
Abstract
Nearly 25 years after the discovery of the human immunodeficiency virus type 1 (HIV-1) effective control of the AIDS pandemic remains elusive. At the root of this challenge is the evolution of this virus to elude immune control. Error-prone nature of replication and retro-transcription is the hallmark of this virus. This fidelity of replication in HIV-1 is due to the absence of proof-reading/repair and post-replicative error correction mechanisms that normally operate during replication of DNA viruses. Advances in sequencing technology and expanded disease surveillance have allowed researchers to characterize the variation in HIV-1 around the world and within individual patient overtime. Although HIV-1 has been classified into distinct subtypes, the classification does not reflect dynamic genetic evolution of HIV-1 through which new strains are constantly emerging. The resultant viral diversity has implications for differential rates of disease progression in different geographical areas, differential responses to antiretroviral therapy (including the development of resistance), and vaccine development. In this review evolution of HIV-1 in India is discussed.
Similar content being viewed by others


Introduction
Since the unfolding of the AIDS epidemic in the early 1980s, there has been an increasing interest in the emergence and evolution of infectious diseases. It has become extremely important to investigate the factors that allowed new infections like HIV to appear, or older ones to reappear and then to track their spread through populations. These tracks form part of science of molecular epidemiology.
Traditionally serology has been used to trace the spread of infectious diseases. These days comparative analysis of gene sequence data is being undertaken to study spread of infectious diseases. Meaning thereby, phylogenetic-trees have become an important analytical tool to track the spread of infections through populations. Since DNA sequences provide the most detailed information possible for any organism in evolutionary studies, this information is recognized as an invaluable document of history of life on earth.
There are two types of HIV: a highly virulent global type (HIV-1) and a somewhat less virulent strain HIV-2 found mostly in West Africa. Both these viruses impose major burdens on the health and economic status of many developing countries. Many African monkeys are commonly infected at high frequencies with HIV like viruses known as simian immunodeficiency viruses (SIVs). The SIVs are widespread in a large number of African simian primates where they do not appear to cause disease. Phylogenetic analyses indicate that these SIVs are the reservoirs for the human viruses, with SIVsm from the sooty mangabey monkey the most likely source of HIV-2, and SIVcpz from the common chimpanzee the progenitor population for HIV-1. Sootey mangabey monkeys are likely the direct source of HIV-2 since these are West African monkeys and HIV-2 also is found predominantly in West Africa. On the other hand, since chimpanzees are rarely infected with SIVcpz in the wild they are less likely to be the direct source of HIV-1. However, it is possible that an unknown SIV from other monkey species may be the ancestor of both HIV-1 and SIVcpz. Nonetheless, separation of HIV-1 and HIV-2 on phylogenetic tree suggests that they must have entered human populations on different occasions.
In infected humans the process of HIV evolution takes place at a very fast pace, with the virus continually fixing mutations by natural selection which allows it to escape from host immune responses. In SIV-infected monkeys the pace is not that intense, since a weaker immune response generates less selective pressure on the virus. This difference in virus–host interaction, together with a wide co-receptor usage such that HIV strains are able to infect cells with both CCR5 and CXCR4 chemokine receptors, may be responsible for increased virulence of HIV in humans compared to SIV in other primates.
HIV-1 genetic diversity over time is driven by two factors namely the high error rate of the viral reverse transcriptase and the rapid turn over of HIV-1 in infected individuals. Recombination events, pressures generated by the host immune responses, and antiviral drugs further contribute to differential viral genetic evolution.
Globally circulating strains exhibit an extraordinary degree of genetic diversity, which may influence several aspects of their biology such as infectivity, transmissibility and immunogenicity. Molecular analyses of various HIV isolates reveal sequence variations over many parts of the viral genome. Sequences derived from these HIV-1 strains have historically been classified on the basis of their Phylogenetic relationship. The groups were originally named M (major), O (outlier), and N (non-M, non-O) [23]. The last two groups (N and O) remain essentially restricted to West Africa, whereas the M group comprises a number of viruses that dominate the global AIDS epidemic. Since HIV-1 M group began its expansion in humans roughly 70 years ago, it has diversified rapidly, now comprising a number of different subtypes and circulating recombinant forms (CRFs). Based on the sequence of the envelope glycoproteins, genetic subtypes including CRFs have been identified in group M, whereas subtypes within group O remain unidentified. Subtypes are genetically defined lineages that can be resolved through phylogenetic analysis of the HIV-1 M group viruses as well-defined clades or branches in a tree. CRF describes a recombinant lineage that plays an important role in the HIV-1 epidemic. The CRF members must share an identical mosaic structure, that is, they have descended from the same recombination event/s.
HIV-1 Genetic Subtypes
On the basis of their Phylogenetic relationships, group M viruses have been classified into nine subtypes or clades (A to K; except E and I). Virus strains representing the genetic subtypes E and I have not yet been found. The viruses originally identified as subtype E (the predominant group of viruses involved in heterosexual transmission in Thailand) and I (a small group of viruses from Mediterranean region) are now considered inter-subtype recombinants and have been termed CRF01_AE and CRF04_cpx, respectively.
Disproportionate spread of different lineages of group M viruses has been taken to indicate that specific biological differences may exist among various subtypes. Therefore, the phylogenetic analysis of subtype sequences remains an important molecular epidemiological tool with which we may track the course of group M pandemic. Existence of viral subtypes or clades may be the result of a “founder effect” in which certain variants of the virus become founders of a sub-epidemic because they happen to be involved in an extensive transmission chain. In this scenario, the subtypes may be similar biologically even though they are genetically very different. Alternatively, it is likely that certain characteristics of subtypes allowed them to out-compete less-fit viral variants.
Despite the lack of clear correlation between subtypes and overt biological characteristics, other more subtle phenotypic distinctions have been reported such as the pattern of co-receptors usage. Firstly there is growing evidence that the subtype C has a preponderance of “non-syncytia inducing” viruses which bind to CCR5 receptors in addition to CD4 receptors present on the target cells and lack “syncytia inducing” viruses that use CXC4 and CD4 receptors to infect the target cells. Secondly distinctive RNA secondary structure in the important regulatory domain, TAR, is a property associated uniquely with subtype A and viruses having AE mosaic. Thirdly different subtypes differ in susceptibilities to antiretroviral drugs. In addition, difference between subtypes is reflected in subtype specific pattern of genetic variation. There is an elevated rate of non-synonymous substitution in the third variable loop of subtype D viruses, compared with other subtypes [11].
Most interesting feature of these subtypes is geographic predilection of their distribution worldwide. It is possible that these subtypes may have spread through different populations at different times and by different routes. For example, subtype A is composed of further two subtypes (A1 and A2), both of which appear to have a widespread geographic distribution [5] and is commonly found in sub-Saharan Africa and Russia where it is predominantly transmitted through heterosexual intercourse [1]. It may be one of the oldest of all subtypes. In contrast, subtype B is associated with the HIV epidemic among homosexual men and injecting drug users in North and South Americas, Europe, Japan and Australia. The most prevalent HIV-1 subtype in the global epidemic is subtype C which is dominant in India, Ethiopia, South Africa, Zimbabwe, Botswana and China and is transmitted through heterosexual intercourse. Interestingly HIV-1 viral lineages of O and N are mainly confined to West Africa and phylogenetically these are separated from the other HIV-1 sequences suggesting multiple entry of the viruses into humans.
Subtype C: The Expanding Pandemic
One of the most dramatic changes in the HIV/AIDS has been the rapid emergence and devastating spread of subtype C viruses. HIV-1 C accounts for 56% of all circulating viruses and is the most commonly transmitted subtype worldwide. The subtype C epidemic has now become the most predominant subtype in Southern African countries and Indian subcontinent where HIV prevalence is the highest in the world. The proportionate increase in C viruses relative to other HIV strains suggest that subtype C may be more easily transmitted or that it has a higher level of “fitness” at the population level. One possible explanation is that founder effects relating to the ongoing introduction of subtype C into new population groups with different host factors, or different social and sexual practices, may be responsible for the rapid spread. However, founder and host effects cannot account for the fact that C viruses are overtaking preexisting virus subtypes in several different geographical regions. It is increasingly evident that additional (non-host) viral factors are also contributing to the rapid spread of HIV-1 C.
Viral studies indicate that subtype C has distinct genetic and phenotypic properties that differentiate it from other HIV-1 subtypes. Subtype C viruses have an extra NF-κB binding site in the long terminal repeat [8], a prematurely truncated Rev protein or a 5-amino-acid insertion in Vpu [4] that may influence viral gene expression, altering transmissibility and pathogenesis of C viruses. Factors related to C viral entry and pathogenesis such as CCR5 and non-syncytium-inducing properties of C isolates, may also contribute to the increased spread of C viruses. Interestingly, though both subtype B and C are spreading exponentially in Brazil, the subtype C growth rate is about twice that of subtype B there; thus providing evidence of a different epidemic potential between two HIV-1 subtypes [19].
Geographical Distribution of HIV-1 Subtypes
Epidemiological and Phylogenetic studies have also shown that HIV-1 clades are unequally spreading throughout the world [27]. The HIV epidemic in Africa began in the late 1970s and, during the late 1980s, gradually spread to the South of the continent. Though Africa is considered home of all HIV-1 subtypes, their spread to other continents is attributed to some groups of individuals, particularly travelers, who contribute to the initiation of local epidemics worldwide. These groups include, in particular, immigrants, IV drug users, tourists, truck drivers, military troops and seamen. The global view on the contribution of travel to HIV-1 spread usually derives from the prevalence of non-B subtypes in various countries [25]. The prevalence of non-B infections has indeed markedly increased in recent years in several European countries [2, 25]. Recent immigrants from areas of high HIV-1 endemicity and European travelers have been shown to contribute in large part to the increase in the prevalence of non-B infections in western and northern Europe. There is no doubt that it is high-risk human behavior and not occupation that determines HIV-1 infection risk.
Predominance of subtype B in the western countries including Japan and Australia is attributed to transmission among homosexual men and is generally thought to be spreading separately from that among IV drug users and heterosexual individuals. However, in India subtype C predominance even among homosexual men and IV drug users suggests interplay of host genetic factors and the virus in determining geographical distribution of HIV-1 subtypes.
Among the HIV-1 group M viruses, HIV-1 subtype C is by far the most prevalent HIV in the world and is linked to heterosexual transmission. It was first discovered in North east Africa in the early 1980s [16, 20] and has since moved to the southern parts of Africa. In addition, the subtype C epidemic has spread to East and Central Africa where it is becoming predominant subtype [18, 28]. From Africa, it has spread to India, Brazil and South and Central China where it appears to have been introduced from India [30]. In England and Wales, preponderance of subtype C infections has been observed among HIV-infected heterosexual STI clinic attendees, particularly in younger age groups, suggesting recent acquisition of this viral strain [24].
The Indian Scenario
In India HIV infection was first reported in 1986 in six commercial sex workers in the State of Tamil Nadu and since then it has been reported from all the States and Union Territories. India now holds dubious distinction of accounting second largest number of HIV infections in the world following South Africa. With an estimated 2.5 million people living with HIV infection in adult population (15–49 years) by 2008, India accounts for 13% of global HIV prevalence [17].
Tracking the epidemic and implementing effective programmes is made difficult by the fact that there is no one epidemic in India. Rather, there are several localized sub-epidemics reflecting the diversity in social-cultural patterns and multiple vulnerabilities present in the country. Though the overall national prevalence is low, six states have reached high prevalence (>1%): Manipur, Nagaland, Andhra Pradesh, Tamil Nadu, Karnataka and Maharashtra. Certain districts in Goa and Gujarat have also reported high prevalence.
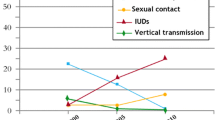
Sexual transmission is driving the AIDS epidemic in India. This route accounts for about 86% of HIV infections in the country. Remaining 14% are accounted for by other routes namely blood transfusion, mother–child transmission and IV drug use, particularly in North East India. Over one-third of all HIV infections occur in young people in the age group 15–24 years.
Early studies have indicated the presence of both HIV-1 and HIV-2 in India [3, 6]. Subsequent studies further emphasized a predominance of subtype C strains in India, which were found to cluster with South African isolates [13, 29]. Other HIV-1 subtypes, A and B, have been reported in India between 1980s and early 1990s among the recipients of blood and blood products and IV drug users, respectively, suggesting multiple introductions of HIV-1 in this country. Subtype A strains were found to be related to Central and East African subtype and subtype B strains obtained from Manipur were related to subtype B sequences circulating in Thailand [13]. However, recent studies have clearly shown that subtype C strains have displaced subtype B in the IV drug users in that part of India [7, 15].
The trends across the country show that there is no explosive HIV epidemic in India as a whole. However, there are serious sub-national epidemics in various parts of the country with rapid spread and evidence of high prevalence of HIV among both Sexual Transmitted Infections (STI) and antenatal clinic attendees in different sites located in States of Andhra Pradesh, Maharashtra, Tamil Nadu, Gujarat, Pondicherry, Assam, Bihar, Chhattisgarh, Delhi, Haryana, Himachal Pradesh, Kerala, Orissa, Goa and Manipur. In high prevalence states the epidemic appears to be spreading gradually from urban to rural areas and from high-risk behavior groups to the general population. The epidemic continues to shift towards women with an estimated 39% of the infected being women [17].
An explosive epidemic driven by intravenous drug use has unfolded in the state of Manipur (North East India) bordering Myanmar and is close to the Golden Triangle composed of Thailand, East Myanmar and West Laos and is the hub of international drug trafficking. A recent study documents two-third of HIV infections in this region of India are caused by subtype C and subtype B (Thai B) accounts for 20% of infections [15]. The presence of multiple subtypes circulating in Manipur suggests the likelihood of recombinant viruses evolving in this region. Indeed, this has been corroborated by a recent study which reported presence of B/C recombinants from this region [26]. Apart from north-eastern states there are also sporadic reports of the presence of A/C and B/C recombinants from West and South India [12, 22].
The occurrence of HIV-1 recombination in nature is borne out by the identification of genomes that are recombinants between different HIV-1 subtypes [14]. Some of these recombinant viruses have become fixed in the human population and are referred to as CRFs, and in at least a few cases CRFs have become the predominant strain in specific geographic areas of infection such as A/E recombinants in Thailand and B/C recombinants in parts of Southeast Asia and China. HIV-1 recombinants are estimated to contribute to 10–40% and 10–30% of the infections in Africa and Asia, respectively. The identification of subtypes and CRFs provides a means of tracking dissemination of the pandemic worldwide.
To delineate the molecular features of HIV-1 strains circulating in India, Phylogenetic analyses of sequences of Indian subtype C isolates along with a small number of subtype sequences from other countries revealed that almost all sequences from India form a distinct lineage within subtype C (CIN). Overall CIN lineage sequences were more closely related to each other (level of diversity, 10.2%) than to subtype C sequences from Botswana, Burundi, South Africa, Tanzania and Zimbabwe (range 15.3–20.7%). Suggesting thereby, much of the current Indian epidemic is descended from a single introduction into the country [21]. In an assessment of the Phylogenetic relationships among subtype C sequences from eleven different countries including India, an overall star-like phylogeny was observed [10]. Unlike sequences from South Africa and Botswana, which are scattered in numerous lineages, almost all sequences from India formed a monophyletic lineage, which is lying close to the sequences. Sequences from India generally clustered together more than sequences from other countries.
Genetic characterization of the virus during the early seroconversion stage is crucial as the virus isolated is closely related to the transmitted strain and hence immunologically naive. Phylogenetic analyses of Indian subtype C envelope sequences obtained from early seroconverts indicated that the Indian sequences not only clustered within the C clade but also clustered away from the African subtype C sequences [9]. Moreover, a recent study demonstrating lower diversity within immunodominant epitopes and a tight clustering of Indian isolates [11] suggested that production of a vaccine particularly against Indian subtype C may not be an unattainable and daunting task.
References
Bobkov AF, Kazennova EV, Selimova LM, Khanina TA, Ryabov GS, Bobkova MR, Sukhanova AL, Kravchenko AV, Ladnaya NN, Weber JN, Pokrovsky VV. Temporal trends in the HIV-1 epidemic in Russia: predominance of subtype A. J Med Virol. 2004;74:191–6.
Couturier E, Damond F, Roques P, Fleury H, Barin F, Brunet JB, Brun-Vézinet F, Simon F. HIV-1 diversity in France, 1996–1998. The AC 11 laboratory network. AIDS. 2000;14:289–96.
Dietrich U, Grez M, von Briesen H, Panhans B, Geissendörfer M, Kühnel H, Maniar J, Mahambre G, Becker WB, Becker ML, et al. HIV-1 strains from India are highly divergent from prototypic African and US/European strains, but are linked to a South African isolate. AIDS. 1993;7:23–7.
Gao F, Robertson DL, Carruthers CD, Morrison SG, Jian B, Chen Y, Barré-Sinoussi F, Girard M, Srinivasan A, Abimiku AG, Shaw GM, Sharp PM, Hahn BH. A comprehensive panel of near-full-length clones and reference sequences for non-subtype B isolates of human immunodeficiency virus type 1. J Virol. 1998;72:5680–98.
Gao F, Vidal N, Li Y, Trask SA, Chen Y, Kostrikis LG, Ho DD, Kim J, Oh MD, Choe K, Salminen M, Robertson DL, Shaw GM, Hahn BH, Peeters M. Evidence of two distinct sub-subtypes within the HIV-1 subtype A radiation. AIDS Res Hum Retroviruses. 2001;17:675–88.
Grez M, Dietrich U, Balfe P, von Briesen H, Maniar JK, Mahambre G, Delwart EL, Mullins JI, Rübsamen-Waigmann H. Genetic analysis of human immunodeficiency virus type 1 and 2 (HIV-1 and HIV-2) mixed infections in India reveals a recent spread of HIV-1 and HIV-2 from a single ancestor for each of these viruses. J Virol. 1994;68:2161–8.
Gupta RM, Prasad VVSP, Rai A, Seth P. Analysis of HIV type 1 subtype C full-length gag gene sequences from India: novel observations and plausible implications. AIDS Res Hum Retroviruses. 2005;21:1066–72.
Jeeninga RE, Hoogenkamp M, Armand-Ugon M, de Baar M, Verhoef K, Berkhout B. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J Virol. 2000;74:3740–51.
Kalpana A, Srikanth T, Abhay J, Sushama J, Swarali K, Ramesh P. gp120 sequences from HIV type 1 subtype C early seroconverters in India. AIDS Res Hum Retroviruses. 2004;20:889–94.
Khan IF, Vajpayee M, Prasad VV, Seth P. Genetic diversity of HIV type 1subtype C env gene sequences from India. AIDS Res Hum Retroviruses. 2007;23:934–40.
Korber B, Theiler J, Wolinsky S. Limitation of a molecular clock applied to considerations of the origin of HIV-1. Science. 1998;280:1868–71.
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol. 1998;73:152–60.
Maitra A, Singh B, Banu S, Deshpande A, Robbins K, Kalish ML, Broor S, Seth P. Subtypes of HIV type 1 circulating in India: partial envelope sequences. AIDS Res Hum Retroviruses. 1999;15:941–4.
Malim MH, Emerman M. HIV-1 sequence variation: drift, shift and attenuation. Cell. 2001;104:469–72.
Mandal D, Jana S, Bhattacharya SK, Chakrabarti S. HIV type 1 subtypes circulating in eastern and northeastern regions of India. AIDS Res Hum Retroviruses. 2002;18:1219–27.
McCormack GP, Glynn JR, Crampin AC, Sibande F, Mulawa D, Bliss L, Broadbent P, Abarca K, Pönnighaus JM, Fine PE, Clewley JP. Early evolution of the human immunodeficiency virus type 1 subtype C epidemic in rural Malawi. J Virol. 2002;76:12890–9.
National AIDS Control Organization website. www.naco.nic.in..
Neilson JR, John GC, Carr JK, Lewis P, Kreiss JK, Jackson S, Nduati RW, Mbori-Ngacha D, Panteleeff DD, Bodrug S, Giachetti C, Bott MA, Richardson BA, Bwayo J, Ndinya-Achola J, Overbaugh J. Subtypes of human immunodeficiency virus type 1 and disease stage among women in Nairobi, Kenya. J Virol. 1999;73:4393–403.
Salemi M, de Oliveira T, Soares MA, Pybus O, Dumans AT, Vandamme AM, Tanuri A, Cassol S, Fitch WM. Different epidemic potentials of the HIV-1B and C subtypes. J Mol Evol. 2005;60:598–605.
Salminen MO, Johansson B, Sönnerborg A, Ayehunie S, Gotte D, Leinikki P, Burke DS, McCutchan FE. Full-length sequence of an Ethiopian human immunodeficiency virus type 1 (HIV-1) isolate of genetic subtype C. AIDS Res Hum Retroviruses. 1996;20:1329–39.
Shankarappa R, Chatterjee R, Learn GH, Neogi D, Ding M, Roy P, Ghosh A, Kingsley L, Harrison L, Mullins JI, Gupta P. Human immunodeficiency virus type 1 env sequences from Calcutta in eastern India: identification of features that distinguish subtype C sequences in India from other subtype C sequences. J Virol. 2001;75:10479–87.
Siddappa NB, Dash PK, Mahadevan A, Desai A, Jayasuryan N, Ravi V, Satishchandra P, Shankar SK, Ranga U. Identification of unique B/C recombinant strains of HIV-1 in the southern state of Karnataka, India. AIDS. 2005;19:1426–9.
Simon F, Mauclere P, Roques P, Loussert-Ajaka I, Muller-Trutwin MC, Saragosti S, Georges-Courbot MC, Barre-Sinoussi F, Brun-Vezinet F. Identification of a new human-immunodeficiency virus type 1 distinct from group M and group O. Nat Med. 1998;4:1032–57.
Tatt ID, Barlow KL, Clewley JP, Gill ON, Parry JV. Surveillance of HIV-1 subtypes among heterosexuals in England and Wales, 1997–2000. J Acquir Immune Defic Syndr. 2004;36:1092–9.
Thomson MM, Najera R. Travel and the introduction of human immunodeficiency virus type 1 non-B subtype genetic forms into Western countries. Clin Infect Dis. 2001;32:1732–7.
Tripathy SP, Kulkarni SS, Jadhav SD, Agnihotri KD, Jere AJ, Kurle SN, Bhattacharya SK, Singh K, Tripathy SP, Paranjape RS. Subtype B and subtype C HIV type-1 recombinants in the northeastern state of Manipur, India. AIDS Res Hum Retroviruses. 2005;21:152–7.
UNAIDS (2004). AIDS Epidemic Update. www.unaids.org.
Vidal N, Peeters M, Mulanga-Kabeya C, Nzilambi N, Robertson D, Ilunga W, Sema H, Tshimanga K, Bongo B, Delaporte E. Unprecedented degree of human immunodeficiency virus type 1 (HIV-1) group M genetic diversity in the Democratic Republic of Congo suggests that the HIV-1 pandemic originated in Central Africa. J Virol. 2000;74:10498–507.
Voevodin A, Crandall KA, Seth P, Al Mufti S. HIV type 1 subtypes B and C from new regions of India and Indian and Ethiopian expatriates in Kuwait. AIDS Res Hum Retroviruses. 1996;12:641–3.
Yu XF, Chen J, Shao Y, Beyrer C, Lai S. Two subtypes of HIV-1 among injection-drug users in southern China. Lancet. 1998;351:1250.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Seth, P. Evolution of HIV-1 in India. Indian J. Virol. 21, 3–7 (2010). https://doi.org/10.1007/s13337-010-0001-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13337-010-0001-4