS)
The metal spray capillary tip of the electrospray probe was set about 10 mm from the mass spectrometer sampling-cone orifice through which gaseous ions entered the instrument. Instead of a solvent, a steam of high purity helium, regulated by a needle valve, was passed through the metal capillary held at a high voltage. Typically, the helium flow rate was maintained at 30 mL/min, and the capillary voltage was set at 3 kV (or adjusted to obtain the best ion flux).
The source temperature was held at 100°C. Cone voltage was typically set as 10–25 V. For MS/MS experiments, pressure of argon gas in the collision cell was held at 5.1 × 10–5 Pa. The heater in the desolvation gas line was used to increase the temperature of the nitrogen desolvation gas when necessary (Gas 2, Figure 1). Typically, the desolvation temperature was set between 150 and 300°C. The cone gas function in the Z-spray source was not utilized. The addition of a co-axial nebulizer gas was also not used for most experiments. However, for the gas-phase deuterium exchange experiments, a glass pipette with a cotton wool plug wetted with a few drops of D2O was connected to the nebulization gas line (Gas 1, Figure 1). The flow rate was typically set between 6 and 60 L/h. A mixture of pentacosane and (Z)-3-pentacosene (about 10 μg each), or a cuticular hydrocarbon extract from fruit flies (equivalent to about 4–5 insects) was added to the glass sample vial of the solids probe. Solvent was allowed to evaporate under room temperature before the sample vial was inserted into the holding port of the solids probe. The spectrum of candle wax volatiles was obtained by holding a small piece of wax with a pair of tweezers and exposing the sample to the helium plasma. Although the sample was not directly heated, it was exposed to a flow of hot “desolvation” gas at a rate of 200 L/hr (the heater was set to 300°C).
For accurate mass measurement experiments, the mass spectrometer was run under the W-mode, and the instrument was calibrated using water cluster ions. Lock-mass corrections were made to all acquired spectra.
3 Results and Discussion
Upon application of a high potential (+1 to +4 kV) to the metal capillary tube while passing a flow of helium through it (instead of a solution), a cone-shaped purple-colored glow discharge was visible, indicating the formation of a zone of helium plasma at the tip of the capillary (Figure 2). The length of the glow regime decreased as the helium flow rate was reduced and disappeared when the helium flow was stopped. The length of the glow cone increased to a maximum of about 3–4 mm as the helium flow was increased. A complex series of competing chemical reactions were expected to occur in the vicinity of the observed discharge. According to proposed mechanisms for other systems [20], the excited helium atoms (23S1) in a so-called “metastable” state transfer energy to ionize molecules of ambient gases by Penning ionization [13]. This ionization is expected because excited 23S1 helium atoms bear an internal energy of 19.8 eV, which is higher than the ionization energies of common atmospheric gases. In this way, particularly atmospheric moisture is known to undergo ionization by “metastable” helium very efficiently [20].

Glow discharge observed from a HPIS operated under capillary voltages of 4.0 (a), 3.0 (b), and 2.0 kV (c), respectively. A = capillary anode; PC = positive column; O = entrance orifice of the mass spectrometer; C = baffle plate
Upon exposure of vapors emanating from several saturated aliphatic hydrocarbons ranging from C-23 to C-44 to the helium plasma generated by the setup shown in Figure 1, respective (M – H)+ ions were generated from each sample (Figure 3). Although several ionization pathways are possible in DART-like sources, the dominant positive-ion formation mechanism is deemed to occur via proton transfers from ionized water or charged water clusters to analyte molecules of higher proton affinity. However, n-alkanes, because of their low proton affinity, are not known to accept protons from protonated atmospheric moisture. Instead, saturated hydrocarbons are subjected to nominal hydride ion removal [21]. Although the exact nature of the hydride-abstracting species remains elusive, the observation that relative intensities of (M – H)+ peaks correlate well with that of the background NO+ peaks has indicated that the reactive hydride-removing species could be NO+ [20, 21]. For the reason that the ionization mechanisms taking place in a helium-plasma ionization source are similar to those happening in a DART source, it is not surprising that the major peaks from HPIS-MS of alkanes represent ions derived by hydride removal from neutral precursors.

Mass spectra of saturated hydrocarbons obtained by HPIS, coupled to a Quadrupole-Time-of-Flight (Q-TOF) mass spectrometer: (a) tricosane, (b) pentacosane, (c) dotriacontane, (d) hexatriacontane, (e) tetracontane, and (f) tetratetracontane. Samples introduced by a heated solids probe. To record spectra (a)–(c), the probe temperature was held at 140°C for 20 s, and increased at 10°C/min to 200°C and kept for 2 min while the desolvation gas temperature was maintained at 200°C. To record spectra (d)–(f), the probe temperature was held at 200°C for 2 min, and increased at 50°C/min to 350°C, and kept for 2 min, while the desolvation gas temperature was maintained at 300°C. Desolvation gas flow rate was 200 L/h for all experiments
The ionization efficacy of alkanes and alkenes in a helium plasma source at different discharge voltages was investigated using (Z)-9-heneicosene, heptadecane, and pentacosane as representative analytes. For the two alkanes, at similar sample amounts, the initial signals representing [M – H]+ ions were observed at a capillary voltage of about 1.5-2.0 kV (Supplementary Figure S-1). More intense signals were obtained when the voltage was increased to 3.0–4.0 kV. For the alkene (Z)-9-heneicosene, on the other hand, the onset of ionization by proton transfer was observed at much lower capillary voltages. Although no signals were observed at capillary voltages below 0.9 kV, intense signals representing the [M + H]+ ion at a S/N ratio > 100:1 were obtained at 1.0 kV (Supplementary Figure S-1). Evidently, the ionization efficacies of alkanes and alkenes are significantly different from each other. Apparently, the proton addition to alkenes, by secondary ions, occurs at a lower density of metastable helium because this reaction happens even at 0.9 kV (the upper-most curve in Supplementary Figure S-1). In contrast, the hydride removal pathway (by secondary ions such as NO+) is triggered only at a higher density of metastable helium. This supposition is supported by the observation that peaks for (M – H)+ ions from saturated hydrocarbons are observed only at higher capillary voltages (the two lower-most curves in Supplementary Figure S-1). Because the differences in volatility of samples also play a role, heptadecane (a compound of lower boiling point) and pentacosane (a compound of higher boiling point) were chosen for the comparison of their ionization efficiencies with that of (Z)-9-heneicosene.
In addition to the peaks for the [M – H]+ ions generated by hydride removal, a few minor peaks that represented oxidation products of the hydrocarbon analytes were also observed. Such oxidation products have been reported from alkanes previously [20]. The oxidation artifacts formed under high-voltage desorption electrospray ionization conditions from hydrocarbons have been characterized by subjecting the products to betaine aldehyde derivatization followed by mass spectrometry. The major oxidation products from alkanes have been recognized to be alcohols and hydroxy ketones [5]. The spectra produced by the reactive DESI method appear very complex due to the presence of products from multiple oxidations and dehydrogenations. In contrast, the spectra produced by the HPIS-MS procedure (Figure 3a–f), even from complex mixtures of analytes, are rather simple (Figure 4).

A mass spectrum recorded on a HPIS-Q-TOF mass spectrometer from the mixture volatile hydrocarbons emanating from a candle wax sample. The sample was exposed to a heated desolvation gas flow (200 L/h at 300°C) without any direct heating (peak labels indicate the number of carbon atoms in each hydrocarbon)
It is known that in DART and other AP ion sources reactive species such as NO+, O +•2 are formed [20]. The species NO+ in particular can initiate a cascade of oxidation and other reactions with many ambient gaseous molecules. Because NO+ is an ion of very high hydride ion affinity (247 kcal/mol), it can abstract hydride ions from many molecules [21]. Cody has reported that the relative intensities of the peaks attributable to hydride-abstraction and oxidization products in DART spectra correlated well with the abundance of NO+ in the background [20]. The physical position of the sample placement, and ion source parameters (such as the temperature of the hot desolvation gas) affect the relative abundances of oxidative products in the plasma. For example, we noted in the spectra of pentacosane, the intensities of the peaks at m/z 367 and 381, for the C25H51O+, and C25H49O +2 ions, respectively, depend on the physical position of the sample placement from the capillary tip (Supplementary Figure S-2). Interestingly, the intensities of the oxidation-product peaks dropped drastically when the sample probe was moved closer to the capillary orifice (Supplementary Figure S-2a and c), and increased when the solids probe was moved away from the capillary orifice (Supplementary Figure S-2b and d). Moving the sample vial back and forth showed that the peak intensity changes occur in a reproducible manner (Supplementary Figure S-2). Although we do not have a definitive rationalization yet, it is evident that the gas-phase ion chemistry that occurs at different positions of the plasma plume is different. Moreover, higher desolvation gas and solids probe temperatures favored the formation of more oxidation products. At relatively low solids-probe (<160°C) and desolvation-gas (<200°C) temperatures the abundance of oxidation products were minimal (see Figure 3a and b). On the other hand, when higher probe and desolvation gas temperatures were used to promote the transfer of less volatile molecules to the gas phase, the abundance of oxidation products also increased (Figure 3e and f, and Supplementary Figure S-3). To obtain chemical formulas of these oxidation products, accurate masses of the ions were measured using water-cluster ions as reference peaks. For example, accurate mass data obtained for m/z 577 and 591 ions derived from tetracontane (C40H82) confirmed the formulas of these ions to be C40H81O+ and C40H79O +2 , respectively (Supplementary Table 1). Moreover, product ion spectra recorded from m/z 351 (C25H +51 ) and 367 (C25H51O+) ions derived from pentacosane (C25H52) showed that tandem mass spectrometry is an easy method to distinguish peaks for hydrocarbons from those representing oxidation products (Supplementary Figure S-4).
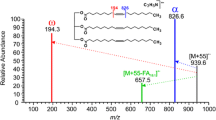
With the use of a heated solids probe, even a peak for the ion representing the hydride-abstracted product of tetratetracontane (C44H90) was obtained (Figure 3f). Such high-mass hydrocarbon ions have not been obtained by APCI or reactive DESI methods. The versatility of the HPIS-MS was further demonstrated by exposing a candle wax sample to the helium plasma. A bouquet of mass spectral peaks representing (M – H)+ ions from C11- to C36-saturated hydrocarbons [and (M + H)+ ions, if any monounsaturated alkenes were present] was obtained by exposing a sample of solid candle wax held with a pair of tweezers to a flow of desolvation gas heated to 300°C (Figure 4). Although the absolute detection limits for saturated hydrocarbons were not determined, the vapor emanating from 0.01 ng of benzophenone was detected at a S/N ratio > 10:1 by the HPIS-Q-TOF MS method (data not shown). The hydrocarbon spectra produced in this way resemble those obtained under field ionization conditions. In fact, the overall HPIS procedure imparts very little internal energy to the ions because hydrocarbons are ionized primarily by secondary chemi-ionization and not by direct Penning ionization. Consequently, the spectra recorded showed only a few, or no, peaks for fragment ions. Nevertheless, with the use of a tandem mass spectrometer, the ions obtained could be mass-isolated and subjected to collision-induced dissociation (CID). Generally, more structural information could be obtained in this way (Supplementary Figure S-5). Of course for some ions this procedure is of little practical value. For example, the CID product ion spectra recorded from protonated (Z)-3-pentacosene and that of (Z)-7-pentacosene were virtually identical (Supplementary Figure S-6).
In an elegant modification of the DART procedure, near-instantaneous analysis of volatile compounds emanating from live insects had been performed [22]. However, our GC-MS analysis of fruit-fly volatiles showed the presence of many saturated hydrocarbons (Figure 5a), none of which had been identified by the DART-MS procedure. In fact, this is an inherent limitation of the DART and APCI procedures for hydrocarbons. Under DART and APCI conditions, atmospheric moisture and ambient gases are ionized with high efficiency. Protonated molecules produced in this way under positive-ion generating APCI conditions, then ionize saturated hydrocarbons by the hydride-removal procedure to produce (M – H)+ ions. In contrast, unsaturated hydrocarbons undergo protonation to form [M + H]+ under similar APCI conditions [21]. Figure 6a and b show respective spectra obtained from (Z)-3-pentacosene, and pentacosane by HPIS-MS. Both compounds produced a peak at m/z 351.4 for the [M + H]+ and [M – H]+ ions of (Z)-3-pentacosene, and pentacosane, respectively. The mass spectrum that was obtained from a 1:1 mixture of (Z)-3-pentacosene and pentacosane, however, showed only one peak (Figure 6c). The ions originating from the two different mechanisms should be isomeric. Because these ions are represented by one molecular formula (C25H +51 ), the composite nature of this peak cannot be demonstrated by high-resolution mass spectrometry. However, we able to demonstrate that the m/z 351 peak obtained in this way represents a mixture of isomeric ions by a gas phase deuterium exchange experiment. A spectrum recorded from (Z)-3-pentacosene while exposing its vapors to D2O during ionization showed a new intense peak at m/z 352.4 for a deuteriated ion (Figure 6a, inset “a”). In contrast, spectra from any saturated hydrocarbon did not show a mass shift when spectra were recorded from samples exposed to D2O vapor (Figure 6b, inset “b” shows a spectrum from pentacosane exposed to D2O). The 1:1 mixture of (Z)-3-pentacosene and pentacosane, which generated only one major peak for the composite mixture of isomeric m/z 351 ions, gave two major peaks when a spectrum was recorded under D2O exchange conditions (Figure 6d). Evidently, gas-phase deuterium exchange procedure provides a method to differentiate ions originating from protonation from those resulting from hydride loss. Thus, our results indicate that the mass spectral peaks assigned as those from monounsaturated hydrocarbons of fruit flies by Yew et al. could well be composites of both saturated and monounsaturated compounds [22] (Figure 5c).

Mass spectral analysis of cuticular hydrocarbons of fruit flies. A chromatogram obtained by GC-MS analysis of an extract (DB-5 coated fused-silica column; 25 m × 0.20 mm; 100°C for 3 min, 5°C/min to 285°C, 15 min isothermal) (a), and the m/z 300–335 range of an HPIS-Q-TOF mass spectrum recorded from an evaporated cuticular hydrocarbon extract introduced by a heated solids probe at ambient conditions (b), and that under D2O vapor (c)

Mass spectra obtained by HPIS-Q-TOF mass spectrometer from (Z)-3-pentacosene (a), pentacosane (b), and a 1:1 mixture of (Z)-3-pentacosene and pentacosane under ambient conditions (c), or exposed to D2O vapor. The insets (a) and (b) show spectra recorded after exposing (Z)-3-pentacosene and pentacosane samples to D2O vapor in the HPIS, respectively
Finally, we would like to add that HPIS consumes much less helium than conventional helium-mediated sources such as DART [1] and FA-APGD [23, 24]. Typical helium flow required for HPIS is about 20–30 mL/min, whereas other helium-mediated sources require about 1–3 L/min of helium. Moreover, in other sources, helium is passed over a sharp needle or a plate to generate the plasma [25–31], and the turbulence generated by the high flow could not only degrade the discharge stability, but also to some extent even hamper analytes interacting well with the plasma. In HPIS, helium is passed at a much lower flow rate through the capillary, and the charge is applied to the capillary. The cone-shaped discharge formed in this way is highly stable. The use of the atmospheric pressure HPIS is relatively simple, and solid samples can be introduced without having to go through a vacuum lock system.
4 Conclusions
A simple modification is described to convert any electrospray ionization source to a helium-plasma ionization source. Samples are directly introduced at atmospheric pressure. Aliphatic hydrocarbon spectra were obtained without any prior sample preparation or purification. In this way, not only hydrocarbons, but practically any compound with an appreciable vapor pressure can be ionized at ambient pressures.
References
Cody, R.B., Laramée, J.A., Durst, H.D.: Versatile new ion source for the analysis of materials in open air under ambient conditions. Anal. Chem. 77, 2297–2302 (2005)
Takats, Z., Wiseman, J.M., Cooks, R.G.: Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 306, 471–473 (2004)
Venter, A., Nefliu, M., Cooks, R.G.: Ambient desorption ionization mass spectrometry. Trends Anal. Chem. 27, 284–290 (2008)
Chan, C.-C., Bolgar, M., Miller, S.A., Attygalle, A.B.: A Combined Desorption-Ionization by Charge Exchange (DICE) and Desorption-Electrospray Ionization (DESI) source for mass spectrometry. J. Am. Soc. Mass Spectrom. 22, 173–178 (2011)
Wu, C., Qian, K., Nefliu, M., Cooks, R.G.: Ambient analysis of saturated hydrocarbons using discharge-induced oxidation in desorption electrospray ionization. J. Am. Soc. Mass Spectrom. 21, 261–267 (2010)
Chan, C.C., Bolgar, M.S., Miller, S.A., Attygalle, A.B.: Desorption Ionization by Charge Exchange (DICE) for sample analysis under ambient conditions by mass spectrometry. J. Am. Soc. Mass Spectrom. 21, 1554–1560 (2010)
Carazzato, D., Bertrand, M.J.: Characterization of a glow discharge ion source for the mass spectrometric analysis of organic compounds. J. Am. Soc. Mass Spectrom. 5, 305–315 (1994)
Mason, R., Milton, D.: Glow discharge mass spectrometry of some organic compounds. Int. J. Mass Spectrom. Ion Processes 91, 209–225 (1989)
McEwen, C.N., McKay, R.G., Larsen, B.S.: Analysis of solids, liquids, and biological tissues using solids probe introduction at atmospheric pressure on commercial LC/MS instruments. Anal. Chem. 77, 7826–7831 (2005)
McEwen, C.N., Gutteridge, S.: Analysis of the inhibition of the ergosterol pathway in fungi using the Atmospheric Solids Analysis Probe (ASAP) method. J. Am. Soc. Mass Spectrom. 18, 1274–1278 (2007).
Zhao, J., Zhu, J., Lubman, D.: Liquid sample injection using an atmospheric pressure direct current glow discharge ionization source. Anal. Chem. 64, 1426–1433 (1992)
Shelley, J.T., Wiley, J.S., Chan, G.C.Y., Schilling, G.D., Ray, S.J., Hieftje, G.M.: Characterization of direct-current atmospheric-pressure discharges useful for ambient desorption/ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 20, 837–844 (2009)
Penning, F.M.: Über ionisation durch metastabile atome. Naturwissenschaften 15, 818 (1927)
Lovelock, J.E.: A sensitive detector for gas chromatography. J. Chromatogr. A 1, 35–46 (1958)
Hsu, H.J., Kuo, T.L., Wu, S.H., Oung, J.N., Shiea, J.: Characterization of synthetic polymers by electrospray-assisted pyrolysis ionization-mass spectrometry. Anal. Chem. 77, 7744–7749 (2005)
Roussis, S.G.: Automated tandem mass spectrometry by orthogonal acceleration TOF data acquisition and simultaneous magnet scanning for the characterization of petroleum mixtures. Anal. Chem. 73, 3611–3623 (2001)
Roussis, S.G., Proulx, R.: The characterization of basic petroleum extracts by high-resolution mass spectrometry and simultaneous orthogonal acceleration time-of-flight-magnet scanning tandem mass spectrometry. Energ Fuels 18, 685–697 (2004)
No, M.H., Kim, E., Lee, J.S., Jung, H.: Hydrocarbon compound type analysis of lube base oil by GC-MSD: Advantages on replacement of the AGHIS magnetic sector type mass spectrometer. Energy Fuels 21, 3341–3345 (2007)
Chan, C.C., Bolgar, M.S., Miller, S.A., Attygalle, A.B.: Evading metal adduct formation during desorption-ionization mass spectrometry. Rapid Commun. Mass Spectrom. 24, 2838–2842 (2010)
Cody, R.B.: Observation of molecular ions and analysis of nonpolar compounds with the direct analysis in real time ion source. Anal. Chem. 81, 1101–1107 (2009)
Harrison, A.G.: Chemical Ionization Mass Spectrometry, 2nd edn. CRC Press, Boca Raton, FL (1992)
Yew, J.Y., Cody, R.B., Kravitz, E.A.: Cuticular hydrocarbon analysis of an awake behaving fly using direct analysis in real-time time-of-flight mass spectrometry. Proc. Natl Acad. Sci. U.S.A. 105, 7135–7140 (2008)
Andrade, F.J., Shelley, J.T., Wetzel, W.C., Webb, M.R., Gamez, G., Ray, S.J., Hieftje, G.M.: Atmospheric pressure chemical ionization source. 1. Ionization of compounds in the gas phase. Anal. Chem. 80, 2646–2653 (2008)
Andrade, F.J., Shelley, J.T., Wetzel, W.C., Webb, M.R., Gamez, G., Ray, S.J., Hieftje, G.M.: Atmospheric pressure chemical ionization source. 2. Desorption-ionization for the direct analysis of solid compounds. Anal. Chem. 80, 2654–2663 (2008)
Faubert, D., Paul, G.J.C., Giroux, J., Bertrand, M.J.: Selective fragmentation and ionization of organic compounds using an energy-tunable rare-gas metastable beam source. Int. J. Mass Spectrom. Ion Processes 124, 69–77 (1993)
Faubert, D., L’Heureux, A., Peraldi, O., Mousselmal, M., Sanchez, G., Bertrand, M.J.: Metastable Atom Bombardment (MAB) ionization source: Design, optimization and analytical performances. In: Advanced Mass Spectrometry, 15th International Mass Spectrometry Conference, pp. 431–432. Wiley, Chichester, UK (2001)
Tsuchiya, M., Kuwabara, H.: Liquid ionization mass spectrometry of nonvolatile organic compounds. Anal. Chem. 56, 14–19 (1984)
Tsuchiya, M.: Liquid ionization mass spectrometry for studies of hydrogen-bonded clusters and of a mixture that contains non-volatile and less polar organic compounds. Mass Spectrom. Rev. 17, 51–69 (1998)
Tsuchiya, M.: Development, principle, and application of liquid ionization mass spectrometry. Anal. Sci. 14, 661–676 (1998)
Hiraoka, K., Fujimaki, S., Kambara, S., Furuya, H., Okazaki, S.: Atmospheric-pressure penning ionization mass spectrometry. Rapid Commun. Mass Spectrom. 18, 2323–2330 (2004)
McLuckey, S.A., Glish, G.L., Asano, K.G., Grant, B.C.: Atmospheric sampling glow discharge ionization source for the determination of trace organic compounds in ambient air. Anal. Chem. 60, 2220–2227 (1988)
Acknowledgment
The authors acknowledge support for this research by funds from Stevens Institute of Technology. They thank David Silverstein, Director, Center for Writing and Communications, Stevens Institute of Technology, for editing the language of this paper.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 3354 kb)
Rights and permissions
About this article
Cite this article
Yang, Z., Attygalle, A.B. Aliphatic Hydrocarbon Spectra by Helium Ionization Mass Spectrometry (HIMS) on a Modified Atmospheric-Pressure Source Designed for Electrospray Ionization. J. Am. Soc. Mass Spectrom. 22, 1395–1402 (2011). https://doi.org/10.1007/s13361-011-0149-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-011-0149-1