
Abstract
Quantitative trait locus (QTL) mapping often yields associations with dissimilar loci/genes as a consequence of diverse factors. One trait for which very limited agreement between mapping studies has been observed is resistance to white mold in soybean. To explore whether different approaches applied to a single data set could lead to more consistent results, haplotype-trait association and epistasis interaction effects were explored as a complement to a more conventional marker-trait analysis. At least 10 genomic regions were significantly associated with Sclerotinia sclerotiorum resistance in soybean, which have not been previously reported. At a significance level of α = 0.05, haplotype-trait association showed that the most prominent signal originated from a haplotype with 4-SNP (single nucleotide polymorphism) on chromosome 17, and single SNP-trait analysis located a nucleotide polymorphism at position rs34387780 on chromosome 3. All of the peak-SNPs (p-value < 0.05) of each chromosome also appeared in their respective haplotypes. Samples with extreme phenotypes were singled-out for association studies, 25–30% from each end of the phenotypic spectrum appeared in the present investigation to be the most appropriate sample size. Some key genes were identified by epistasis interaction analysis. By combining information on the nearest positional genes indicated that most loci have not been previously reported. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses suggest potential candidate genes underlying callose deposition in the cell wall and mitogen-activated protein kinase (MAPK) signaling pathway-plant, as well as plant-pathogen interaction pathway, were activated. Integration of multi-method genome-wide association study (GWAS) revealed novel genomic regions and promising candidate genes in novel regions, which include Glyma.01g048500, Glyma.03g129100, Glyma.17g072200, and the Dishevelled (Dvl) family of proteins on chromosomes 1, 3, 17, and 20, respectively.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
White mold (WM) in soybean is a global soybean disease that emerged in the mid-80 s and spread across countries such as the USA, Canada, Argentina, Turkey, Japan, and India (Hoffnan et al. 1998; Ploper 1999). Particularly in North American and Canadian cropping areas, WM has become one of the major factors affecting soybean production. The disease is caused by infection with Sclerotinia sclerotiorum (Lib.) de Bary. In 1986, the incidence of this pathogen in Canadian legume production reached 25% (Tu 1986). In recent years, WM has continued to spread, causing an increased harm and serious threats to soybean yield (Koenning and Wrather 2010; Peltier et al. 2012). Taken together, the selecting resistant soybean cultivars is the most economical and effective way to control the disease. Although no soybean line that is completely resistant to Sclerotinia has been identified to date, some are known to exhibit partial resistance, a property shown to be a quantitatively inherited trait conferred by multiple genes (Kim et al. 1999; Li et al. 2010; Song et al. 2017). However, the fact that quantitative traits are highly sensitive to environmental factors impedes the accurate selection of target traits under field conditions, thus making it difficult to identify resistant germplasm resources. Therefore, the use of molecular markers to selectively develop disease-resistant cultivars is essential.
Since the first localization of QTL for Sclerotinia resistance of soybean by Kim and Diers within the progeny of a biparental cross in 2000 (Kim and Diers 2000), many additional studies have attempted to map loci conferring Sclerotinia resistance (Arahana et al. 2001; Li et al. 2010; Guo et al. 2008; Hanet al. 2008; Vuong et al. 2008; Huynh et al. 2010). In the last decade, with the development of next-generation sequencing technologies, genome-wide association studies (GWAS) using SNPs have become the most widely used approach to map traits of interest. Subsequent to the progress made by GWAS in human disease research, this approach has been used in the detection of candidate genes for disease resistance.
Currently, the use of molecular markers for selectively cultivating Sclerotinia-resistant soybean has not yet been implemented in practice. Treating the leaves, roots, and stems with oxalic acid and subsequently evaluating the degree of wilting and the level of soluble pigment still represent the main method for selecting soybean cultivars with Sclerotinia resistance (Li et al. 2016). In 2010, Li et al. (2011) used SSR markers to discover QTL conferring soluble pigment (SP) content variations that are related to WM resistance and identified linkage groups D1a + q (Gm01), B1 (Gm11), and A2 (Gm08). In a more recent GWAS for WM resistance-associated SP content, 25,179 SNPs from 330 soybean genotypes allowed the detection of a prominent signal on chromosome 13, followed by signals on Gm06, Gm10, and Gm11; 4 candidate genes related to WM disease response and the biosynthetic pathways of anthocyanin were located in the vicinity (< 60 kb) of significant SNPs (Zhao et al. 2015). In 2014, Bastien et al. (2014) used 7864 SNPs of 130 Canadian soybean genotypes by TASSEL3.0 software to elucidate QTL (in order of significance) on Gm15, Gm01, Gm20, and Gm19. In 2015, Iquira et al. (2015) used 8297 SNPs from 101 samples and located QTL (in order of significance) on Gm03, Gm08, and Gm20. Because of differences in soybean type, sample size, phenotype measurement method, marker type and density, and analytical algorithm method, QTL for WM resistance show large variations across studies.
One weakness of working with individual markers/SNPs is that these are not inherited independently; rather, these have a propensity to be inherited in clusters of physically close loci, thus resulting in linkage disequilibrium (LD). Markers that are in high LD within a particular set of materials form haplotypes. The diversity of haplotypes in a genomic region is not limited to the two alleles typically seen for a single SNP locus. As a consequence, haplotypes may more accurately capture the underlying allelic diversity at loci controlling a trait of interest. When used in association tests, haplotypes may therefore better capture the association to the studied trait (Song et al. 2017).
At the same time, most complex traits are determined by the interactions between multiple genes. This is especially true for the phenotypes controlling disease susceptibility, for which associations based on single loci are usually not sufficient to explain or simulate complex traits. Although these alleles are associated with the disease, most of them merely have small effects when considered individually. Gene–gene interactions are often considered to be the cause of the unexplained genetic variations in complex phenotypes, and these interactions are sometimes overlooked in GWAS (Kanishka et al. 2011).
In addition, a challenge in characterizing a complex disease such as WM is the accurate assessment of the degree of resistance. One possible way to facilitate the identification of reliable QTL is to divide the population into two groups expressing extreme phenotypes and discarding those with intermediate phenotypes (Anderson et al., 2010). The use of extreme samples taken from 10 to 35% on either side of the phenotypic distribution curve has previously been reported to be effective for the purpose of identifying associated QTL (Zhang et al. 2011).
To further investigate the QTL conferring partial resistance to WM in soybean, In this study, we employ 20,691 SNPs to reanalysis a previously published dataset that used 7864 SNPs analysis (Bastien et al. 2014), and performed haplotype-trait association analyses, as well as epistatic interaction analysis using both the full genotype set (n = 126) and only extreme phenotypes (n = 76), which comparing single SNP-trait association tests only by Bastien et al. The detected QTL were compared with the results of other studies, and candidate genes were identified by GO enrichment and KEGG pathway enrichment analyses.
Materials and methods
Soybean lines
A panel of 126 soybean lines originate from the article “ genome wide association mapping of sclerotinia sclerotiorum resistance in soybean with a genotyping-by-sequencing approach” (Bastien et al. 2014), in which representative of the diversity present in a private breeding program (Semences Prograin Inc.) in Eastern Canada (see list in Supplementary Table S1).
Phenotype data
Phenotype data (the stem lesion length) were provided by Bastien(Bastien et al. 2012).
Genotype data
Single-end sequencing was performed on three lanes of an Illumina HiSeq2000 (at the McGill University-Génome Québec Innovation Center in Montreal, QC, Canada). Data quality control is essential to GWAS. In the present investigation, the r2 value for linkage disequilibrium, the LOD, and the confidence interval were set to 0.8, 3, and 95%, respectively. HWE (Hardy–Weinberg equilibrium) was not considered. A total of 20,691 SNPs passed the MAF (minor allele frequency) lower limit of 5%; for association, α = 0.05 was used as the cut-off significance, which corresponded to a non-adjusted p-value of 2.42 × 10−6 (0.05/20,691). The 126 samples provided a total of 2,607,066 loci, among which 213,559 (8.192%) were heterozygous.
Population structure
STRUCTURE 2.3.4 (http://taylor0.biology.ucla.edu/structureHarvesteroybase.org/tools.php) for population structure analysis with the MCMC algorithm and NJ algorithm was used to perform clustering analysis, and phylogenetic reconstruction was conducted using MEGA5.05 (http://www.megasoftware.net/). SPSS was used for principal component analysis (https://www.ibm.com/analytics/us/en/technology/spss/). The Bayesian clustering algorithm implemented in STRUCTURE 2.3.4 was used to simulate population genetic structure. According to the Q value distribution of each population, it is considered that the sample has a single blood relationship when the Q value of a sample in a population is greater than or equal to 0.6, and it is considered that the sample has mixed sources when Q value is less than 0.6. To obtain an estimate of the most probable number of population (K), K values from 1 to 10 were simulated with 20 iterations for each K, using 10,000 burn-in periods followed by 10,000 Markov Chain Monte Carlo iterations. Delta K was plotted against K values and the best number of clusters was determined following the method proposed by Evanno et al. (2005), and population structure diagram was obtained by the Structure Harvester platform (Earl and Vonholdt 2012).
Association analysis
For complete phenotype set, single SNP-trait association tests, with additive effects only and additive effects plus dominant effects of GLM (general linear model) or MLM (mixed linear model) model, were run in TASSEL v.5.0 (http://www.maizegenetics.net/tassel) in max(T) permutation mode using a permutation test with 10,0000 times, and for haplotype analysis using PLINKv1.07 (http://www.softpedia.com/get/Science-CAD/PLINK.shtml), the command line “–blocks” was used to assign SNPs to their respective haplotypes. Then, analysis for haplotype-trait association was performed, and haplotype-trait association analysis only for additive effects.
The frequency distribution of the genotypic segregation in the two groups (high-value group and low-value group, namely extreme phenotypes that were distributed at both ends of 126 phenotypic spectrum, it can be seen as reduced data set, we treat it as a quality character, assuming a disease phenotype as 1 = unaffected, 2 = affected, 0 = miss) will thus deviate from the Mendelian law. Using a chi-square test to assess this deviation for either or both groups, it was then possible to infer whether the marker is linked to the QTL. In the analysis with extreme phenotypes as implemented in HAPLOVIEW4.2 (http://www.broadinstitute.org/haploview), because sample size was reduced without a decrease in the number of markers used, an increase in the number of false positives resulted from higher inflation; p-values were thus adjusted through a 10,000-time permutation test approach. With the 95% CI of D’ value bound between 0.70 and 0.98 for adjacent SNPs to infer haplotype blocks, then carry out haplotype-phenotype association tests of extreme phenotype set.
SNP × SNP epistatic interaction analysis
PLINK v1.07 has different modes for specifying which SNPs are tested. To increase the power of epistasis detection, three analytical methods were used: (1) all SNP-by-all SNP two-locus epistasis test between all of the 205,284,045 SNP pairs; (2) SNP-by-SNP epistasis test between only significant SNPs pre-filtered by genome-wide association analyses; and (3) SNP-by-all epistasis test between significant SNPs and all SNPs. CYTOSCAPE v3.7.1 was used to visualize the interaction network.
Results
LD decay and haplotype construction
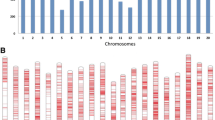
To compare the results of QTL mapping analyses conducted either using individual SNP markers or haplotypes, we first needed to assemble a catalogue of haplotypes for our association panel comprising 126 soybean lines. Genotyping-by-sequencing was used to obtain SNP data for this panel of soybean lines and these were then used to construct haplotypes based on the observed LD. At r2 > 0.2, 84,255 pairs of SNPs were found to be in LD. Among the 84,255 marker pairs in LD, an average r2 of 0.73 was observed; 46,322 pairs (55.0%) had r2 ≥0.8, and 7841 pairs (9.3%) showed an r2 value of 1. The distribution of LD along each chromosome was uneven. The average physical distance between pairs of loci decreased as their LD increased. Pairs with r2 < 0.8 were 154.1 kb apart, pairs with r2 ≥ 0.8 were 139.7 kb apart, and pairs with r2 = 1 were separated by 99.6 kb. The 20 chromosomes yielded a total of 2,858 predicted haplotypes (Fig. 1), with chromosome 18 possessing the most haplotypes (229) and chromosome 11 possessing the least (57). The largest haplotype was composed of 36 SNPs, whereas the smallest haplotype comprised only 2 SNPs, and a haplotype consists of 5 SNPs on average. The average length of the haplotype is 64.4 kb; the longest haplotype spanned 200.0 kb, whereas the shortest encompassed only 2 bp. The distribution of SNPs along each chromosome was uneven, where chromosome 18 contained the largest number of markers (1596), whereas chromosome 11 included the fewest (488).

Distribution of SNP/haplotype: (a) Number of SNP on each chromosome. (b) Number of haplotype on each chromosome
Population structure analysis
Population genetic structure was performed based on the Bayesian method. Delta K reached a maximum value at K = 2, suggesting that the 126 soybean lines were divided into two subgroups (consisting of 58 and 68 samples) (Fig. 2).

Population structure diagram of the 126 soybean lines. Note: Red: group I; Green: group II. Vertical lines on the X-axis refer to each variety. The proportion of each color represents probability rate with which a given genotype belongs to each group
In addition, the contribution rates of the first three PC (principal component) were 6.24%, 5.44%, and 5.16%, respectively, and the 126 lines cannot be clearly grouped by PC method, while the 126 soybean genotypes into two groups using NJ (neighbor joining). However, the genetic distance between these two groups was not significant, indicating that the samples have a low level of genetic stratification.
Genome-wide association study approaches
The results showed that the resistance to Sclerotinia infection among the 126 soybean lines was significantly different, in which varied from 28.6 to 192.4 mm, with the traits of continuous distribution and quantitative inheritance.
Complete phenotype set association analysis
Single SNP-trait association analysis
Six different modes were tested: (1) GLM; (2) GLM (PC); (3) GLM (Q); (4) MLM (K); (5) MLM (PCA: principal component analysis + K); and (6) MLM (Q + K). In this study, the number of principal components is selected according to Q-Q Plot (quantile–quantile plots) (Figure S1), and Q-Q Plot revealed that the model using the first 79 PCs, which altogether captured 85% of the population structure, produced results that correlated most closely with the expected p-values (TableS2, S3). Thus, the first 79 PCs were used to capture population structure in the association analyses. To demonstrate the reliability of our results, significance level was evaluated by Bonferroni correction and permutation tests with 10,0000 permutations. The strongest signal came from the SNP at position rs34387780 on chromosome 3 by GLM, followed by those from markers on chromosomes 20, 1, 4, and 17 (Table 1).
The loci showing the most significant association signals included one SNP located at position rs34387780 on chromosome 3 and five other markers in close vicinity (Fig. 3).

Manhattan plot of the association between single SNP-Trait. Note: P-values for the association of each single nucleotide polymorphism with phenotype are shown on the y-axis. The SNPs are plotted on the x-axis according to their chromosomal location. Blue line: α = 0.05; red line: α = 0.01 (additive effects only)
Haplotype-trait association analysis
For haplotype-trait association analysis, one signal from chromosome 17 represented the most significant association and accounted for 17.56% of the phenotypic variations. Less significant haplotypes were also found on chromosome 1 (Table 2).
Extreme phenotypes set association analysis
Single SNP-trait association analysis
The present investigation attempted using extreme phenotype groups for association analysis. We utilized 10%, 20%, and 30% of the 126 soybean lines as cut-off thresholds for defining membership to the two extreme sample groups. Results from association analyses showed that all significant signals detected using both the 10%- and 20%-extreme groups were represented in those identified using the 30%-extreme groups; hence, only the latter was used for further analyses. The strongest association was found on chromosome 1, followed by those on chromosomes 20, 10, 17, 13, and 4 (Table S4).
Haplotype-trait association analysis
The strongest association was identified for a haplotype formed by 12 SNPs on chromosome 1; haplotypes on chromosomes 13, 4, 10, 20, and 17 were also detected as having significant association to soybean lesion length (Table S5).
The analyses using extreme phenotype groups (both single SNP-trait and haplotype-trait) revealed that the peak-SNPs in the single SNP-trait analysis also appeared in their respective haplotypes. Because the method using extreme groups suffered from a diminished number of soybean genotypes, the analysis using haplotypes had proven to be advantageous by reducing the degree of freedom; hence, the adjusted p-value threshold for the haplotype-based analysis was lowered to α = 0.01 for subsequent candidate gene selection.
The results of QTL mapping for S. sclerotiorum resistance in soybean using the abovementioned methods are shown in Fig. 4.

Prioritizing candidate genes. Note: Triangle represents the position of haplotype, red font indicates QTL is within a gene, and green font indicates that the QTL is outside a gene
SNP × SNP epistatic interaction analysis
With methods (2) and (3), the use of different p-value thresholds for declaring significant association and for defining significant SNP groups yielded different results. With a single-SNP cut-off p-value of 1 × 10−4, neither method (2) nor method (3) produced significant epistatic interactions. Thus, we used the ALL × ALL epistasis mode by PLINK v1.07 and performed a total of 205,284,045 valid SNP × SNP tests. A total of 220 SNPs were involved in the interaction network (cutoff p-value < 0.07) (Table S5), with 112 SNPs in the first three subnetworks. Multiple interactions were found between Gm01 and Gm15 and within Gm06 (Fig. 5). In the first sub-network, there were 9, 17, and 24 proteins interacting with SNP Chr1-rs54719664, Chr1-rs54799844, and Chr1-rs4719984 based on shortest path search, respectively, of which high-node genes in the network may regulate other genes or be regulated by other genes. Among these, Chr1-rs54719664 is closely linked to the Glma.01g216800 and Glma.01g216900 gene, which were annotated as cytochrome P450, family 87, subfamily A, and polypeptide 2. Chr1-rs54799844, a SNP locus within the Glma.01g217900 gene, was annotated as transcription initiation factor TFIIE, beta subunit. Chr1-rs4719984 is closely linked to Glma.01g043300 and Glma.01g043400 genes, which were annotated as WRKY DNA-binding protein 72 and cystathionine beta-synthase (CBS) protein, respectively.

The top three epistasis interaction subnetworks
For further analysis, the asymptotic p-values produced by the all SNP-by-all SNP interaction analysis that were smaller than 1 × 10−8 were considered (Table 3).
Gene assignment and functional annotations
We integrated the results of the commonly detected loci/regions, including genes by epistasis interaction analysis (P < 10−7) and the candidate genes potentially involved in the defense mechanism of soybean to Sclerotinia infection (Table 4).
These can be classified into genes encoding signal transduction molecules, transcriptional regulators, disease resistance proteins, and proteins of unknown functions. These positional candidate genes were further prioritized using GO enrichment analysis by the SoyBase Analysis Tools (http://soybase.org/tools.php) (Fig. 6) and KEGG web platform (https://www.genome.jp/kegg/tool/map_pathway2.html) (Fig. 7), including 241 genes in GO annotation, which resulted in different significantly enriched GO terms and KEGG pathways relevant to S. sclerotiorum-related mechanisms. Among the enriched GO terms and pathways were callose deposition in cell wall-related such as “GO:0,052,543”; signal transduction-related such as “GO:0,009,737 ~ response to abscisic acid stimulus” and “GO:0,005,245 ~ voltage-gated calcium channel activity”; biosynthetic process-related such as “GO:0,006,537 ~ glutamate synthase” and “GO:0,009,833 ~ Cellulose synthase family protein”; and response to stress-

The results of GO functional enrichment analysis

The results of KEGG pathways enrichment analysis
related such as GO:0,004,735 ~ pyrroline-5-carboxylate reductase.”
Discussion
The phenotypic data identification is very important in genome-wide association studies, because the determination of the phenotypic data of 126 soybean lines used in this study had previously been performed in a greenhouse to ensure equal selective pressure, environmental variability that affects phenotypic variation can be ignored, and that ensure the reliability of phenotypic value, those disease resistance lines such as Karlo RR, PRO 275, and Toma, PS73, Nattosan, and Supra as susceptible ones; however, Williams 82 was in the middle phenotypic value on stem lesion lengths in response to Sclerotinia infection.
Quantitative traits are controlled by many genes that participate in multiple signaling pathways. QTL analysis based on genome-wide essentially consists of a statistical measure that is based on probabilistic criteria and identifies a set of genomic segments that may influence a given quantitative trait. In theory, the reliability of the analysis is thus positively correlated with both sample size and the number of polymorphic loci. The associated regions revealed herein are more abundant and different from those identified in previous investigations. Although we analyzed the same soybean samples used by Bastien et al. (2014), differences in analytical method and marker density have marked repercussions on the outcome of GWAS. In terms of marker density, the mean inter-SNP distance was 43.3 kb in the present study, whereas Bastien et al. reported 238.4 kb. Hence, this 5.5-fold increase in marker density leads to the discovery of association loci/regions that have not been previously identified by Bastien et al. Among the 4 QTL detected by Bastien et al. [Gm15:13,651,235 (V1.0)–13,666,875 (V2.0), Gm01:29,185,984 (V1.0)–27,657,519 (V2.0), Gm20:39,698,515 (V1.0)–40,820,870 (V2.0), and Gm19:50,557,054 (V1.0) – 50,677,474 (V2.0)], only the SNPs on chromosomes 15 and 1 appeared in the QTL mapping results of this study, but these showed low association to WM (Gm15: Punadjusted-value = 0.06; Gm1: Punadjusted-value = 0.5189). The other two SNPs (on chromosomes 20 and 19), similar to the four QTL identified by Iquira et al. (2015), were not included in the results of this study. The present study used stem lesion length after inoculation with S. sclerotiorum as phenotype, whereas Li et al. (2016) and Zhao et al. (2015) based their association analysis on soluble pigment content in soybean stems because sample type, sample size, method of obtaining phenotype, marker density, and analytical method were dissimilar, and different QTL maps were also generated.
Furthermore, previous studies have only analyzed the association between single-locus and trait, whereas the present study also investigated the genome-wide distribution of haplotypes in soybean and their association with lesion length due to Sclerotinia infection. All of the peak-SNPs appeared in the corresponding haplotypes; the 6 SNPs from positions rs34387780 to rs34395745 formed a haplotype on chromosome 3. Because some of these SNPs are heterozygous in some materials, we observed slight differences in the p-values obtained using single-SNP and the corresponding haplotype. Some studies were conducted for genomic regions spanning 100–300 kb around single association loci (Zhao et al. 2015), effectively introducing the concept of haplotypes. However, the association of haplotype-trait may increase the detection rate of true disease-related QTL because haplotypes encompass the genetic information of multiple SNPs. It can be seen from the genes identified through the haplotype-based association that the disease-related genes tend to form a gene cluster, which acts synergistically in disease response. In addition, the analysis of the haplotype-phenotype association improves our understanding of this disease response pathway, in which haplotypes are likely to work as functional units. In addition, by being able to reduce the degree of freedom, haplotypes yielded better statistical and analytical effects than single-SNPs.
The strongest epistasis interaction was observed between the vacuolar-type H ( +)-ATPase C (V-H + -ATPase C) gene on chromosome 6 and the S-adenosyl-L-methionine-dependent methyltransferases superfamily protein (SAM-Mtases) gene on chromosome 18. The V-H + -ATPase C subunit is the most sensitive subunit in the plant’s response to stress. During periods of stress, the number of C subunits and their mRNA levels drastically and rapidly increase (Xu et al. 2011). The S-adenosyl-L-methionine–dependent methyltransferases superfamily protein (SAM-Mtases) gene has direct effects on the synthesis of many secondary metabolites, such as lignin and flavonoids, and it plays an important role in plant physiological processes involved in hormonal growth and insecticidal, antibacterial, and disease resistance behaviors. Furthermore, arginine decarboxylase, on chromosome 6, is a key enzyme in polyamine biosynthesis. The epistatic interaction between the two genes on chromosomes 6 and 18, coupled with the polyamine oxidase gene identified on chromosome 13, suggests that the polyamine biosynthetic pathway participates in the resistance of soybean to Sclerotinia infection (Scandiani et al. 2015).
Molybdenum is one of the essential trace elements in plants; its physiological functions of molybdenum in plants are mainly achieved through molybdenum-containing enzymes. Studies in wheat indicate that molybdenum is associated with abiotic stress and activity of resistant enzymes (Al-Issawia et al. 2016). The interaction analysis on chromosomes 7 and 8 suggests that molybdenum cofactors, which can be combined with many molybdenum enzymes that induce physiological and anti-stress functions, play a role in the infection of soybean by Sclerotinia through Ca2+-signal transduction.
Cytochrome P450 is involved in plant responses to abiotic and biological stressors (Jarsch and Ott 2011; An et al. 2017). Remorin is a family of N-terminal proline-rich membrane proteins; in particular, the proline content of group 1b remorin protein in dicotyledonous plants is twice that of group 1a remorin in other organisms (Raffaele et al. 2007). Additionally, the identification of pyrroline-5-carboxylate reductase (P5CR), the enzyme that catalyzes the final step of proline synthesis through single-SNP association analysis, is also corroborated by the fact that remorin is a proline-rich protein. The interaction between plant remorin protein and bacteria has already been investigated (Lefebvre et al. 2010; Tóth et al. 2012). Using maize as a model, Jamann et al. (2016) were the first to confirm that an interaction exists between remorin and fungus. Previous studies have shown that members of the copine family may play a role in Ca2 + signaling; thus, given the interaction detected between the genes on chromosomes 1 and 15, these genes may be implicated in the response of soybean to Sclerotinia infection.
Therefore, it is very important to perform interaction analysis between SNP pairs. With epistatic interaction analysis, some genes that respond to plant abiotic stress and participate in anti-stress defense responses were uncovered. A relatively large body of evidence supports the involvement of P450 expression in host–pathogen interactions (Motallebi et al. 2015). None of the identified single-SNP association loci appeared in loci that were detected by epistatic interaction analysis; this is probably because only two-locus epistasis test models of association were used, whereas the complex phenotype of Sclerotinia resistance results from the interaction of multiple loci.
Enrichment analysis indicated that calcium-binding protein, leucine-rich repeat receptor-like protein kinase, protein kinase superfamily protein, small GTP-binding protein, transcription factors (WRKY, zinc finger protein, heat shock transcription factor), and glycosyltransferases (levansucrase and GDP-fucose protein O-fucosyltransferase) are involved in plant signal transduction. Cellulase synthase, glutamate synthase, P5CR, lycopene β-cyclase, polyamine oxidase, SAM-Mtases, glucose methylcholine oxidoreductase family protein, glutathione-S-transferase, cytochrome P450, NBS-LRR disease resistance protein, and Argonaute family protein probably participate in the disease resistance mechanisms of soybean to Sclerotinia infection. Among these, calcium-binding protein, SAM-Mtases, cytochrome P450, WRKY, glutathione-S-transferase, leucine-rich repeat receptor-like protein kinase, glycosyltransferase, and protein kinase superfamily protein were detected multiple times in the association analysis. The Ca2 + signaling pathway is a very important transduction pathway that plays an important regulatory role in soybean’s resistance to S. sclerotiorum (Zhou et al. 2013); this is corroborated by the repeated detection of CaM and CDPK. The effect of SAM-Mtases on disease resistance may be mediated by the synthesis of plant lignin and their participation in the regulation of flavonoid metabolism; the SAM-Mtases gene is hence expected to be one of the important candidate genes for plant disease resistance. P450s have been isolated from host–pathogen interactions, some of which are as toxins and some as stress signals that activate anti-disease responses. WRKY transcription factors form a large family of plant transcription factors. Studies have shown that WRKY proteins are a key component of plant stress response. In plants, the WRKY transcription factor is most abundant in soybean. WRKY belongs to a broad class of transcriptional regulators, plays an important role in a variety of signal transduction pathways, and has been reported in various studies on plant responses to adverse conditions (Wen et al. 2017). The expression of WRKY6, WRKY8, and WRKY11 was significantly upregulated in Arabidopsis after inoculation with S. sclerotiorum; the WRKY gene may thus achieve its anti-Sclerotinia control function by activating multiple pathways (Zhao 2012.). The role of the Ca2+ signaling pathway and the WRKY11 gene in the plant’s anti-Sclerotinia property has been demonstrated (Wang 2015). Glutathione S-transferase (GST) on chromosomes 17 and 1 may be extensively involved in the lipid peroxide decomposition during oxidative-stress caused by soybean’s response to Sclerotinia infection (Ferreira et al. 2010). Through modification of plant hormones and hypersensitivity responses, glycosyltransferase can antagonize essential pathogenic factors to maximize disease resistance (Meng et al. 2013). Consequently, the three glycosyltransferases on chromosome 1, galactosyltransferase, levansucrase, and GDP-fucose protein O-fucosyltransferase may affect the resistance of soybean to Sclerotinia by various glycosylation reactions.
Plants do not have a strong immune system; thus, their own defense mechanism is hence particularly important, with the cell wall is the first barrier of plant against pathogen infection Studies have confirmed that epidermal and cortical cells of resistant varieties make use of cytoplasmic disintegration and enhanced cell walls to delay pathogen impregnation and to increase pathogenic resistance (Valera 2014); this is consistent with the results of GO analysis in this study. During pathogenesis, Sclerotinia induces cellulose fiber degradation to soluble disaccharide through secretion of cellolase, and then degradation of cellobiose to glucose by β-glucosidase. Experiments show that the pathogenicity of the S. sclerotiorum strain in rape is positively correlated with its cellulase activity (Mao et al. 2011). At least 8 of the genes identified on chromosome 3 are associated with disease resistance such as Glyma.03g128300 ~ glutamate synthase, Glyma.03g128900 ~ lycopene β-cyclase, and Glyma.03g129100 ~ pyrroline-5-carboxylate reductase, which are associated to disease resistance and resistance to oxidative stress (Antoniou et al. 2017; Kim et al. 2012; Khedr et al. 2003). We have verified that polymorphisms in the P5CR gene of Brassica napus are associated with resistance to S. sclerotiorum (Zhou et al. 2020).
It is well known that the R gene is the first class of disease resistance-conferring genes isolated from plants and that the leucine-rich repeat is the conserved domain of the R gene. The effects of Ras small GTPase signaling in plant disease resistance have been reported. In addition, the Dvl protein is a key protein of the classical Wnt/β-catenin signaling pathway. Not only does it mediate Wnt signal transduction from the cell membrane to intracellular domain, but also it participates in the downstream intra-nuclear formation of transcription complexes. The β-catenin signal constitutes part of an important signaling pathway that is related to the interaction between animals and diseases. It also upregulates the expression of GDP-fucose protein O-fucosyltransferase (FUT) (He et al. 2017; Wang et al. 2013). In this study, whether the identified Dvl and FUT proteins are involved in the response of soybean to Sclerotinia requires further confirmation because the association of Wnt signaling pathways to plant responses to pathogens has not been reported to date.
Conclusion
It is necessary to perform haplotype association, epistatic interactive analysis, and post-GWAS to better understand the mechanism of induced disease resistance in plants. There is also a need to confirm the results using multiple models and to select QTL that have been repeatedly identified. Analysis of genes linked to the obtained association loci suggests that signals for plant anti-stress and anti-disease processes exhibit a high degree of relatedness and influence soybean’s response to Sclerotinia infection by a complex mechanism. The present comparative analysis was based on the use of a higher quantity of markers and a variety of analytical methods. S. sclerotiorum resistance is influenced by many genes that are involved in multiple processes, including the response to candidate genes participating in the signaling pathway of soybean’s response to S. sclerotiorum infection, and conferring resistance to WM was discovered. A total of 10 genomic regions in 7 chromosomes were detected, of which 5 tagSNPs were identified these were the peak SNPs at positions rs5589867, rs34387780, rs5734897, rs42091969, and rs37847000 on chromosomes 1, 3, 17, 20, and 13, respectively. Because the resistance of soybean to Sclerotinia belongs to quantitative trait inheritance, and thus meta-analysis may be appropriate to provide a deeper and more integrated knowledge of QTL and soybean signal transduction during WM infection.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author.
References
Al-Issawia M, Rihanb HZ, Al-Shmganic H, Fullerd MP (2016) Molybdenum application enhances antioxidant enzyme activity and COR15a protein expression under cold stress in wheat. Journal of Plant Interactions 11:5–10
An JP, Li R, Qu FJ, You CX, Wang XF, Hao YJ (2017) Ectopic expression of an apple cytochrome P450 gene MdCYPM1 negatively regulates plant photomorphogenesis and stress response in Arabidopsis. Biochem Biophys Res Commun 483:1–9
Anderson CA, Pettersson FH, Clarke GM, Cardon LR, Morris AP, Zondervan KT (2010) Data quality control in genetic case-control association studies. Nat Protoc 5:1564–1573
Antoniou C, Chatzimichail G, Xenofontos R et al (2017) Melatonin systemically ameliorates drought stress-induced damage in Medicago sativa plants by modulating nitro-oxidative homeostasis and proline metabolism. J Pineal Res 62:27
Arahana VS, Graef GL, Specht JE, Eskridge SJR, KM, (2001) Identification of QTLs for resistance to Sclerotinia sclerotiorum in soybean. Crop Sci 41:180–188
Bastien M, Sonah H, Belzile F (2014) Genome wide association mapping of Sclerotinia sclerotiorum resistance in soybean with a genotyping-bysequencing approach. The Plant Genome 7:1–13
Bastien M, Huynh TT, Giroux G et al (2012) A reproducible assay for measuring partial resistance to Sclerotinia sclerotiorum in soybean. Can J Plant Sci 92:279–288
Earl DA, Vonholdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Ferreira LC, Cataneo AC, Remaeh LMR et al (2010) Nitric oxide reduces oxidative stress generated by lactofen in soybean plants. Pestic Biochem Physiol 97:47–54
Guo XM, Wang DC, Gordon SG, Helliwell E, Smith T, Berry SA (2008) Genetic mapping of QTLs underlying partial resistance to Sclerotinia sclerotiorum in soybean PI 391589A and PI 391589B. Crop Sci 48:1129–1139
Han F, Katt M, Schuh W, Webb DM (2008) QTL controlling Sclerotinia stem rot resistance in soybean. US Patent 7250:552
He X, Zhang W, Yan C et al (2017) Chemical biology reveals CARF as a positive regulator of canonical Wnt signaling by promoting TCF/β-catenin transcriptional activity. Cell Discov 3:17003
Hoffnan DD, Hartman GL, Mueller DS (1998) Yield and seed quality of soybean cultivars infected with Sclerotinia sclertiorum. Plant Dis 82:826–829
Huynh TT, Bastien M, Iquira E, Turcotte P, Belzile F (2010) Identification of QTLs associated with partial resistance to white mold in soybean using field-based inoculation. Crop Sci 50:969–979
Iquira E, Humira S, François B (2015) Association mapping of QTLs for Sclerotinia stem rot resistance in a collection of soybean plant introductions using a genotyping by sequencing (GBS) approach. BMC Plant Biol 15:5
Jamann T, Luo X, Morales L, Kolkman J, Chung C, Nelson R (2016) A remorin gene is implicated in quantitative disease resistance in maize. Theor Appl Genet 129:591–602
Jarsch IK, Ott T (2011) Perspectives on remorin proteins, membrane rafts, and their role during plant–microbe interactions. Mol Plant Microbe Interact 24:7–12
Kanishka B, McCarthy MI, Morris AP (2011) Rapid testing of gene-gene interactions in genome-wide association studies of binary and quantitative phenotypes. Genet Epidemiol 35:800–808
Khedr AHA, Abbas MA, Wahid AAA, Quick WP, Abogadallah GM (2003) Proline induces the expression of salt-stress-responsive proteins and may improve the adaptation of Pancratium maritimum L. to salt-stress. J Exp Bot 54:2553–2562
Kim HS, Diers SCH, BW, (1999) Evaluation of soybean cultivars for resistance to Sclerotinia stem rot in field environments. Crop Sci 39:64–68
Kim HS, Diers BW (2000) Inheritance of partial resistance to Sclerotinia stem rot in soybean. Crop Sci 40:55–61
Kim SH, Ahn YO, Ahn MJ, Lee HS, Kwak SS (2012) Down-regulation of β-carotene hydroxylase increases β-carotene and total carotenoids enhancing salt stress tolerance in transgenic cultured cells of sweet potato. Phytochemistry 74:69–78
Koenning SR, Wrather JA (2010) Suppression of soybean yield potential in the continental United States by plant diseases from 2006 to 2009. Plant Health Progress 11(1)
Lefebvre B, Timmers T, Mbengue M, Moreau S, Hervé C (2010) A remorin protein interacts with symbiotic receptors and regulates bacterial infection. Proc Natl Acad Sci 107:2343–2348
Li D, Sun M, Han Y, Teng W, Li W (2010) Identification of QTL underlying soluble pigment content in soybean stems related to resistance to soybean white mold (Sclerotinia sclerotiorum). Euphytica 172:49–57
Li YH, Smulders MJM, Chang RZ, Qiu LJ (2011) Genetic diversity and association mapping in a collection of selected Chinese soybean accessions based on SSR marker analysis. Conserv Genet 12:1145–1157
Li Z, Wang S, Meng F, Liu J, Yi Z, Zhang H et al (2016) Soluble pigment level in stems determining partly-resistance to white mold in soybean. Soybean Science 35:481–488
Mao W, Hou YM, Liu ZW (2011) Analysis of several enzymes activity induced by Sclerotinia sclerotiorum and oxalic acid in rapeseed. Journal of Dalian Polytechnic University 30:39–42
Meng Q, Yin H, Hu J, Du Y (2013) Research progress of plant glycosyltransferase in plant resistance on disease. Letters in Biotechnology 24:290–293
Motallebi P, Niknam V, Ebrahimzadeh H et al (2015) Methyl jasmonate strengthens wheat plants against root and crown rot pathogen Fusarium culmorum infection. J Plant Growth Regul 34:624–636
Peltier AJ, Bradley CA, Chilvers MI, Malvick DK, Mueller DS, Wise KA et al (2012) Yield loss and control of Sclerotinia stem rot of soybean. Journal of Integrated Pest Management 3:1–7
Ploper LD (1999) Management of economically important diseases on soybean in Argentina. In: Proceedings of the World Soybean Research Conference VI, Chicago, Illinois, USA, pp 269–280
Raffaele S, Mongrand S, Gamas P, Niebe lA, Ott T, (2007) Genome-wide annotation of remorins, a plant-specific protein family: evolutionary and functional perspectives. Plant Physiol 145:593–600
Scandiani MM, Luque AG, Razori MV et al (2015) Metabolic profiles of soybean roots during early stages of Fusarium tucumaniae infection. J Exp Bot 66:391–402
Song W, Zhao X, LX Xu et al (2017) Identification of QTL and consistency QTL with resistance to Sclerotinia sclerotiorum in soybean. Chinese Journal of Oil Crop Sciences 39(6):763–770
Tóth K, Stratil TF, Madsen EB et al (2012) Functional domain analysis of the remorin protein LjSYMREM1 in Lotus japonicus. PLoS One 7:e30817
Tu JC (1986) Integrated disease control of white mould (Sclerotinia sclerotiorum) in navy bean (Phaseolus vulgaris). International Symposium on Crop Protection 39:731–740
Valera RE (2014) Physiological, anatomical and molecular characterization of partial resistance against Sclerotinia sclerotiorum in soybean. The University of Guelph, Guelph, Ontario, Canada, pp 1–171
Vuong TD, Diers BW, Hartman GL (2008) Identification of QTL for resistance to Sclerotinia stem rot in soybean plant introduction 194639. Crop Sci 48:2209–2214
Wang JP (2015) Molecular mechanisms underlying effect of mycelial age on pathogenicity of Sclerotinia sclerotiorum and plant resistance to this fungus. Dissertation, Zhejiang University
Wang S, Yin J, Chen D et al (2013) Small-molecule modulation of Wnt signaling via modulating the Axin-LRP5/6 interaction. Nat Chem Biol 9:579–585
Wen F, Wu X, Liao L, Liu X, Li P (2017) Research advances on physiological function of WRKY transcription factor in plant stress resistance. Guihaia 37:69–79
Xu C, Zheng L, Gao C, Wang C, Liu G, Jiang J et al (2011) Ovexpression of a vacuolar H+-ATPase c subunit gene mediates physiological changes leading to enhanced salt tolerance in transgenic tobacco. Plant Mol Biol Report 29:424–430
Zhang Q, Yao GX, Hu GL, Tang B, Chen C, Li ZC (2011) Identification of QTLs for grain traits in rice using extreme materials in grain size. Acta Agron Sin 37:784–792
Zhao L (2012) Functional Analysis of Arabidopsis WRKY and AG01 genes in regulation of resistanceto Sclerotinia sclerotiorum. Dissertation, Zhejiang University
Zhao X, Han Y, Li Y, Liu D, Sun M, Zhao Y (2015) Loci and candidate gene identification for resistance to Sclerotinia sclerotiorum in soybean (Glycine max L. Merr.) via association and linkage maps. Plant J 82:245–255
Zhou J, Sun A, Xing D (2013) Modulation of cellular redox status by thiamine-activated NADPH oxidase confers Arabidopsis resistance to Sclerotinia sclerotiorum. J Exp Bot 64:3261–3272
Zhou WY, Zhang XJ, Sun XM, Ma Z, Zhang Y (2020) Study of clone and polymorphism of P5CR in B. napus. Genomics and Applied Biology 39:5678–5683
Acknowledgements
The authors would like to thank François Belzile, Maxime Bastien, and Martine Jean. We thank dataset of the SoyBase (https://soybase.org/) and thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This work was funded by the Sci-technological Project of Shaanxi Province (2019NY-041) and the Department of Education Project of Shaanxi Province (18JK0170).
Author information
Authors and Affiliations
Contributions
Yu Zhang performed the association analysis and wrote the manuscript. Yuexing Wang and Wanying Zhou participated in the development of the GBS genotyping protocol in soybean. Wanying Zhou, Shimao Zheng, and Runzhou Ye analyzed some of the data and performed read sequence analysis, SNP calling, and imputation to generate the SNP dataset. Runzhou Ye revised the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Communicated by: Izabela Pawłowicz
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Wang, Y., Zhou, W. et al. Detection of candidate gene networks involved in resistance to Sclerotinia sclerotiorum in soybean. J Appl Genetics 63, 1–14 (2022). https://doi.org/10.1007/s13353-021-00654-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-021-00654-z