
Abstract
Statins, widely prescribed for cardiovascular diseases, are also being eyed for management of age-related macular degeneration (AMD). Poor bioavailability and blood-aqueous barrier may however limit significant ocular concentration of statins following oral administration. We for the first time propose and investigate local application of atorvastatin (ATS; representative statin) loaded into solid lipid nanoparticles (SLNs), as self-administrable eye drops. Insolubility, instability, and high molecular weight > 500 of ATS, and ensuring that SLNs reach posterior eye were the challenges to be met. ATS-SLNs, developed (2339/DEL/2014) using suitable components, quality-by-design (QBD) approach, and scalable hot high-pressure homogenization, were characterized and evaluated comprehensively for ocular suitability. ATS-SLNs were 8 and 12 times more bioavailable (AUC) in aqueous and vitreous humor, respectively, than free ATS. Three-tier (in vitro, ex vivo, and in vivo) ocular safety, higher corneal flux (2.5-fold), and improved stability (13.62 times) including photostability of ATS on incorporation in ATS-SLNs were established. Autoclavability and aqueous nature are the other highlights of ATS-SLNs. Presence of intact fluorescein-labeled SLNs (F-SLNs) in internal eye tissues post–in vivo application as eye drops provides direct evidence of successful delivery. Perinuclear fluorescence in ARPE-19 cells confirms the effective uptake of F-SLNs. Prolonged residence, up to 7 h, was attributed to the mucus-penetrating nature of ATS-SLNs.

Graphical abstract
Similar content being viewed by others
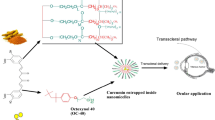

Introduction
Age-related macular degeneration (AMD) is a progressive degenerative macular disease attacking the region of highest visual acuity, the macula. It becomes a leading cause of irreversible blindness in people over 50 years of age especially those living in industrialized countries [1, 2]. More than 30 million people across the globe have the condition, and the incidence of AMD is expected to be on the rise over the next 40 years [3]. Recent and widely used therapy for AMD involves intravitreal injection of anti-vascular endothelial growth factor (VEGF) antibodies, viz. bevacizumab (Avastin), ranibizumab (Lucentis), or anti-VEGF receptor—aflibercept (Eylea) and siRNA [4,5,6]. Intravitreal injections at times may result in retinal detachment, hemorrhage, high intraocular pressure, and endophthalmitis [7]. AMD is a chronic ocular disease and thus requires long-term, repetitive dosing, sometimes extending through the entire lifetime of the patient. General frequency of administration for intravitreal injection of anti-VEGF antibodies is 4 weeks. Since the therapy is invasive and painful and requires hospitalization along with a companion, hence, it results in a significant reduction in quality of life. Moreover, injection administration by an expert will add to the complexity and cost of the therapy. All these factors contribute to a reduced patient compliance [8]. Working out a non-invasive treatment option using a suitable agent, as topical drops, is the future endeavor of scientific community for management of AMD. However, it is very difficult to achieve therapeutic drug levels in the vitreous, retina, or choroid following topical application of drugs/drug solution [8]. This is because of the unique anatomical and physiological constraints to drug delivery existing within the eye. The novel drug delivery systems like nanosized particles are now reported to reach the internal sections of the eye [9, 10] following topical application in the form of eye drops via a variety of mechanisms and uptake pathways.
Several epidemiological and clinical studies indicate that cardiovascular disease (CVD) and AMD have similar pathology because of their shared risk factors (including analogous aging changes in vascular intima and Bruch’s membrane (BM) [11], smoking, hypertension, and serum lipid status) and genetic predisposition [12]. BM lining lying below the retinal pigment epithelium (RPE) forms the inner margin of the choriocapillaris and is considered the structural analog of the vascular intima of arteries [13]. Furthermore, protein molecular composition of drusen of the eye and arteriosclerotic plug and their modification, oxidation, and aggregation leading to CVD and/or AMD respectively are also similar [2]. Based on these observations and the pleiotropic nature of statins, statin intervention highly supported for CVD is also being assessed for application in AMD [14]. Statins possess actions far wider than their presently claimed and widely exploited cholesterol-lowering properties for which they are often prescribed [15] and are now established to show anti-inflammatory [2], anti-oxidative [16], anti-tumor [17], and immunomodulatory [18] effects. Hall et al. showed a markedly lower rate of exudative AMD among a cohort of patients on HMG Co-A reductase inhibitor (“statin”) [19]. However, subsequent clinical studies show inconsistent results on the benefit of statin therapy in AMD [20,21,22]. Reasons assigned for these conflicting reports include small sample size [23], non-uniform dose among the study subjects [23, 24], and studies combining wet or dry AMD cases together. Further, it is indicated that the dose of statin is directly related to its efficiency [25]. The PROVE-IT study [26] and REVERSAL and ASTEROID trials showed benefit of aggressive over moderate-intensity dosage therapy of statins [27, 28].
Considering the purported beneficial role of statins in control of AMD and importance of high dose concentration, we propose that topical application of statins directly to the eye as drops, will achieve high local concentration resulting in a significant and consistent therapeutic effect.
ATS is among one of the first statins to be introduced and has the advantage of a long and safe clinical experience. However, the large molecular mass exceeding 500 Da, poor solubility, and high first pass metabolism result in a poor oral bioavailability of only 12% [29, 30]. Existence of blood-aqueous humor or blood-retinal barrier will further restrict its availability in the ocular tissues. Insolubility (0.00495 mg/ml) and photodegradation of ATS are also serious challenges to permeability and development of ATS as ocular drops [31]. Generally single or a mixture of surfactants is required for solubilization of any insoluble entity and for its encapsulation into nanocarriers. ATS is prone to degradation, including oxidation, in surfactant solutions such as polysorbate 80, the most widely employed and effective surfactant, which also makes nanoformulation development difficult. The oxidation so caused is not reversed even upon use of anti-oxidants [32]. Hence, surfactant and solvents need to be selected carefully for formulating ATS as a solution or nanodispersion.
Presently we employed a suitably designed solid lipid nanoparticle (SLN) system which was optimized statistically using Design Expert® 8.0.1 (Stat-Ease Inc.) for important formulation parameters, viz. particle size and entrapment efficiency, after a careful selection of components. Following optimization, the final formulation was characterized extensively for (i) particle size, zeta potential, size distribution, and morphology; (ii) nature of formulated SLNs using DSC, FTIR, and XRD; and (iii) stability including photostability upon storage. The system was also evaluated for parameters, viz. pH, refractive index, osmolarity, in vivo safety, and sterility, which are important considerations for an ophthalmic formulation. Ex vivo corneal permeability, ocular residence, and in vivo pharmacokinetic profile of ATS-SLNs were compared with the corresponding free drug dispersion. Fluorescein-labeled SLN system was used to confirm the capacity of SLNs to target posterior eye tissue involved in the AMD pathology. Cell line studies were used (i) to confirm the safety of formulation to corneal and retinal cell lines; and (ii) for uptake and (iii) apoptosis studies. In vivo safety was established in rabbits using acute and repeat dose toxicity studies as per Organisation for Economic Co-operation and Development (OECD) guidelines.
To conclude, we conceive the proposed application of statins as eye drops for control of AMD, picking up ATS as a model statin. We used nanotechnology to develop an ophthalmologically compatible system with capacity to deliver up to the macula. A proof of concept is provided for the same.
Methods
Materials
Atorvastatin calcium and Poloxamer 188 (P 188) were a kind gift from Ranbaxy Labs. Ltd., Gurgaon; Compritol® 888 ATO was obtained as a gift sample from Gattefosse, France; and Phospholipon 90 H (P 90H) was gifted by Lipoid, Germany. All other reagents used in the study were of analytical grade.
Selection of components and preliminary screening studies
Eighteen formulations with varying concentration of selected components, viz. Compritol® 888 ATO, P 90H, P 188, and PEG 400, were prepared in order to predefine the minimum and maximum concentration of each component (Supplementary Table 1) that can be employed for stable formulation. Various process parameters like stirring speed and time and number of cycles and pressure of high-pressure homogenizer were also screened and fixed for subsequent studies.
Preparation of solid lipid nanoparticles
The lipid phase, comprising molten Compritol® 888 ATO (lipid), ATS, and PEG 400, was heated to 75 °C and added to the hot aqueous phase comprising P 188 and P 90H heated to the same temperature under continuous stirring at high speed (9000 rpm) for 8 min using a high-speed stirrer homogenizer (WiseTis, HG-15D, Daihan, Korea) to result in a coarse emulsion. The latter was passed through a high-pressure homogenizer (HPH) (EmulsiFlex-C3, Avestin, Canada), at 1000 bar for 3 cycles. The formed o/w emulsion was cooled to room temperature to result in SLN dispersion containing both the encapsulated and free drug. In all the formulations, an accurately weighed amount of the drug and excipients was added as mentioned in Supplementary Table 1 and Table 1. Water was weighed accordingly so as to result in 70 g w/w batches.
Note: Fluorescein sodium (0.25% w/v)–loaded SLNs (F-SLNs) were also prepared similarly except that ATS was replaced with fluorescein sodium. Free fluorescein was removed from the system by dialysis against water containing PEG 400 (500 ml for 5 ml of SLNs) using presoaked (12 h in deionized water) dialysis bag (cutoff 7 kDa MW), fastened at both the end.
ATS suspension (SUS) was used as a control for ATS-SLNs. For preparation of suspension, 0.25% w/v of ATS was added to water containing 0.2% w/v sodium carboxymethyl cellulose as a dispersing agent and stirred for 3 h.
Systematic optimization of ATS-SLNs as per experimental design
Preoptimization studies revealed that the concentrations of all the components including the lipid and surfactants or co-surfactants were critical material attributes (CMAs) to obtain SLNs with desired properties, i.e., particle size and entrapment efficiency (Table 1), taken as critical quality attributes (CQAs). A central composite design with α = 1 was employed as per the standard protocol [33] using the Design Expert® 8.0.1(Stat-Ease Inc.). The ratios of Compritol® 888 ATO/P 90H concentration (X1) and P 188/PEG 400 concentration (X2) were taken as the independent variables, studied at 3 levels (i.e., − 1, 0, + 1) and optimized for the dependent (response) variables, i.e., particle size (Y1) and percentage entrapment efficiency (Y2) as shown in Table 1. Values for central point (0, 0, 0) were generated in sextuplet while the values for other formulation and processing variables were kept constant throughout the study. In total, 13 experimental runs were conducted and Table 1 describes the employed factor combination, coded levels, and the obtained responses. The order of experimental runs was randomized to deflect any possible sources of experimental bias.
Data analysis
Second-order polynomial models with added interaction terms were generated using the Design Expert® 8.0.1 (Stat-Ease Inc.), to ascertain and quantify the correlation between the CMAs and CQAs [34]. The general non-linear polynomial equation generated by the central composite design (CCD) is given below:
where Y is the measured response variable; A0 is the intercept; A1–A5 are the regression coefficients; A1 and A2 are the main effect of X1 and X2; A3 is the interaction of the main factors; A4 and A5 are the quadratic effects of the independent variables; and E is the error. The response surface analysis was carried out employing 3D-response surface plots for understanding the factor-response relationship and plausible interaction(s) among them.
Optimized formulation and its validation
Optimum formulation was identified by “trading-off” various CQAs using numerical optimization and desirability function approaching 1. Validation was carried out by preparing four formulations including the optimized formulation as check points. Linear correlation plots between observed and the predicted responses for these formulation check points were constructed, and percent prediction error between the observed and the calculated values was determined to ratify the prognostic ability of the experimental methodology.
Characterization of SLNs
Morphological evaluation using optical and transmission electron microscope
SLNs were observed after suitable dilution (10×) with distilled water under an optical microscope (Nikon Eclipse i90, Japan). For observation under transmission electron microscope (TEM), ATS-SLNs were stained with 2% phosphotungstic acid (PTA) in phosphate buffer (pH 6.8) for 5 min, after which the excess PTA was removed. Samples were spread on a carbon-coated copper grid and observed under TEM (H 100, Hitachi Ltd., Japan) at a voltage of 80 kV, for morphological parameters like size, sphericity, and aggregation.
Hydrodynamic diameter, polydispersity index, and zeta potential
Mean diameters of ATS-SLNs in the dispersion (10× dilution) and polydispersity index (PDI) were determined using photon correlation spectroscopy (Beckman Coulter, Delsa™ Nano C, Switzerland). Zeta potential of undiluted ATS-SLN dispersion was measured using Beckman Coulter, Delsa™ Nano C, at 25 °C and the electric field strength of 23.2 V/cm.
Total drug content and entrapment efficiency
SLN dispersion (1 ml) was dissolved completely in a mixture of chloroform:methanol (2:1), and the absorbance of the obtained solution, determined UV spectrophotometrically (using previously validated UV method; Supplementary Table 2) at 246 nm, was used to determine total drug content (TDC) (chloroform helps to dissolve the lipid matrix, disrupt formed SLNs, and release the entrapped drug). TDC is a measure of free and entrapped drug in SLN formulation.
Entrapment efficiency (EE) was determined by dialyzing ATS-SLN dispersion (1 ml) in a dialysis bag (7 kDa MW cutoff) immersed in 75 ml methanol kept under stirring to remove and dissolve all the free and unentrapped drug [35, 36]. After 30 min, SLNs remaining in the bag were removed and disrupted with suitable quantity of chloroform:methanol (2:1). The amount of entrapped drug was determined in the solution spectrophotometrically. The time and volume of dialysis were optimized by analyzing release of equivalent quantity of free drug.
In vitro release
In vitro release of ATS from ATS-SLN dispersion was performed across the presoaked (12 h in deionized water) dialysis bag (cutoff 7 kDa MW), fastened at both the ends and enclosing 0.5 ml ATS-SLNs (n = 3). Simulated tear fluid, pH 7.2, containing 2% ethanol was used as the dissolution medium (50 ml), maintained at 37 ± 0.5 °C, and stirred at 50 rpm. The procedure was repeated for ATS-SUS, under similar conditions. Aliquots of the dissolution medium were withdrawn at suitable time interval and replaced with the same volume of fresh medium to maintain the sink conditions. Withdrawn samples were suitably diluted and analyzed spectrophotometrically.
FTIR, DSC, and XRD studies
All studies were conducted by standard procedures which are elaborated in the Supplementary data.
Stability studies
At 5 ± 3 °C (refrigerated conditions)
ATS-SLN samples were stored in amber-colored, tightly closed, screw-capped vials, at 2–8 °C for 12 months. Samples were withdrawn periodically and analyzed for any aggregation, drug degradation in terms of TDC and leakiness (as EE).
Photostability
ATS-SUS and ATS-SLNs were exposed to conditions described under ICH guidelines for photostability. This included not less than an overall illumination of 1.2 million lux hours with an integrated near ultraviolet energy of not less than 200 W h2 for 10 days at 25 °C in a photostability chamber (Binder, Germany), both in clear and in amber glass containers.
Comprehensive testing as ocular drops
pH and osmolarity
pH was measured using CyberScan, Eutech pH 510, at 25 °C. Advanced® Model 3250 Osmometer (MA, USA) was used to determine the osmolarity of the developed ATS-SLNs.
Refractive index and viscosity
Refractive index of ATS-SLNs and water taken as control was determined using Abbe’s refractometer. Viscosity of ATS-SLNs was determined using a modified Ubbelohde viscometer at 25 ± 0.1 °C.
Porcine cornea permeability studies
Freshly procured porcine cornea was mounted on a previously described diffusion cell [35, 37]. The receptor compartment comprised freshly prepared glutathione bicarbonate ringer (GBR) containing 2% ethanol as the diffusion medium (20 ± 2 ml), stirred continuously (50 rpm) at 37 ± 0.2 °C. Ethanol was added in the diffusion medium to maintain sink conditions [38]. ATS-SLNs (0.2 ml) placed on the porcine cornea represent the donor side. Samples were withdrawn at various time points with replacement and were analyzed spectrophotometrically at 246 nm after suitable dilution. The apparent corneal permeability coefficient (Papp) and steady-state flux of both ATS-SLNs and ATS-SUS were determined, as reported previously [35, 37]. At the end of the experiment, each cornea (free from any adjoining tissue, viz. sclera) was weighed (Ww). Then, it was soaked in 1 ml methanol, dried overnight at 90 °C, and reweighed (Wr). The corneal hydration level (HL%) was calculated by the following equation:
Both eyes of each animal were removed. One eye received ATS-SUS while the contralateral eye cornea was used to determine permeability of ATS-SLNs [33].
Sterilization
ATS-SLNs were autoclaved at 121 °C (15 psi pressure) for 20 min and change in TDC, particle size, and percentage entrapment was noted.
Sterility testing
Sterility testing studies were performed, as per the Indian Pharmacopoeia [39], on the final autoclaved formulation, using the direct inoculation method (detail described in Supplementary data).
Safety of ATS-SLNs
Cytotoxicity studies
The viability of HCLE (human cornea limbal epithelial), R28 (retinal neuronal), ARPE-19 (a human retinal pigment epithelium), and RCE (rabbit corneal epithelium) cells when exposed to ATS-SLN formulation, was determined by a cell proliferation assay using MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) and WST-1 reagent (Roche Molecular Biochemicals). Test procedures are described in the Supplementary data.
Human corneal–limbal epithelial (HCLE) cells were kindly provided by Dr. Ilene Gipson. This is an established cell line [40] that has been authenticated by short tandem repeat DNA analysis [41]. To establish HCLE cells, primary cultures of human corneal–limbal cells derived from corneal donor rims were immortalized by abolition of p16 control and p53 function before immortalization by expression of human telomerase reverse transcriptase. Corneoscleral donor rims consisted of peripheral and limbal corneas that were left after removal of central corneal buttons for transplantation. These tissues were obtained in accordance with good clinical practice, the Institutional Review Board and informed consent regulations of the Schepens Eye Research Institute and Massachusetts Eye and Ear Infirmary, and the tenets of the Declaration of Helsinki.
Retinal R28 cells, obtained from Kerafast, were originated from a post-natal day 6 rat retinal culture immortalized with the 12S E1A gene of the adenovirus in a replication-incompetent viral vector [42]. Sprague Dawley rats were used in accordance with the Association for Research in Vision and Ophthalmology statement for the Use of Animals in Ophthalmic and Vision Research. The rat and retinal-specific gene expression of these cells has been confirmed by microarray analysis.
In vivo toxicity inrabbits as per OECD guidelines [43]
(detail described in Supplementary data)
Ocular tolerance evaluation
To examine the effect of ATS-SLNs on ocular structure and integrity, the left eyeball was removed from rat eyes 0.5 h, 1 h, and 2 h post administration of ATS-SLNs to the left eye of the rat. The right eye of these animals was taken as control. The eye balls were washed with saline and fixed with 8% w/w formalin solution. The material was dehydrated with an alcohol gradient, put in melted paraffin, and solidified in block form. Cross sections (< 5 μm) were cut, stained with hematoxylin and eosin (H and E), and microscopically observed (Nikon Eclipse 90i, Japan) for any pathological effects [44].
Cellular uptake in retinal cell lines
Retinal pigment epithelial cells (ARPE-19) obtained from ATCC, were maintained in 50% Dulbecco’s modified Eagles medium (DMEM) and 50% F12 medium supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin G, and 100 μg/ml streptomycin, in a humidified incubator at 37 °C in 5% CO2. Cells were grown on coverslip and incubated with fluorescein-labeled SLNs (F-SLNs), at concentration of 3 μg/ml or equivalent free fluorescein solution in standard medium for 2 h. Nuclei and cytoskeleton were stained with DAPI and TRITC-labeled phalloidin, respectively. Briefly, TRITC-labeled phalloidin (P1951 Sigma; final staining solutions in aqueous physiological buffers are in the concentration range of 0.1–100 mM) was applied to the cells to visualize cell morphology. Nuclei were stained with the fluorescent Hoechst 33342 dye (DAPI) 10 μg/ml (Invitrogen) for 15 min at RT [45].
Effect of ATS-SLNs on apoptosis
UV-induced
ARPE-19 and RCE (procured from ECACC) cells were grown as described in Supplementary data. After 2 h, plates were dried, exposed to UVC rays (60 mJ/cm2 for ARPE19 and 25 mJ/cm2 for RCE) in a Stratalinker 1800 apparatus (Stratagene, La Jolla, CA), and fresh medium containing the drugs was added. After 24 h (for ARPE-19 and RCE, respectively), cells were analyzed for viability by the WST-1 reagent (Roche Molecular Biochemicals) according to the manufacturer’s protocol: briefly, the medium was replaced with a complete RPMI 1640 medium without phenol red with 10% WST-1 reagent. Plates were incubated for 1.5 h at 37 °C in a 5% CO2, humidified atmosphere, vigorously shaken 1 min at room temperature, and analyzed by a microplate reader at 450 nm (Bio-Rad, Hercules, CA).
Apoptosis assay by FACS
For apoptosis analysis, 9 × 105 cells were seeded in 60-mm petri dishes. In the first instance, the cells were pretreated with ATS-SLN, ATS-SUS, and blank SLN for 2 h, and washed twice in PBS, after which PBS was removed and cells were irradiated as previously reported. In the other case, cells were given various treatments (ATS-SLN, ATS-SUS, blank SLN) immediately after irradiation. Fresh media as such or containing ATS-SLN, STS-SUS, and blank SLN were added, and cells were returned to incubator for 24 h prior to FACS or Western blot (cytochrome C release) analysis. Cells treated with 20 mM H2O2 in cell medium for 20 min were used as a positive control. FACS analysis was performed by an Annexin V/propidium iodide–based method using a commercially available kit (Annexin V-FITC apoptosis detection kit ab14085): briefly, after treatments, cells were collected (1–5 × 105 cells) by centrifugation and resuspended in 500 μl of 1× binding buffer, and incubated with 5 μl of Annexin V-FITC and 5 μl of propidium iodide at room temperature for 5 min in the dark. Cells were analyzed by a FACS CANTO apparatus (Becton Dickinson, USA).
Apoptosis assay by cytochrome C release
Cells were treated as for FACS analysis, collected, and lysed in Nonidet P-40 lysis buffer (25 mM Tris, pH 7.5, 100 mM KCl, 0.25 mM dithioerythritol, 10 mM MgCl2, 1% Nonidet P-40, protease inhibitor mix from Roche Molecular Biochemicals) for 30 min on ice. After centrifugation for 15 min at 12,000g, supernatants were harvested and proteins quantified by the BCA Protein Assay Reagent (Pierce, Thermo Fisher, USA). Proteins (30 μg/lane) were electrophoresed through SDS polyacrylamide 12.5% gel and electroblotted onto nitrocellulose membrane (Invitrogen, Thermo Fisher Scientific, USA) using an iBlot 2 Dry Blotting System® (Invitrogen, Thermo Fisher Scientific, USA). The non-specific signals were blocked with Odyssey Blocking Buffer (LI-COR, Lincoln, NE, USA) and incubated overnight at 4 °C with 0.2 μg/ml of anti-cytochrome c monoclonal antibody (sc-13560 Santa Cruz) and anti-tubulin antibody (Sigma-Aldrich, Saint Louis, MO, USA). The membrane was washed and subsequently incubated with anti-mouse Alexa Fluor® antibody (1:8000) from Thermo Fisher (USA). Detection was carried out by an Odyssey Infrared Imaging System (LI-COR, Lincoln, NE, USA).
Apoptosis by time-lapse assay
The cumulative apoptotic events were scored by time-lapse videomicroscopy using a Zeiss inverted phase-contrast microscope equipped with a 10× objective and Panasonic CCD cameras. After cell detachment from the substrate, an apoptotic event was counted the moment the cell had shrunk completely and bubble formation started, as previously reported [46].
Pharmacokinetic study
Male rabbits, weighing approximately 1.5 kg, were purchased from Lala Lajpat Rai University of Veterinary and Animal Sciences (Hisar, India). Prior to experiments, the rabbits were housed in standard cages and allowed free access to food and water. All the animal study protocols were approved by the Institutional Animals Ethics Committee, PU, Chandigarh (vide letter no. IAEC/411, dated September 11, 2013), and guidelines as per the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) were followed.
A total of 32 rabbits were used for the study. ATS suspension and ATS-SLNs (150 μlFootnote 1 each; 150 μl × 2.5 mg/ml = 375 μg of ATS) were instilled into both eyes of two rabbits each, for each time point of the study (n = 4). The time points selected were 0.25, 0.5, 1, 2, 6, 12, 24, and 48 h. Sample collection, preparation (extraction procedure), and data analysis details are described in Supplementary data. A previously validated HPLC method was used to determine the concentration of ATS in aqueous and vitreous humor (described in Supplementary data).
Residence time
A drop (30 μl) of F-SLNs was instilled into the conjunctival fornix of the right eye of female rabbits. Animals were allowed to move freely after administration, and at different sampling times (0.25, 0.5, 1.0, 1.5, 2.0, 3.0 up to 7 h), 50 μl of simulated tear fluid (STF; pH 7.2) was introduced into the precorneal cavity followed by forced blinking (five times). STF was withdrawn and released repeatedly into the cavity using a suitable posteum pipette, followed by withdrawing the sample and observing under the fluorescence microscope (Axioscope A1, Carl Zeiss, Germany).
Proof of concept of effective delivery to the posterior eye
A drop (30 μl) of the F-SLNs was administered into the fornix of the right eye of rats. Animals were sacrificed at 0.5, 1, and 2 h post administration; eyes were enucleated immediately and stored at − 20 °C. Frozen eyes were embedded in Cryomatrix™ and cut into 10-μm-thick sections using a cryostat (IEC Minotome 3398, American Instrument Exchange, Inc., MA, USA). Fixed, complete eye sections were observed using a fluorescence microscope (Axioscope A1, Carl Zeiss, Germany) and confocal laser scanning microscope (Nikon EclipsTi, NIS Element, Japan) using a 63× oil immersion objective and an aperture of 1 Airy unit. All protocols were approved by the animal ethics committee (IAEC) of Panjab University, Chandigarh (IAEC/411, dated November 9, 2013).
Statistical analysis
All results are expressed as the mean ± standard deviation. The results were analyzed for statistical significance by a one-way analysis of variance (ANOVA) test followed by the Tukey test or the Student pair t test, whichever applicable.
Results and discussion
Statins are currently being eyed for use in management of a variety of ocular pathologies of vitreoretinal origin, including AMD [2, 22, 47]. However results with clinical studies were only moderately promising [2, 22, 24, 48] and were based on the clinical evidence and/or the pharmacodynamic effects [21, 49]; and none of the studies confirmed whether significant drug reached the ocular tissues so as to elicit a measurable physiological response. It is important that following oral administration, sufficient statin crosses the blood-aqueous barrier and reaches the internal eye to produce an effect. Statins are poorly bioavailable (5–14%; ATS 12%), and it may be noted that only a small proportion (generally 1/3 to 1/5) of the concentration of any drug in plasma, reaches the aqueous/vitreous humor [50, 51]. Although retinal availability of lipophilic drugs like statins following systemic administration can also occur via the choroid tissue, the high molecular weight (> 500 Da) is a limitation for this pathway [52].
We presently developed an eye drop formulation of ATS for achieving high local concentration in the ocular tissue. Challenges to achieving this were manifold: (i) insolubility (0.00495 mg/ml) of ATS, (ii) circumventing ocular barriers to reach the posterior eye including multiple drainage pathways, and (iii) high molecular weight of ATS (557.63 Da) which limits its permeation.
Significant work is published on solubility and bioavailability enhancement of ATS following oral administration. Solid dispersion of ATS using various polymers, viz. PEG 4000, PVK 30, and Kollicoat IR, has been prepared and reported [53, 54]. In addition to poor solubility, extensive first-pass metabolism limits the oral bioavailability of ATS to < 15% and it is argued that solid dispersions can only improve the former, but once absorbed, ATS will still undergo first-pass effect. Hence, it was proposed that encapsulation within a slow release nanoparticulate system can manage both solubility and metabolism issues. Polymeric nanoparticles of ATS with sufficient encapsulation (nearly 70%) and extended release were thus prepared by some groups [55,56,57,58]. Sripriyalakshmi et al. prepared ATS-loaded albumin nanoparticles by desolvation method, using glutaraldehyde (8%) as cross-linker and ethanol as a precipitating agent [59]. Kumar et al. developed ATS-SLNs through hot homogenization technique using trimyristin and soy-phosphatidylcholine as lipids. For preparing lipid drug matrix, a mixture of chloroform–methanol was used [60]. Similarly ATS-SLNs by solvent injection technique are also reported [61]. However, use of cross-linking agents and organic solvents and a residual contamination with these can be a concern in ocular delivery. Polymerization initiation, formation of toxic monomers and degradation products, and large polymer aggregates are also a limitation with polymeric systems [62, 63]. Other problems include lack of large-scale production method [64] and a suitable sterilization method, e.g., autoclaving.
To address all these limitations, an optimized ATS-loaded solid lipid nanoparticulate system was developed presently which is an aqueous system comprising ingredients employed at concentrations approved to be safe by USFDA [65] for ocular use. Further, the methodology used, i.e., high-pressure homogenization, is scalable and industrially viable.
Prescreening studies
SLNs comprise a nanosized lipidic core stabilized by a layer of surfactants. Selection of lipid and surfactant and their concentration are thus an important consideration for development of SLNs. However, high proneness of ATS to degradation, including oxidation in surfactant solutions especially polysorbate 80 (the most commonly employed surfactant in pharmaceutical formulations), was a serious challenge to its solubilization for preparing SLNs [32]. A series of surfactants and solvents, viz. Transcutol P, propylene glycol, Poloxamer (P) 188, PEG 300, PEG 400, and PEG 600, were thus tried in the initial prescreening studies for formation of ATS-SLNs. Minimum volume of surfactant required to completely dissolve the required quantity of ATS (0.25% w/v) along with protection from oxidation and hydrolysis was taken as criteria for their selection. However, safety and biocompatibility of selected components at their employed concentrations with ocular tissues and their amenability to steam sterilization were also a consideration [66].
Highest solubility of ATS was noticed in PEG 400 (56 mg/ml); hence, it was selected as a solubilizer. To improve drug loading and entrapment efficiency, it is important that the drug is incorporated in the SLN matrix in a soluble form [67] and for that purpose, an optimized concentration of PEG 400 was employed. Among the surfactants, not only did a 4% solution of P 188 enhance the solubility of ATS by 100 times (0.04 mg/ml), but also ATS was found to be stable (7 days) in this solution; hence, it was selected as the surfactant of choice. Both P 188 and PEG 400 can help achieve small size, high loading, and significant entrapment. Both these agents are permeation enhancers [68, 69], and PEG also has a P-gp efflux–inhibiting property. In addition, PEG is a surfactant-supporting polymer which is known to reduce the gelation of SLNs [61]. P 188 protects against oxidation by getting adsorbed on the surface of lipidic nanoparticles due to its large hydrophilic blocks providing barrier for free radical chain reaction [70].
Even though ATS is a lipophilic molecule (log P = 4.41), yet it was not soluble in any of the lipids, viz. Compritol® 888 ATO, glyceryl monostearate, stearic acid, and Precirol® ATO 5, screened by us. Considering our prior experience with Compritol® 888 ATO for developing SLNs for a variety of drug molecules, its ability to form stable SLNs of sufficiently small size and its biocompatibility with ocular tissue [71], it was selected presently as the lipid matrix. Soya lecithin (Phospholipon 90H) was used as a co-surfactant and its addition improved the entrapment efficiency and stability of ATS-SLNs [72]. It is reported that oxidative stress is the initial pathological mechanism in the development of AMD which leads to oxidation of phospholipids present in the photoreceptors [73]. Excess accumulation of oxidized phosphatidylcholine was observed in immunohistochemistry of the eyes with AMD [13]. Hence, it is proposed that the anti-oxidant nature [74] of P 90H (i) will help in reducing the oxidative stress of AMD eyes and (ii) may also restore the level of unoxidized phospholipid in the photoreceptors.
Screening studies and optimization of ATS-SLNs
Based on the above-discussed facts and the results of Table 1 and Supplementary data, and on our prior experience in the lab, a range of concentration of various components, viz. Compritol® 888 ATO, P90H, P 188, and PEG 400, was selected. SLN dispersions, which were stable for more than a week, were characterized for TDC, EE, and particle size. On the basis of stability of dispersion, the minimum and maximum of each component were selected. The selected CMAs were varied in the range of 1–5% w/w for Compritol® 888 ATO, 1–2% w/w for P 90H, 1.5–5% w/w for P 188, and 2–4% for PEG 400, in the optimization studies. Drug concentration varied from 0.2 to 0.5% w/w, and it was observed that an increase beyond 0.25% w/w tended to decrease the EE; hence, ATS concentration was fixed at 0.25% for further optimization. Based on preliminary experiments, various process variables were also fixed at particular levels like—the number of HPH cycles was fixed at 3, because 4 or higher number of passage through HPH resulted in an increase in size by 20–30 nm per cycle; likewise, stirring speed and time of stirring for preparation of the coarse preemulsion was fixed at 9000 rpm for 8 min. Further increase in stirring speed and time resulted in a decrease in entrapment efficiency, and below the selected levels, bigger particles were observed.
Systematic optimization of ATS-SLNs
FDA recommends that the design of pharmaceutical products [75] should involve a “quality-by-design” approach; hence, an optimized ATS-SLN formulation was designed presently employing CCD as per standard protocol (Table 1) using a desirability approach.
Regression analysis of experimental design
In the analysis of optimization experiment, a second-order function (response surface) was used to characterize the effect of independent variables (X1—Compritol® 888 ATO/P90H; X2—P 188/PEG 400) on the particle size and entrapment efficiency (responses). Produced data for the responses were regressed to obtain the coefficients including interaction term [76]. Regression analysis was facilitated by coding of the factor in order to simplify the analysis. The generated polynomial mathematical equation for the responses is represented and tabulated in Table 2 along with the significance (p value).
Polynomial mathematical equations defined by CCD are as follows:
The sign and magnitude of the effects in each equation signify the relative influence of each factor on the response. A negative sign signifies an antagonistic effect while a positive sign indicates a synergistic effect. Negative correlation of particle size was observed with the increase in the ratio of P 188/PEG 400. Combined effect (interaction term) of both CMAs also exhibited a negative influence on particle size. Further, p value (< 0.05) of individual factors demonstrated that the ratio of Compritol® 888 ATO/P 90H as such, as well as in its square term, has a significant contribution to the variability in particle size. For EE, the most significant factors were quadratic term of Compritol® 888 ATO/P 90H ratio and interaction of both CMAs which had p values < 0.005. Both CMAs as such and their combined effect showed positive correlation with EE, but their quadratic terms found a negative influence on EE (Table 2).
Table 3 represents the result of ANOVA test on the quadratic regression model and details the model summary statistics for the selected significant quadratic models. The quadratic model was statistically significant (p < 0.001) in both cases while the lack of fit was insignificant (p > 0.05). It can be observed from Table 3 that all responses had r2 value > 0.9, which indicates a good correlation between the experimental and the predicted responses. Further, the selected models explain a significant proportion of variability in the observed responses (F ratio test, p value < 0.001). Adequate precision measures the signal-to-noise ratio and a value > 4 indicates good reliability [33].
For further establishing the appropriateness of the selected models, several diagnostic plots like normal plot of residuals and Box–Cox plot (not shown), were used to establish its appropriateness and goodness of fit of the data. The plots also confirmed that CCD matrix data was free from outlier variables and did not require any transformation.
The relationship between formulation variables for preparing ATS-SLNs and the resulting response is summarized graphically (Fig. 1) by the response surface plots obtained from the modeling steps. The particle size was found to decrease with the increase in Compritol® 888 ATO/P 90H ratio from 0.5 to 2.5. Beyond 2.5, SLNs of higher particle size were observed as shown in Fig. 1a. This increase in particle size beyond a certain lipid concentration is probably due to an increase in viscosity of developed system resulting in agglomeration of particles [77]. Increase in the P 188/PEG400 ratio resulted in a decrease in particle size up to mid-level and after that, particle size increased. Similar concentration-dependent surfactant and co-surfactant effect of P 188 on particle size of paclitaxel-loaded polymeric nanoparticles has been reported [58]. It seems that the hydrophobic propylene chain of P 188 aligns on the lipid/water interface along with PEG and P90H to cover the melted lipid droplets more efficiently, thus reducing the interfacial tension at the lipid–water interface, resulting in a lower particle size. Further, P 188 may also offer steric stabilization to formed particles, thus avoiding their aggregation. However, as the concentration of P 188 increases beyond a certain level (3.25% in this case), the hydrophilic (PEG) chains of one particle of P 188 may interact with those of the other particles. These inter-particle interactions can result in agglomeration at higher concentrations [78].

3D responses for (a) particle size and (b) entrapment efficiency
Entrapment efficiency of SLNs initially increased with an increase in the Compritol® 888 ATO/P 90H ratio, while a further increase (> 2.5) resulted in a decrease in percentage EE as shown in Fig. 1b. The initial increment in EE can be attributed to availability of a sufficient Compritol® 888 ATO to completely encapsulate ATS (fixed at 0.25% w/v) complemented with an adequate quantity of P 90H. Compritol® 888 ATO is a complex lipid composed of mixtures of mono-, di-, and triglycerides which form less perfect crystals, and thus accord space to accommodate drug molecules [79]. P 90H is also reported to create space for drug incorporation, and the monomolecular P 90H layer stabilizes the particle interface [80]. A decrease in percentage EE at a high ratio of Compritol® 888 ATO/P 90H can be attributed to the availability of a lesser quantity of P 90H with respect to Compritol® 888 ATO.
Increase in percentage EE was also noticed with an increase in the ratio of P 188/PEG 400. However, at an extreme ratio, i.e., from 2.0 to 2.5, a significant decrease in the percentage EE of the prepared SLNs was observed. PEG 400 has a high affinity for water and since ATS had a very high solubility (56 mg/ml versus 0.00495 mg/ml in water) in PEG 400, hence at a high P 188/PEG400 ratio, the proportion of PEG 400 being small will partition towards water-carrying ATS with it resulting in its low entrapment in the lipid matrix [81].
Selection of optimum formulation and validation
Minimum particle size and maximum EE were set as the selection criteria for finding optimum formulation. Based on these criteria, the software generated a single, most suitable solution, comprising the Compritol® 888 ATO/P 90H ratio 2.5 and P 188/PEG400 ratio 1.73 (Table 4). To check the reliability of the developed mathematical models, the response of the optimal SLN formulation and three additional checkpoint formulations covering the entire range of the experimental domain was recorded. For each of these test runs, the experimentally determined response was compared with the response predicted by the mathematical model. Table 4 lists the test conditions of the optimum and the random check points and their experimental and predicted values for both the response variables, along with the calculated percentage prediction error. Supplementary Fig. 1 (a, b) shows linear (r2 > 0.9) correlation plots between the observed and predicted response variables, indicating excellent goodness of fit. Low percentage prediction error (− 6.5 to 4.5 for Y1 and − 4.35 to 1.11 for Y2) and significant r2 confirm robustness of the mathematical model and high prognostic ability of response surface methodology.
Characterization of SLNs
Morphology of ATS-SLNs using optical and transmission electron microscopy
Optical inspection indicated the ATS-SLNs to be small in size and round in shape, with no aggregation or irregularities (Fig. 2). Similar results were obtained with TEM. The ATS-SLNs were found to be spherical in shape (Fig. 3), and the size of observed particles invariably varied between 70 and 185 nm. The TEM photograph at a higher magnification indicates differently transmitting areas in a single SLN particle (Fig. 3b). These may be due to P 90H, ATS, and PEG interspersed in the lipid matrix with uniform distribution (no core or shell structure is observed) as also indicated by a sustained release (Fig. 4) of ATS with no burst release. A surfactant layer enclosing the solid–lipid matrix is also observable in Fig. 3b. The former explains the larger hydrodynamic diameter [82] observed as measured by differential light scattering (DLS) (“Particle size and zeta potential”).

Optical photomicrograph (400× magnification) of SLNs

TEM micrographs of ATS-SLNs at (a) 60,000x and (b) 300,000x magnification

In vitro drug release of ATS-SLNs and ATS-SUS in simulated tear fluid (pH 7.2) containing 2% ethanol
Particle size and zeta potential
The developed ATS-SLNs showed an average particle size (n = 6) of 256.3 ± 10.5 nm and PDI of 0.26 ± 0.02 (n = 6) (Supplementary Fig. 2). The PDI < 0.3 indicates a narrow particle size distribution. Small size offers a greater surface area for association and permeation through the cornea and conjunctiva [83]. Ample evidence of achieving therapeutic concentration of actives in the posterior eye tissue (including the retina) using these systems is indicated [84]. However, size is shown to influence the extent of absorption/permeation and disposition through the ocular static and dynamic barriers. Sakurai et al. studied the intraocular permeation efficiency of fluorescein-labeled polystyrene nanospheres of varying size (50, 200, 2000 nm). They observed that nanospheres with the particle size 200 nm could penetrate up to the retina while the particle size 2000 nm could not reach the retina and was seen in the vitreous cavity and trabecular meshwork only [85]. Similarly, uptake of PLGA particles in rabbit conjunctival epithelial cells was found to be higher for 100-nm particles in comparison with the corresponding 800-nm and 10,000-nm particles [86]. Size-dependent in vivo corneal uptake of indomethacin-loaded poly(epsilon-caprolactone) colloidal particles also indicates higher permeation of nanosized particles than microparticles after topical instillation into the albino rabbit eye [87].
Generally for ophthalmic formulations, particles ≤ 500 nm are indicated for efficient vitreal mobility [88] and permeation through ocular barriers. Moreover, a size-dependent disposition of nanoparticles studied by Amrite et al. indicated that very small particles (20 nm) undergo rapid clearance from the periocular tissue as compared with the larger particles (200 nm and 2 μm) which are retained for longer periods of up to 2 months. Systemic clearance via periocular blood or lymphatic circulation was indicated as a probable reason for rapid clearance of small particles [83, 89]. Thus, > 20–200–500 nm may be considered an optimum size for ocular formulations. In our case, 50% of particles are ≤ 250 nm and 90% particles are below 530 nm, establishing the suitability of ATS-SLNs for effective ocular uptake. Further, no particles of size < 30 nm (frequency of 1.2% at 36 nm; Supplementary Fig. 2(a)) were observed. The micrometric granular pattern and perinuclear finer fluorescence observed presently in the ARPE-19 cell line studies using F-SLNs as discussed in “Cellular uptake” (Fig. 12) also confirm that probably SLNs prepared presently with sizes 200–500 nm reach the nuclear membrane while the bigger particles (observed as a granular pattern) remain on the outside of the cells.
Usually a zeta potential of ≥ ± 25 mV is recommended for achieving stable dispersions. ATS-SLNs prepared presently by us, however, had a near neutral charge (zeta potential = − 2.65 mV) as shown in Supplementary Fig. 2(b) and were found to be stable with no significant change (p < 0.001) in particle size, TDC, and EE at 5 ± 3 °C for up to 1 year (“Stability studies”). The suitability of neutral nanoparticles for efficient retinal delivery of therapeutics with a capacity to evade reticuloendothelial system (RES) uptake is indicated in literature [90, 91].
Total drug content and entrapment efficiency
The drug content of the developed ATS-SLNs was 2.35 ± 0.05 mg/ml which was 94.00 ± 1.21% of what was originally incorporated (2.5 mg/ml) indicating an efficient production process.
A sufficiently high entrapment efficiency of 73.1 ± 1.52% (n = 6) was obtained.
In vitro release
The drug release profile of ATS-SLNs through dialysis membrane at 37 ± 0.5 °C (Fig. 4) showed a prolonged release with only 58.45% drug being released up to 96 h. Free drug on the other hand was released completely (100%) within 20 h. Fitting of release data to the various mathematical models like zero-order, first-order, Higuchi, and Korsmeyer–Peppas models indicated a first-order drug release (r2 = 0.986) pattern of ATS-SLNs. Higuchi release model (r2 = 0.932) was best fitted for ATS-SLNs at all the time points, indicating that drug release occurs by diffusion [92]. The latter was confirmed by the release exponent value (n) of 0.264, observed from the Korsmeyer–Peppas model indicating Fickian diffusion of ATS from ATS-SLNs. An almost 10% release observed at the first time point (15 min) is attributable to the unentrapped drug (~ 26%) in ATS-SLN dispersion.
FTIR, DSC, and XRD studies
FTIR
The FTIR spectra for blank SLNs and ATS-SLNs along with its various components are shown in Fig. 5. ATS showed intense absorption bands at 3363 cm−1 (O-H stretch), 3246 cm−1 (aromatic N-H stretch), 1650 cm−1 (aromatic C=O stretch), 1431 cm−1 (C-F aromatic stretch), 1315 cm−1 (C-O stretch of carboxylate group), 1217 cm−1 (C-N stretching of amide), and 695 cm−1 (aromatic out of plane bend), respectively [93].

FTIR spectra of (a) ATS, (b) P90H, (c) P 188 (Poloxamer 188), (d) Compritol® 888 ATO, (e) Blank SLNs, and (f) ATS-SLNs
Compritol® 888 ATO exhibited characteristic peaks at 2918 cm−1 and 2851 cm−1 (alkanes stretch) and 1741 cm−1 (C=O ester stretch). In the FTIR spectra of ATS-SLNs, peaks corresponding to ATS disappeared indicating its efficient entrapment in the lipid matrix, while the peaks corresponding to 2918 cm−1 (alkane stretch) and 2851 cm−1 (seen in Compritol® 888 ATO and P 90H) respectively are observable in both blank and ATS-SLNs. A peak corresponding to 1107 cm−1 for P 188 (1111 cm−1) representing C-O bond stretch [94] was observed for both blank and ATS-SLNs. This observation indicates that P 188 forms a layer on the surface of developed particles. The similarity of the FTIR spectra obtained for drug-loaded SLNs to that exhibited by blank SLNs, reconfirms efficient encapsulation of ATS within the lipidic core of developed SLNs.
DSC
Melting endotherm of ATS, corresponding to its melting point appeared at 164.81 °C (134.4 J/g), and Compritol® 888 ATO also showed a sharp peak at 73.1 °C again corresponding to its melting point, with an enthalpy of 109.3 J/g (Fig. 6). ATS-SLNs showed a broad endothermic shift to 86.93 °C and a significantly lowered heat flow of 15.03 J/g (Fig. 6), indicating incorporation of ATS into the lipid matrix.

Thermogram of (a) ATS, (b) P 188, (c) Compritol® 888 ATO, (d) PEG 400, (e) P90H, (f) blank SLNs, (g) ATS-SLNs
Lower enthalpy for ATS-SLNs versus pure lipid indicates a reduction in particle size. Shift and broadening of endothermic peak of PEG 400 at 102.69 to 92.09 °C in blank SLNs with lower enthalpy (26.45 to 21.50 J/g) indicates interaction of PEG 400 with the lipid matrix. However, the peak corresponding to PEG 400 disappears completely in ATS-SLNs. This could mean that PEG effectively solubilizes ATS and helps in its incorporation within the lipid matrix of SLNs.
XRD
Powder X-ray diffraction study of pure drug (ATS) and lyophilized ATS-SLN formulation was performed to confirm the amorphous or crystalline nature of SLNs. ATS exhibited characteristic peaks between 2θ of 6.1 and 48.1 (Fig. 7), while these peaks disappeared in the XRD pattern of lyophilized ATS-SLN formulation, indicating loss of ordered lipid matrix [95] and the amorphousness of the formulation. Presence of some broad and diffuse peaks especially those corresponding to P 188 and P 90H were however observed as expected because P 188 covers the surface of the particles.

Powder X-ray diffraction patterns of (a) ATS-SLNs, (b) P 90H, (c) P 188, (d) Compritol® 888 ATO, (e) ATS
Stability studies
Stability studies under refrigerated conditions
No significant change (p < 0.001) in entrapment efficiency, total drug content, and particle size (except at 12 months) of ATS-SLNs was observed upon storage under refrigeration for 12 months, confirming their long-term stability (Table 5). TDC of ATS-SUS was however reduced to almost half (45.5%) at 12 months even under refrigerated condition. The significant improvement (13.62 times) in stability of ATS highlights importance of its incorporation into SLNs.
The SLN dispersion was also found stable to lyophilization (without cryoprotectant) with an insignificant (10–20 nm) change in particle size following redispersion of lyophilized formulation. Lyophilization usually results in an increase in size (100–200 nm) on reconstitution [96]. The lower tendency of present formulation to form aggregates during lyophilization may be due to the presence of 4% P 188, which completely covers the SLNs and thus stabilizes them. P 188 is reported to provide a stabilizing coat (outer shell) to SLNs [70].
Photostability studies
Total drug content of ATS-SLNs did not change significantly (p < 0.05; Table 6) before and after exposure to UV light during photostability studies, as per ICH guidelines, in an amber glass vial as well as clear glass vial. ATS-SUS however showed a significant (p < 0.05) drug loss (~ 30%) upon UV exposure. ATS is reported to undergo photooxidation especially at the pyrrole ring in its structure [31]. Protective shield provided by the enclosure within the lipid matrix of SLNs coupled by the anti-oxidant property of P 90H [74] and PEG 400 [97] may be responsible for protection of ATS against photodegradation.
Comprehensive testing as ocular drops
pH and osmolarity
The pH and osmolarity of ATS-SLN dispersion was 6.85 ± 0.5 and 210 ± 4.5 mOsm/l, respectively. Ocular drops with pH significantly different from tears (7.2) can elicit increased lacrimation due to irritation and pain. This can wash off and dilute the instilled drug from the eye fornix, resulting in a poor bioavailability. pH 7.2 ± 0.2 is defined as an optimum pH for maximum comfort in ophthalmic preparations [98]. The pH of the developed formulation was near neutral 6.85 ± 0.5 and did not require addition of any buffering agent.
Normal osmolarity of tears is 290–300 mOsm/l [99]. Hypotonic solution as obtained presently for ATS-SLNs (210 ± 4.5 mOsm/l) is advocated to be more suitable for ocular delivery in comparison with hypertonic or isotonic solutions [100]. Evaporation from the tear film tends to increase the osmolarity of the applied solution, and hyperosmotic drops can damage the ocular surface epithelia [79].
Refractive index and viscosity
Refractive index of ATS-SLNs (1.36) was found close to that of water (1.33) indicating that the developed SLN formulation would be appropriate for ocular use. Furthermore, for minimum interference with vision, a refractive index close to that of ocular tissues (1.34–1.41) as obtained presently is a desirable feature [101].
Optimal viscosity (ῂ) recommended for ocular drops is ≤ 20 cP. Higher values may result in reflex tearing and blinking in order to regain the original viscosity of the lacrimal fluid (1.05–5.97 cP). The viscosity for ATS-SLNs dispersion was 8.12 cP. The latter will not induce any excessive blinking or tearing while simultaneously retaining the dispersion on the surface of the eye for a longer period.
Stability upon sterilization
The developed ATS-SLNs were stable on autoclaving with no significant change (p < 0.05) in entrapment efficiency and particle size (Table 7). Sterilization is an important consideration in ocular delivery. Terminal sterilization is usually preferred as other methods involve post-sterilization handling, which requires trained personnel and aseptic handling, both of which add to the production cost. The achieved autoclavability of the developed system could be due to the selected components, viz. P 188, P90H, and PEG 400. These components change their performance with temperature and hence are harmonious to steam sterilization [102]. PEG being a plasticizer increases the rigidity of the system facilitating stability to autoclaving and resists gelation [80]. Lipidic particles are established to be physically stable to autoclaving conditions. Capacity to resist change in particle size and entrapment efficiency after autoclaving is especially reported for P 188 [103] and other Phospholipon-based SLNs [35]. SLNs are anticipated to melt during autoclaving followed by their recrystallization upon cooling [104, 105].
Sterility testing
The developed and autoclaved blank formulation did not show any growth of fungus and bacteria until 14 days and were found to be sterile. The positive control and validation test confirmed the suitability of media and absence of any inhibitory activity of the formulation.
Corneal permeability studies
ATS-SLNs showed a significant improvement in apparent permeation coefficient (Papp) (2.2-fold), total amount permeated (1.6-fold), and percentage drug permeated (1.8-fold) at 4 h (Fig. 8) as compared with aqueous suspension of free ATS taken as control (Supplementary Table 3). Steady-state flux value increased by 2.5 times in comparison with ATS-SUS. ATS-SLNs showed significantly greater percentage permeation (Fig. 8) than ATS-SUS, at all times (p < 0.05).

Percentage amount of ATS-SLNs and ATS-SUS permeated through the porcine cornea at various time intervals (n = 6)
The near normal corneal hydration (75–80%) confirmed that the ATS-SLNs did not damage the corneal endothelium or epithelium [106].
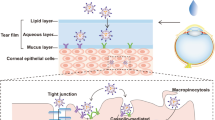
Since the target tissue for AMD is the macula, which is part of the retina, the topically applied drug should traverse the cornea and aqueous and vitreous humor to reach the retina. Corneal epithelium (which consists of several layers of cells and tight junctions created by cell adhesion proteins) acts as a major barrier to the drug entry into the eye. Significant improvement in flux (2.5 times) and total amount of ATS permeated (3.33 μg/min/cm2; 584 μg) when applied as ATS-SLNs versus ATS-SUS (1.33 μg/min/cm2; 373 μg) indicate improved permeation, attributed to the nanosize (256 nm) and composition of SLNs. Intercellular pore size of corneal epithelium is in the range of 200–300 nm, indicating the feasibility of paracellular transport of developed ATS-SLNs. Since the outermost (6–7 cells thick) epithelial layers of the cornea are lipophilic in nature, so free ATS which is also lipophilic showed significant permeation even when applied as ATS-SUS.
It may be noted that the ATS-SUS was prepared in 0.2% w/v sodium carboxymethyl cellulose used as a dispersing agent, and the latter has a strong mucoadhesive power which will result in prolonged retention of ATS-SUS in cul-de-sac of the eye [107].
Establishing safety of ATS-SLNs
Cytotoxicity studies
No statistically significant (p > 0.05) cytotoxicity of ATS-SLNs was observed (Supplementary Fig. 3) when administered to HCLE (at concentrations 1.5, 3, and 6 μg/ml); R28 (at concentrations 1.5 and 3 μg/ml); RCE (at concentrations 1, 100, and 1000 ng/ml); and ARPE-19 (1, 100, and 1000 ng/ml) cell lines after incubation for 24 h.
However, statistically significant (p < 0.01) cell viability loss was found at a high concentration of 6 μg/ml of ATS-SLNs in R28 cell lines. It may however be noted that the test concentration of 6 μg/ml is much more than the maximum concentration (Table 8; pharmacokinetic study discussed subsequently) of drug achieved in vivo, in aqueous (1.11 ± 0.16 μg/ml) and vitreous humor (1.43 ± 0.22 μg/ml), respectively, following topical administration.
In vivo safety studies
The score for both (i) acute dermal irritation/corrosion study [108] (Supplementary Table 4) and (ii) acute eye irritation [109] (Supplementary Table 5) was 0, thus demonstrating a non-irritant nature of developed ATS-SLNs and confirming them to be safe for ocular use.
Repeat instillation study was performed in view of the fact that AMD and related pathologies require lifelong treatment and will need daily instillation of statin formulations for effective control of the disease. Similar studies have been reported by us earlier [35, 110]. The score obtained from this study was also 0 (Supplementary Table 6) and proves the system to be safe for repetitive ocular use.
Similarly aggressive therapy, viz. chronic repeat instillation (5 times at 5-min interval), for a period of 1 week was also evaluated, and the SLN formulation was again found to be safe (Supplementary Table 7) for chronic ocular use. Safety was also expected as all the ingredients employed for preparing the SLN formulation are biodegradable, biocompatible, and safe for ocular use [66] at the employed concentrations.
Ocular tolerance evaluation
Corneal, retinal, and conjunctival cross sections post ATS-SLN administration were observed microscopically to investigate its influence on cell structure and tissue integrity. Results confirm that application of ATS-SLNs to the eye did not compromise tissue integrity in anyway. No inflammation or damage was observed in the retina and sclera. The developed formulation left the tissue structure and integrity visibly unaffected (Supplementary Fig. 4). Results confirm good ocular biocompatibility of the developed formulation.
Cytotoxicity studies on corneal and retinal cell lines confirmed the biocompatibility of the developed system even for exposures as long as 24 h. Corneal hydration rates obtained (≥ 80%) in the ex vivo corneal permeation studies also match with those (76–80%) claimed to be normal [106], establishing safety of the formulation. Furthermore, the developed SLNs were also evaluated for the ocular tolerance in rabbits. It is reported in literature that the rabbit eye is more sensitive than the human eye and requires a longer epithelial repair time [111]. Thus, it is rational to expect an even better tolerance by the human eye [71] for the developed SLN formulation. The three-tier (in vitro, ex vivo, and in vivo) data obtained presently helps to conclude safety of the developed formulation.
Residence time
Optical images of fluid withdrawn from corneal surface post instillation of F-SLN drops showed (Fig. 9) fluorescence and/or intact fluorescent SLNs up to 7 h, while corresponding free fluorescein solution (F-SOL) failed to exhibit any fluorescence within 1.5 h of topical instillation. The longer residence of SLNs on the eye may be attributed to their mucus penetration effects. Mucus is the first protective barrier which checks entry of any foreign particles including bacteria into the eye, by entrapping them into luminal mucus layer (LML). The latter is rapidly cleared, preventing contact of such particles with the corneal epithelium. It may thus be said that mucoadhesive particles including nanoparticles will be eliminated from the eye as they are largely immobilized in the LML while mucus-penetrating particles quickly pass through the LML to reach the underlying adherent mucus which is cleared slowly. P 188–based nanosized mucus-penetrating particles (MPPs) of loterprednoletabonate (LE) showed higher therapeutic drug levels in the posterior eye in comparison with conventional nanoparticles of LE [112]. Particles developed by us contain PEG 400 in addition to P 188, and both these polymers have been established as effective mucopenetrating agents [113]. Low molecular weight PEGs are especially studied for their mucus-penetrating effects [114]. P 188 (poly ethylene glycol (PEG)-poly propylene oxide (PPO)-PEG; known as pluronics) can coat lipidic particle surface by adsorption of the hydrophobic PPO segments, leaving a dense brush of uncharged, hydrophilic segments protruding from the particle surface [80]. The latter can help these particles to swim through the mucus and reach the corneal surface. Since the particles were observed on the corneal surface up to 7 h, hence, it may be said that they penetrated the mucus layer to reach and interact with the corneal surface.

Fluorescent images (100x) of lacrimal fluid samples collected at different time points following topical instillation of F-SLNs and F-SOL in the rabbit eye
Cellular uptake
At 2-h post-incubation of F-SLNs (Fig. 10a), at a concentration of 3 μg/ml with ARPE-19 cells, a micrometric granular pattern and perinuclear finer fluorescence were observed around and in the cells. Free fluorescein solution (Fig. 10b) treatment did not show fluorescence in or near the cells. Entry of smaller SLNs (50–100 nm, approx. 10% of total) through the cell membrane to reach the nuclear membrane could explain the perinuclear fluorescence. The granular pattern on the surface of the cells could be due to the bigger particles (> 100 nm; 90%) which do not permeate (Supplementary Fig. 2 particle size distribution) easily. These particles, however, either (i) may undergo erosion to smaller particles before their uptake or (ii) may release ATS which can permeate into cells in association with smaller particles. All particles however probably produce some permeability changes. This fact is supported by an enhanced UV-induced apoptosis (as discussed subsequently in the apoptosis study) observed with blank SLNs.

Confocal images of ARPE-19 cells incubated with F-SLNs (a) and free fluorescein solution (F-SOL) (b). Nuclei and cytoskeleton were stained with DAPI and TRITC-labeled phalloidin, respectively
Effect of ATS-SLNs on apoptosis
This study was designed to assess if ATS-SLNs exert apoptosis and hence induce anti-angiogenesis in the retinal and corneal cells. The latter is the proposed mechanism of action of commonly used AMD medication, viz. the anti-VEGF antibodies. Anti-angiogenic effects of statins were mostly achieved at high concentrations (> 0.1 μmol/l atorvastatin) [123]. UV-irradiated ARPE-19 cells showed a viability loss of about 73% with respect to non-irradiated cells (Fig. 11). This was increased to approx. 93% upon treatment with blank SLNs (100 and 1000 ng/ml). However, the effect was completely abrogated and there was a trend (even if not significant) of ATS-SLNs to counteract the toxic effect of UV. Free ATS (ATS-SUS) did not show any effect both on viability and on apoptosis (Fig. 12). UV-irradiated RCE cells showed a viability loss of about 85% with respect to non-irradiated cells, while no differences were seen following treatments (data not shown). However, no change in viability was observed when antimycin was used instead of UV irradiation as an apoptotic stimulus, both in ARPE-19 and in RCE cells (data not shown).

ARPE-19 cells were treated with 60 mJ/cm2 UVC in the presence of test drugs. After 48 h, viability was evaluated by WST-1 reagent. Data are reported as % of U taken as 100%. C, not irradiated, untreated control; U, irradiated, untreated control. Statistical analysis was performed by the Student t test (mean of three experiments)

ARPE-19 cells were irradiated with 60 mJ/cm2 UVC in the presence of test drugs (2 h pretreatment with 103 ng/ml) and apoptosis recorded using time-lapse apparatus. C, not irradiated, untreated control; U, irradiated, untreated control
Apoptosis was demonstrated to be responsible for UV-induced viability loss by the time-lapse (Fig. 12), FACS (Supplementary Fig. 5), and cytochrome C release (Supplementary Fig. 6) analyses. Time-lapse experiments showed the ability of blank SLNs to enhance the apoptotic effect of UV at times beyond 24 h. SLNs probably produce some permeability changes in ARPE-19 cells, which enhance UV-induced apoptosis. It is reported that opening of permeability transition pore (PTP) in a mitochondrial cell membrane leads to apoptosis [115]. Induced apoptotic effect is however counteracted in the ATS-SLN group. ATS was reported to inhibit PKC inhibitor–induced apoptosis, in adult rat cardiac myocytes [116,117,118].
This experiment indicates that ATS has no apoptotic effect on ARPE-19 cells (Figs. 11 and 12) and RCE cells.
More detailed investigations, which were out of the scope of presently planned studies, are thus indicated to establish the mechanisms by which ATS can treat AMD symptoms. The anti-inflammatory and anti-oxidant effects are also highly indicated.
Pharmacokinetic study
The concentration–time profiles and corresponding pharmacokinetic parameters of ATS-SLN and ATS-SUS after topical administration in aqueous and vitreous humor are shown in Fig. 13 and Table 8, respectively. A higher Cmax (1.6-fold) and AUC (7.0 fold) for ATS-SLNs with respect to the ATS-SUS was noted in aqueous humor. Similarly, 3.0-fold and 11.7-fold higher effects in vitreous humor were observed for Cmax and AUC, respectively. Significantly higher ocular delivery potential of ATS-SLNs in comparison with free ATS may be attributed to improved transcorneal and conjunctival uptake of ATS-SLNs because of their nanosize and penetration enhancing effect of Compritol® 888 ATO and P 188 [69]. Conjunctival pathway is now identified as the key pathway for posterior eye delivery [52]. Prolonged precorneal retention, mucus penetration and improved permeation, and probable p-gp efflux inhibition due to the presence of PEG 400 and P 188 in the ATS-SLNs are the several reasons assigned to improved ocular bioavailability observed with the latter. Similar improvements in concentration of cyclosporine A [119] and gatifloxacin [120] were reported in aqueous and vitreous humor following topical administration of their respectively loaded SLNs.

Comparative concentration–time profile of ATS-SLNs and ATS-SUS in aqueous (a) and vitreous (b) humor following topical application to the rabbit eye
Peak effect was obtained within 60 min of instillation of the last drop of SLNs (five 30-μl drops of ATS-SLNs were instilled into each eye at 5-min intervals) while free ATS showed a peak effect at 30 min for aqueous humor and 60 min for vitreous humor. Early Tmax of ATS-SUS may be due to the strong mucoadhesive nature of sodium carboxymethyl cellulose used as suspending/dispersing agent and the intrinsic lipophilic nature of ATS (log P = 4.41). However, a significantly higher Cmax and a sustained effect (AUC 700% and 1170% in aqueous and vitreous humor) of up to 48 h (versus 6 h for ATS-SUS) observed for ATS-SLNs as also indicated by the extended MRT of 3–3.5-fold, demonstrate the effectiveness of ATS-SLNs. Furthermore, elimination rate constant (Kel) of developed SLN formulation was found to decrease by 3.5 times re-establishing a prolonged effect.
The protective covering provided by SLNs to ATS not only improves its stability to oxidation and photodegradation but probably also prevents its in vivo metabolism [29]. T1/2 of ATS in plasma is reported to be 14 h. However, ATS-SUS exhibited a T1/2 of 2.6 h and 4.7 h respectively in aqueous and vitreous humor. Anti-fungals like ketoconazole, fluconazole, and amphotericin B are also reported to show a higher T1/2 in plasma versus that exhibited in ocular fluids (as the T1/2 depends on the dose, dosage form, and route of administration) [121]. However, the T1/2 was significantly increased (by 3–4 times) to 9.2 and 14.4 h in aqueous and vitreous humor, respectively, following administration of ATS as ATS-SLNs. Furthermore, a sustained concentration of about 250 ng was maintained in the vitreous humor from 12 to 48 h while the value was negligibly low at ≥ 6 h with ATS-SUS. Since the macula is the major target for control of AMD, hence, higher and better sustained concentration of ATS in vitreous following topical application of ATS-SLNs is a highly promising indication. Vitreous humor is formed and removed at a much slower rate (0.45 μg/day) than aqueous humor (2.4 ± 0.6 μl/min), such that it may be assumed that the limiting step for any drug delivery system is to reach the vitreous humor and once that is achieved, it may accumulate in the vitreous humor to a larger extent than in the aqueous humor [51]. However, viscous nature of the vitreous humor can at times limit the access of the carrier system to the posterior part of the eye including the retina and macula. SLNs prepared by us comprise PEG and P 188, both of which are expected to help these SLNs to stream through the vitreous humor due to their neutral surface and mucus-penetrating nature [112].
Literature demonstrates that only 50–80 ng/ml of ATS is found in human plasma and/or serum after oral administration of 40 mg of ATS [122]. For lipophilic drugs, it is indicated that only one-third of the drug in plasma can reach the aqueous and vitreous humor of the eye. Based on this, not more than or equal to 30 ng/ml of ATS is expected to reach the ocular fluids following its oral administration. In contrast, topical administration of 150 μl (equivalent to 375 μg ATS) of ATS-SLNs administered presently to rabbits achieved 1430 ng/ml of ATS in vitreous humor and 1110 ng/ml in aqueous humor which is around 30 times higher than the reported plasma concentrations or 48 times more than the expected ocular concentration. Localized concentration of ATS in vitreous humor will ensure minimal systemic side effects, viz. constipation, flatulence, dyspepsia, and abdominal pain associated with use of ATS. Furthermore, incidence of severe rhabdomyolysis (which at times may be fatal) in a significant patient population leads to discontinuation of oral ATS therapy. Such side effects will be significantly reduced while better therapeutics will be achieved following ocular application of ATS-SLNs.
Proof-of-concept studies with F-SLNs
Histological sections showed diffuse fluorescence in the vitreous humor, retina, sclera, and choroid of the rat eye, post single-drop instillation of the fluorescein sodium–labeled SLNs. Intact particles were also observed in these tissues as indicated by white arrows in Fig. 14.

Fluorescent images of different tissues of the eye (× 100)
Rows 1, 2, and 3 show the sections of the cornea, vitreous humor, and posterior tissues prepared 0.5-h, 1-h, and 2-h post-application, respectively. Red arrows indicate different layers of tissues and white arrows show presence of intact and fluorescing SLNs.
A representative illustration of confocal microscopy (Fig. 15), after 1-h application, substantiates penetration potential of the developed SLN formulation. Significant higher fluorescence was observed in the cornea, sclera, choroid, and retina including the RPE and vitreous humor in comparison with free fluorescein.

Confocal images 1 h after topical application
It is generally observed that a topically dosed drug can transport to the posterior segment from ocular surface through (1) the transcorneal route which can be divided further into (a) transvitreous route—where drug diffuses through the cornea, enters the vitreous cavity, and distributes to the back of the eye; (b) the uvea–scleral route where drug diffuses through the cornea, penetrates the aqueous humor, and continues through the uvea–sclera where it can gain access to the choroid and retina; and the (2) periocular route (conjunctival uptake), where diffusion occurs around the sclera and drugs are absorbed through the sclera [84, 120]. Presence of intact particles observed in the sclera, cornea, retina, and vitreous humor indicates that transport of developed SLNs occurs not only through corneal pathways (Fig. 14) but also via conjunctival/scleral route due to their small size and charge free surface. Higher permeation of ATS-SLNs may also be attributed to PEG-modified surface (both PEG 400 and PEG blocks of P 188) of developed SLNs. The latter have a capability to solubilize cell membrane due to their solubility in both polar and non-polar materials [107]. Presence of ATS-SLNs (even after 6 h) at precorneal site (Fig. 9) indicates their prolonged retention and/or mucus-penetrating nature. Presence of SLNs in the STF withdrawn by washing the rabbit corneal surface may be attributed to the forced blinking (5–6) of the rabbit eye induced by us prior to each sampling point, as this will detach the mucus entangled particles adhering to the corneal surface. Longer retention of presently developed nanosystem was also confirmed by presence of significant fluorescence in the cornea even after 2 h of administration (Fig. 15). Furthermore, the capacity of developed SLNs to reach the vitreous humor, retina, and RPE in intact form (as seen from fluorescence and confocal microscopy) is complemented with higher achieved therapeutic concentration of ATS (as seen from vitreous pharmacokinetic study). Cellular uptake of these particles was also confirmed in the in vitro studies (Fig. 10).
Conclusions
AMD and heart disease (where statins are extensively prescribed) share their pathology and risk factors. Ophthalmologists are trying to establish such off-label roles of orally administered statins in control of AMD. ATS-loaded SLNs were successfully developed and optimized [105] presently, so as to provide enhanced permeation across cornea and higher availability of ATS in both the aqueous and vitreous humor. Further, the localized concentration of ATS in vitreous humor will ensure minimal systemic adverse events. Though the apoptotic effect of ATS could not be demonstrated presently, more detailed cellular and molecular studies at different doses would be required to properly delineate the mechanism of action of ATS for the control of AMD, which was outside the scope of the present study. Enough scientific evidence exists in favor of use of ATS for posterior eye diseases including AMD and diabetic retinopathy.
Established safety in corneal and retinal cell lines followed by in vivo acute and subchronic toxicity studies in rabbits coupled with their autoclavability and aqueous nature, ascertains the suitability of developed system for ocular use. Presence of developed F-SLN intact and/or higher diffuse fluorescence in the posterior eye including the retina following topical administration as drops, gives the direct evidence of their efficient target ability. Thus, the presently developed SLN system has special application for treatment of posterior eye diseases including AMD and diabetic retinopathy which are otherwise catered to by the invasive routes of administration of the active. The painful therapy compromises the quality of life of patients and may lead to depression. Therefore, availability of non-invasive treatment option is an unmet clinical need of high relevance for these posterior eye diseases. Presently reported system has a versatility to incorporate any other active of significance to the posterior eye.
Change history
03 May 2020
In the original version of this article Ana Guzman-Aranguez���s name appeared incorrect.
Notes
Five 30-μl drops at 5-min intervals (150 μl) were instilled into each eye. Time of instilling last drop was taken as 0 time.
References
James CB, Donald SF, Mark P, Joshua DS. Clinical update: statins and the eye: making sense of the data. Eyenet. 2013:1–3.
Peter G, Tianjing L, Elham H. Statins for age-related macular degeneration. Cochrane Database Syst Rev. 2016;4. https://doi.org/10.1002/14651858.CD006927.
Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. 2014;121:2081–90.
Rosenfeld PJ, Brown DM, Heier JS, MARINA Study Group. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1419–31.
Reich SJ, Fosnot J, Kuoki A. Small interfering RNA (siRNA) targeting VEGF effectively inhibits ocular neovascularization in a mouse model. Mol Vis. 2003;9:210–6.
Brown DM, Kaiser PK, Michels M. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1432–44.
Schmidt-Erfurth UM, Richard G, Augustin A, Aylward WG, Bandello F, Corcòstegui B, et al. Guidance for the treatment of neovascular age-related macular degeneration on behalf of the European Society for Retina Specialists’ Guidelines Committee (EURETINA). Acta Ophthalmol. 2007;85:486–94.
Polat O, İnan SO, Doğan M, Küsbeci T, Yavaş GF, İnan UU, et al. Factors affecting compliance to intravitreal anti-vascular endothelial growth factor therapy in patients with age-related macular degeneration. Turk J Ophthalmol. 2017;47(4):205–10.
Fangueiro JF, Calpena AC, Clares B, Andreani T, Egea MA, Veiga FJ, et al. Biopharmaceutical evaluation of epigallocatechin gallate-loaded cationic lipid nanoparticles (EGCG-LNs): in vivo, in vitro and ex vivo studies. Int J Pharm. 2016;502(1):161–9. https://doi.org/10.1016/j.ijpharm.2016.02.039.
Hirani A, Grover A, Lee YW, Pathak Y, Sutariya V. Triamcinolone acetonide nanoparticles incorporated in thermoreversible gels for age-related macular degeneration. Pharm Dev Technol. 2016;21(1):61–7. https://doi.org/10.3109/10837450.2014.965326.
Sivaprasad S, Bailey TA, Chong VN. Bruch’s membrane and the vascular intima: is there a common basis for age-related changes and disease? Clin Exp Ophthalmol. 2005;33:518–23.
Yip JL, Khawaja AP, Chan MP, Broadway DC, Peto T, Tufail A, et al. Cross sectional and longitudinal associations between cardiovascular risk factors and age related macular degeneration in the EPIC-Norfolk eye study. PLoS One. 2015;10(7):e0132565.
Curcio CA, Millican CL, Bailey T, Kruth HS. Accumulation of cholesterol with age in human Bruch’s membrane. Invest Ophthalmol Vis Sci. 2001;42:265–74.
Wilson HL, Schwartz DM, Bhatt HR. Statin and aspirin therapy are associated with decreased rates of choroidal neovascularization among patients with age-related macular degeneration. Am J Ophthalmol. 2004;137:615–24.
Maguire MG, Ying G, McCannel CA, Liu CH, Dai Y. Statin use and the incidence of advanced age-related macular degeneration in the complications of age-related macular degeneration prevention trial. Ophthalmology. 2009;116:2381–5.
Mona FA, Barbara AM, Azza BR. Statins for prevention of diabetic-related blindness: a new treatment option? Exp Rev Ophthalmol. 2011;6(3):269–72.
Hindler K, Cleeland CS, Rivera E, Collard CD. The role of statins in cancer therapy. Oncologist. 2006;11:306–15.
Mach F. Statins as immunomodulators. Transpl Immunol. 2002;9:197–200.
Hall NF, Gale CR, Syddall H, Phillips DI, Martyn CN. Risk of macular degeneration in users of statins: cross sectional study. Br Med J. 2001;323:375–6.
Guymer RH, Chiu AW, Lim H, Baird PN. HMG CoA reductase inhibitors (statins): do they have a role in age related macular degeneration? Surv Ophthalmol. 2005;50:194–206.
Yamada K, Sakurai E, Itaya M, Yamasaki S, Ogura Y. Inhibition of laser-induced choroidal neovascularization by atorvastatin by downregulation of monocyte chemotactic protein-1 synthesis in mice. Invest Ophthalmol Vis Sci. 2007;48:1839–43.
Demetrios GV, Anthony BD, Zoi GK, Jeremy WG, Emmanuel G, John IL, et al. Regression of some high-risk features of age-related macular degeneration (AMD) in patients receiving intensive statin treatment. EBioMedicine. 2016;5:198–203.
Gehlbach P, Li T, Hatef E. Statins for age-related macular degeneration. Cochrane Database Syst Rev. 2015;2:1–12. https://doi.org/10.1002/14651858.CD006927.pub4.
Ma L, Wang Y, Du J, Wang M, Zhang R, Fu Y. The association between statin use and risk of age-related macular degeneration. Sci Rep. 2015;5:18280.
Cannon CP, Braunwald E, McCabe CH. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–504.
Khush KK, Waters D. Lessons from the PROVE-IT trial. Higher dose of potent statin better for high-risk patients. Cleve Clin J Med. 2004;71:609–16.
Nissen SE. Effect of intensive lipid lowering on progression of coronary atherosclerosis: evidence for an early benefit from the reversal of atherosclerosis with aggressive lipid lowering (REVERSAL) trial. Am J Cardiol. 2005;96:61–8.
Nissen SE, Nicholls SJ, Sipahi I. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295:1556–65.
Davidson MH, Robinson JG. Lipid-lowering effects of statins: a comparative review. Expert Opin Pharmacother. 2006;7:1701–14.
Fong CW. Statins in therapy: understanding their hydrophilicity, lipophilicity, binding to 3-hydroxy-3-methylglutaryl-CoA reductase, ability to cross the blood brain barrier and metabolic stability based on electrostatic molecular orbital studies. Eur J Med Chem. 2014;85:661–74.
Lam MW, Mabury SA. Photodegradation of the pharmaceuticals atorvastatin, carbamazepine, levofloxacin, and sulfamethoxazole in natural waters. Aquat Sci. 2005;67:177–88.
Joshi L. Stabilized pharmaceutical composition comprising an HMG-COA reductase inhibitor. US Patent publication no 2009/0247603A1. 1993.
Yadav M, Ahuja M. Preparation and evaluation of nanoparticles of gum cordia, an anionic polysaccharide for ophthalmic delivery. Carbohydr Polym. 2010;81:871–7.
Bhatia A, Singh B, Raza K, Wadhwa S, Katare OP. Tamoxifen-loaded lecithin organogel (LO) for topical application: development, optimization and characterization. Int J Pharm. 2013;444:47–59.
Kakkar S, Karuppayil SM, Raut JS, Giansanti F, Papucci L, Schiavone N, et al. Lipid-polyethylene glycol based nano-ocular formulation of ketoconazole. Int J Pharm. 2015;495:276–89.
Kakkar V, Kaur IP, Kaur AP, Saini K, Singh KK. Topical delivery of tetrahydrocurcumin lipid nanoparticles effectively inhibits skin inflammation: in vitro and in vivo study. Drug Dev Ind Pharm. 2018;44:1701–12.
Aggarwal D, Garg A, Kaur IP. Development of a topical niosomal preparation of acetazolamide: preparation and evaluation. J Pharm Pharmacol. 2004;56:1509–17.
Li X, Nie SF, Kong J, Li H, Ju CY, Pan WS. A controlled-release ocular delivery system for ibuprofen based on nanostructured lipid carriers. Int J pharm Res. 2008;363:177–82.
IP. 2211 Sterility. 2007;1 (The Indian Pharmacopoeia Commission, Ghaziabad, India):52–9.
Gipson IK, Spurr-Michaud S, Argüeso P, Tisdale A, Ng T. Mucin gene expression in immortalized human corneal-limbal and conjunctival epithelial cell lines. Invest Ophthalmol Vis Sci. 2003;44:2496–506.
McDermott AM, Baidouri H, Woodward AM, Kam WR, Liu Y, Chen X, et al. Short tandem repeat (STR) profiles of commonly used human ocular surface cell lines. Curr Eye Res. 2018;43:1097–101.
Seigel GM. Review: R28 retinal precursor cells: the first 20 years. Mol Vis. 2014;20:301–6.
Loma P, Guzman-Aranguez A, Lara MJPD, Pintor J. Diadenosine tetraphosphate improves adrenergic anti-glaucomatous drug delivery and efficiency. Exp Eye Res. 2015;134:141–7.
Pastor-Clerigues A, Serrano A, Milara J, Marti-Bonmati E, Lopez-Perez FJ, Garcia-Montanes S, et al. Evaluation of the ocular tolerance of three tacrolimus topical pharmaceutical preparations by bovine corneal opacity and permeability test. Curr Eye Res. 2016;41(7):890–6.
Margheri F, Manetti M, Serratı S, Nosi D, Pucci M, Matucci-Cerinic M, et al. Domain 1 of the urokinase-type plasminogen activator receptor is required for its morphologic and functional, beta2 integrin-mediated connection with actin cytoskeleton in human microvascular endothelial cells: failure of association in systemic sclerosis endothelial cells. Arthritis Rheum. 2006;54:3926–38.
Papucci L, Schiavone N, Witort E, Donnini M, Lapucci M, Tempestini A, et al. Coenzyme Q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J Biol Chem. 2003;278:28220–8.
Paul MC, Ruth EH, Richard F, Augusto AB. The effect of statins on intraocular pressure and on the incidence and progression of glaucoma: a systematic review and meta-analysis. Invest Ophthalmol Vis Sci. 2016;57:2729–48.
McGwin G, Xie A, Owsley C. The use of cholesterol-lowering medications and age-related macular degeneration. Ophthalmology. 2005;112:488–94.
Jung HJ, Chauhan A. Extended release of timolol from nanoparticle-loaded fornix insert for glaucoma therapy. J Ocul Pharmacol Ther. 2013;29(2):229–35.
María DS, Paula S, Marcelo AA, Carlos EL, Laura M. Plasma and ocular prednisolone disposition after oral treatment in cats. Biomed Res Int. 2013:1–14.
Djebli N, Khier S, Griguer F, Coutant AL, Tavernier A, Fabre G, et al. Ocular drug distribution after topical administration: population pharmacokinetic model in rabbits. Eur J Drug Metab Pharmacokinet. 2016;42:59–68.
Agrahari V, Agrahari V, Mandal A, Pal D, Mitra AK. How are we improving the delivery to back of the eye? Advances and challenges of novel therapeutic approaches. Expert Opin Drug Deliv. 2016;1:1–17.
Choudharya A, Rana AC, Aggarwal G, Kumar V, Zakira F. Development and characterization of atorvastatin solid dispersion formulation using skimmed milk for improved oral bioavailability. Acta Pharm Sin B. 2012;2:421–8.
Mahmoodzadeh E, Vahdati S, Hashemzadeh N, Jouyban A. Solubility of atorvastatin calcium in water-ethanol mixture: solution mediated phase transformation. Res Pharm Sci. 2012;7.
Kumar AN, Deecaraman M, Rani C, Mohanraj KP, Venkateskumar KN. Formulation development and in vitro evaluation of nanosuspensions loaded with atorvastatin calcium. Asian J Pharmacol. 2010;4:28–33.
Chandiran S, Anandakirouchenane E. Enhancement of solubility of atorvastatin calcium by nanosuspension technique. Int J Biopharm. 2014;5:214–7.
Anwar M, Warsi MH, Mallick N, Akhter S, Gahoi S, Jain GK, et al. Enhanced bioavailability of nano-sized chitosan–atorvastatin conjugate after oral administration to rats. Eur J Pharm Sci. 2011;44:241–9.
Tian Q, Ding F, Guo L, Wang J, Wu F, Yu Y. Targeted solid lipid nanoparticles with peptide ligand for oral delivery of atorvastatin calcium. RSC Adv. 2016;6:35901.
Sripriyalakshmi S, Anjali CH, George P, Doss C, Rajith B, Aswathy R. BSA nanoparticle loaded atorvastatin calcium - a new facet for an old drug. PLoS One. 2014;9:e86317.
Kumar PP, Gayatri P, Reddy S, Somagoni J, Rao YM. Atorvastatin loaded solid lipid nanoparticles: formulation, optimization, and in - vitro characterization. IOSR J Pharm. 2012;2(5):23–32.
Yu LM, Lu JJ, Chiu IW, Leung KS, Chan YW, Zhang L, et al. Poly (ethylene glycol) enhances the surface activity of a pulmonary surfactant. Colloids Surf B Biointerf. 2004;36:167–76.
Carvalho A, Lopes I, Goncalves O, Bárbara E, Elisabete M, Oliveira R, et al. Polymeric versus lipid nanoparticles: comparative study of nanoparticulate systems as indomethacin carriers. J Appl Solution Chem Model. 2015;4.
Sharma N, Parshotam M, Senshang L. Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: a co-surfactant study. Asian J Pharmacol. 2016;11:404–16.
Paliwal R, Babu RJ, Palakurthi S. Nanomedicine scale-up technologies: feasibilities and challenges. AAPS. 2014;15:1527–34.
FDA. https://www.accessdata.fda.gov/scripts/cder/iig/getiigWEB.cfm, acesssed 26.04.2017.
Seyfoddin A, Shaw J, Al-Kassas R. Solid lipid nanoparticles for ocular drug delivery. Drug Deliv. 2010;17:467–87.
Souto EB, Mehnert W, Müller RH. Polymorphic behaviour of Compritol1 888 ATO as bulk lipid and as SLN and NLC. J Microencapsul. 2006;23:417–33.
Jiao J. Polyoxyethylated nonionic surfactants and their applications in topical ocular drug delivery. Adv Drug Deliv Rev. 2008;60:1663–73.
Kaur IP, Smitha R. Penetration enhancers and ocular bioadhesives: two new avenues for ophthalmic drug delivery. Drug Dev Ind Pharm. 2002;28:353–69.
Liu M, Zhang J, Shan W, Huang Y. Developments of mucus penetrating nanoparticles. Asian J Pharmacol. 2015;10:275–82.
Gokce EH, Sandri G, Egrilmez S, Bonferoni MC, Guneri T, Caramella C. Cyclosporine A-loaded solid lipid nanoparticles: ocular tolerance and in vivo drug release in rabbit eyes. Curr Eye Res. 2009;34:996–1003.
Attama AA, Reichl S, Müller-Goymann CC. Diclofenac sodium delivery to the eye: in vitro evaluation of novel solid lipid nanoparticle formulation using human cornea construct. Int J Pharm Res. 2008;355:307–13.
Suzuki M, Kamei M, Itabe H, Yoneda K, Bando H, Kume N, et al. Oxidized phospholipids in the macula increase with age and in eyes with age-related macular degeneration. Mol Vis. 2007;13:772–8.
Judde A, Villeneuve P, Rossignol-Castera A, Le Guillou A. Antioxidant effect of soy lecithins on vegetable oil stability and their synergism with tocopherols. J Am Oil Chem Soc. 2003;80:1209–15.
Zhang L, Mao S. Application of quality by design in the current drug development. Asian J Pharmacol. 2017;12:1–8.
Singh B, Chakkal SK, Ahuja N. Formulation and optimization of controlled release mucoadhesive tablets of atenolol using response surface methodology. AAPS. 2006;7:19–28.
Liu CH, Wu CT. Optimization of nanostructured lipid carriers for lutein delivery. Colloids Surf A Physicochem Eng Asp. 2010;353:149–56.
Salmaso S, Caliceti P. Stealth properties to improve therapeutic efficacy of drug nanocarriers. J Drug Deliv. 2013;374252.
Lester M, Orsoni GJ, Gamba G, Taffara M, Mangiafico P, Giuffrida S, et al. Improvement of the ocular surface using hypotonic 0.4% hyaluronic acid drops in Keratoconjunctivitis sicca. Eye. 2000;14:892–8.
Maisel K, Reddy M, Xu Q, Chattopadhyay S, Cone R, Ensign LM, et al. Nanoparticles coated with high molecular weight PEG penetrate mucus and provide uniform vaginal and colorectal distribution in vivo. Nanomedicine. 2016;11:1337–43.
Emmett M, Ana CS. Osmotic effects of polyethylene glycol. Gastroenterology. 1988:933–41.
Hackley VA, Clogston JD. Measuring the hydrodynamic size of nanoparticles in aqueous media using batch-mode dynamic light scattering. Methods Mol Biol. 2011;697:35–52.
Shen Y, Tu J. Preparation and ocular pharmacokinetics of ganciclovir liposomes. AAPS. 2007;9:E371–E7.
Kaur IP, Kakkar S. Nanotherapy for posterior eye diseases. J Control Release. 2014;193:100–12.
Sakurai E, Ozeki H, Kunou N, Ogura Y. Effect of particle size of polymeric nanospheres on intravitreal kinetics. Ophthalmic Res. 2001;33:31–6.
Qaddoumi MG, Ueda H, Yang J, Davda J, Labhasetwar V, Lee VHL. The characteristics and mechanisms of uptake of PLGA nanoparticles in rabbit conjunctival epithelial cell layers. Pharm Res. 2004;21(4):641–8.
Calvo P, Alonso MJ, Vila–Jato JL, Robinson JR. Improved ocular bioavailability of indomethacin by novel ocular drug carriers. J Pharm Pharmacol. 1996;48:1147–52.
Alqawlaq S, Sivak JM, Huzil JT, Ivanova MV, Flanagan JG, Beazely MA, et al. Preclinical development and ocular biodistribution of gemini-DNA nanoparticles after intravitreal and topical administration: towards non-invasive glaucoma gene therapy. Nanomed Nanotech Biol Med. 2014;10:1637–47.
Amrite AC, Edelhauser HF, Singh SR, Kompella UB. Effect of circulation on the disposition and ocular tissue distribution of 20 nm nanoparticles after periocular administration. Mol Vis. 2008;14:150–60.
Bhandari R, Kaur IP. Pharmacokinetics, tissue distribution and relative bioavailability of isoniazid-solid lipid nanoparticles. Int J Pharm. 2013;441:202–12.
Monem AS, Ali FM, Ismail MW. Prolonged effect of liposomes encapsulating pilocarpine HCl in normal and glaucomatous rabbits. Int J Pharm. 2000;198:29–38.
Costa P, Manuel J, Lobo S. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–33.
Panghal D, Nagpal M, Thakur GS, Arora S. Dissolution improvement of atorvastatin calcium using modified locust bean gum by the solid dispersion technique. Sci Pharm. 2014;82:177–91.
Xie Y, Li G, Yuan X, Cai Z, Rong R. Preparation and in vitro evaluation of solid dispersions of total flavones of Hippophae rhamnoides L. AAPS. 2009;10:631–40.
Attama AA, Schicke BC, Paepenmuller T, Muller-Goymann CC. Solid lipid nanodispersions containing mixed lipid core and a polar heterolipid: characterization. Eur J Pharm Biopharm. 2007;67:48–57.
Lemarchand C, Couvreur P, Besnard M, Costantini D, Gref R. Novel polyester-polysaccharide nanoparticles. Pharm Res. 2003;20:1284–92.
Karla JM, Marcela A, Rafael VD. Antioxidant capacity of poly (ethylene glycol) (PEG) as protection mechanism against hydrogen peroxide inactivation of peroxidases. Appl Biochem Biotechnol. 2015;177:1364–73.
Baranowski P, Karolewicz B, Gajda M, Pluta J. Ophthalmic drug dosage forms: characterisation and research methods. Sci World J. 2014:1–14.
Tomlinson A, Khanal S, Kanna R, Diaper C, McFadyen A. Tear film osmolarity: determination of a referent for dry eye diagnosis. Invest Ophthalmol Vis Sci. 2006;47:4309–15.
Aragona P, Di Stefano G, Ferreri F, Spinella R, Stilo A. Sodium hyaluronate eye drops of different osmolarity for the treatment of dry eye in Sjogren’s syndrome patients. Br J Ophthalmol. 2002;86:879–84.
Anjana D, Nair KA, Somashekara N, Venkata M, Sripathy R, Yelucheri R, et al. Development of curcumin based ophthalmic formulation. Am J Inf Diseases. 2012;8:41–9.
Sanna V, Kirschvink N, Gustin P, Gavini E, Roland I, Delattre L, et al. Preparation and in vivo toxicity study of solid lipid microparticles as carrier for pulmonary administration. AAPS PharmSciTech. 2003;5(2).
Goke K, Roese E, Arnold A, Kuntsche J, Bunjes H. Control over particle size distribution by autoclaving poloxamer-stabilized trimyristin nanodispersions. Mol Pharm. 2016;13:3187–95.
Muller RH, Runge S, Ravelli V, Mehnert W, Thunemann AF, Souto EB. Oral bioavailability of cyclosporine: solid lipid nanoparticles (SLN) versus drug nanocrystals. Int J Pharm. 2006;317:82–9.
Kaur IP, Yadav M, inventors; An ocular formulation of statins and a method for its preparation 2339/DEL/2014 dated 18/08/14 (published)2014.
Han S, Shen JQ, Gan Y, Geng H, Zhang X, Zhu C, et al. Novel vehicle based on cubosomes for ophthalmic delivery of flurbiprofen with low irritancy and high bioavailability. Acta Pharmacol Sin. 2010;31:990–8.
Liu S, Dozois MD, Chang CN, Ahmad A, Ng DL, Hileeto D, et al. Prolonged ocular retention of mucoadhesive nanoparticle eye drop formulation enables treatment of eye diseases using significantly reduced dosage. Mol Pharm. 2016;13:2897–905.
OECD. Test No. 404: acute dermal irritation/corrosion. OECD guidelines for the testing of chemicals. 2015:1–8.
OECD. Test No. 405: acute eye irritation/corrosion. OECD guidelines for the testing of chemicals. 2017:1–8.
Kakkar S, Kaur IP. Spanlastics—a novel nanovesicular carrier system for ocular delivery. Int J Pharm. 2011;413:202–10.
Wilson SL, Ahearne M, Hopkinson A. An overview of current techniques for ocular toxicity testing. Toxicology. 2015;327:32–46.
Schopf LR, Popov AM, Enlow EM, Elizabeth ME, James LB, Winston ZO, et al. Topical ocular drug delivery to the back of the eye by mucus-penetrating particles. Transl Vis Sci Technol. 2015;4:11.
Ensign LM, Schneider C, Suk JS, Cone R. Mucus penetrating nanoparticles: biophysical tool and method of drug and gene delivery. Adv Mater. 2012;24(28):3887–94.
Li H, Guissi NE, Su Z, Ping Q, Sun M. Effects of surface hydrophilic properties of PEG-based mucus-penetrating nanostructured lipid carriers on oral drug delivery. RSC Adv. 2016;87:84164–76.
Tempestini A, Schiavone N, Papucci L, Witort E, Lapucci A, Cutrì M, et al. The mechanisms of apoptosis in biology and medicine: a new focus for ophthalmology. Eur J Ophthalmol. 2003;13:S11–8.
Sabe AA, Elmadhun NY, Sadek AA, Dalal RS, Chu LM, Bianchi C, et al. Atorvastatin regulates apoptosis in chronically ischemic myocardium. J Card Surg. 2015;30:218–23.
Wang J, Chen H, Zhou Y, Su Q, Liu T, Wang LL. Atorvastatin inhibits myocardial apoptosis in a swine model of coronary microembolization by regulating PTEN/ PI3K/Akt signaling pathway. Cell Physiol Biochem. 2016;38:207–19.
Tanaka K, Honda M, Takabatake T. Anti-apoptotic effect of atorvastatin, a 3-hydroxy-3-methyl glutryl coenzyme a reductase inhibitor, on cardiac myocytes through protein kinase C activation. Clin Exp Pharmacol. 2004;31:360–4.
Basaran E, Demirel M, Sirmagul B, Yazan Y. Cyclosporine-A incorporated cationic solid lipid nanoparticles for ocular delivery. J Microencapsul. 2010;27:37–47.
Kalam AM, Sultana Y, Ali A, Aqil M, Mishra AKCK, Aljuffali IA, et al. Part II: enhancement of transcorneal delivery of gatifloxacin by solid lipid nanoparticles in comparison to commercial aqueous eye drops. J Appl Polym Sci. 2013;101:1828–36.
Felton T, Troke PF. Tissue penetration of antifungal agents. Clin Microbiol Rev. 2014;27(1):68–88.
Mahmoud Y, Ahmad A, Mahmoud A, Tawfiq A. Simultaneous determination of amlodipine and atorvastatin with its metabolites; ortho and para hydroxy atorvastatin; in human plasma by LC–MS/MS. J Chromatogr B. 2013;917:1–10.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All the animal study protocols were approved by the Institutional Animals Ethics Committee (PU, Chandigarh; vide letter no. IAEC/411, dated September 11, 2013), and guidelines as per the Committee for the Purpose of Control And Supervision of Experiments on Animals (CPCSEA) were followed.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of the article has been revised: Ana Guzman-Aranguez's name has been corrected.
Electronic supplementary material
ESM 1
(DOC 2320 kb)
Rights and permissions
About this article

Cite this article
Yadav, M., Schiavone, N., Guzman-Aranguez, A. et al. Atorvastatin-loaded solid lipid nanoparticles as eye drops: proposed treatment option for age-related macular degeneration (AMD). Drug Deliv. and Transl. Res. 10, 919–944 (2020). https://doi.org/10.1007/s13346-020-00733-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-020-00733-4