
Abstract
Antiphospholipid antibodies (aPL) are associated with the recurrent pregnancy loss and thrombosis that characterizes the antiphospholipid antibody syndrome (APS). Although the ontogeny of these pathogenic antibodies has not been fully elucidated, there is evidence that indicates the involvement of both genetic and environmental factors. The ability of aPL to induce a procoagulant phenotype in APS patients plays a central role in the development of arterial and venous thrombotic manifestations typical of the disease. Inflammation serves as a necessary link between this procoagulant phenotype and actual thrombus development and is an important mediator of the placental injury seen in APS patients with obstetric complications. Recent evidence has indicated a role for abnormal cellular proliferation and differentiation in the pathophysiology of APS, especially in those patients with pregnancy morbidity and other more atypical manifestations that have no identifiable thrombotic cause. The interplay of genetic and environmental factors responsible for aPL development and the mechanisms by which these antibodies produce disease in APS patients is the focus of this review.
Similar content being viewed by others


Introduction
Antiphospholipid antibody syndrome (APS) is an autoimmune multisystemic disorder characterized clinically by recurrent thrombosis and pregnancy morbidity and serologically by the presence of antiphospholipid antibodies (aPL) including anticardiolipin (aCL) and anti-β2 glycoprotein I (Anti-β2GPI) antibodies and lupus anticoagulant (LA) [1–3]. It is now widely accepted that aPL are a heterogenous group of antibodies (Abs) that react with a myriad of phospholipids (PLs), PL-protein complexes and PL-binding proteins. The main antigenic target of these antibodies is recognized to be β2GPI, which along with prothrombin accounts for more than 90% of the antibody-binding activity in APS patients. Other potentially significant antigenic targets include tissue plasminogen activator (tPA), phosphatidylserine (PS), plasmin, annexin 2, activated protein C (APC), thrombin, antithrombin III (AT-III) and annexin V [4–10].
APS was first described in a subset of patients with systemic lupus erythematosus (SLE) and related connective tissue diseases (CTD) that had abnormal LA tests, subsequently being classified as ‘secondary’ APS (SAPS) in the presence of these conditions and ‘primary’ (PAPS) in their absence [11]. In the general population, PAPS is the most common cause of acquired thrombophilia and is a recognized risk factor for the development of deep vein thrombosis (DVT) with or without pulmonary embolism, new strokes in individuals below the age of 50 and recurrent fetal loss [12]. The prevalence of DVT occurrence in the general population is estimated at 2–5%, 15–20% associated with PAPS, suggesting that the prevalence of venous thrombosis associated with PAPS may be as high as 0.3–1% of the general population [12]. APL Abs are present in 30–40% of SLE patients and up to a third of these patients (10–15% SLE patients) have clinical manifestations of APS, especially venous or arterial thromboses [13, 14]. The APS-related thrombotic events range in severity from the relatively benign superficial thrombophlebitis to myocardial infarction, stroke, and catastrophic APS (CAPS) [15]. SAPS also accounts for a significant proportion of recurrent pregnancy loss in SLE patients, indeed, aPL are now regarded as the most frequent acquired risk for a treatable cause of recurrent pregnancy loss and for pregnancy complications (early and severe pre-eclampsia) [13, 16–18].
Apart from the well-recognized arterial and venous thrombotic events typical of APS, these patients can also present with a variety of ‘non-criteria’ manifestations which include thrombocytopenia, nephropathy, livedo reticularis, cardiac valve disease, skin ulcers, diffuse pulmonary hemorrhage and neurological manifestations such as chorea, epilepsy, cognitive dysfunction and transverse myelopathy [19–21]. These manifestations often develop during, and do not improve with, heparin or warfarin anticoagulation therapy highlighting the concept that these manifestations more than likely do not result from the same biological abnormalities that cause arterial and venous thromboembolism in APS patients [19].
The pathophysiology of this disease has its basis in the action of the myriad autoantibodies found in APS patients on their numerous antigenic targets [22]. The origin of these pathogenic autoantibodies still remains a mystery but is likely to be due to the complex interaction of multiple environmental factors in individuals with genetic markers that increase susceptibility to the disease [23, 24]. Perhaps the most studied aspect of the disease is the increased propensity for thrombus formation and this procoagulant phenotype in APS occurs as a result of the synergy of many contributory elements. These include aPL-mediated activation of platelets, monocytes, and endothelial cells and aPL-induced perturbation of natural anticoagulant and fibrinolytic systems [8, 25–28]. Non-thrombotic mechanisms also play a major role in APS and their contribution to the development of obstetric and other more atypical manifestations of the disease has recently been the focus of much research [29, 30]. Abnormal cellular proliferation and differentiation impacting on cell function has been shown to be an important factor in aPL-induced pregnancy morbidity and this is also likely to be one of the main mechanisms involved in the development of those APS disease manifestations for which a thrombotic basis is not clearly identifiable [19, 30–32]. Inflammation is a central pathogenic factor in APS; it serves as a necessary link between the procoagulant phenotype seen in this disease and actual thrombus development and is an essential mediator of placental injury typical of aPL-induced obstetric complications [33, 34]. The proposed genetic and environmental factors contributing to the development of aPL and the mechanisms by which these autoantibodies produce the myriad disease manifestations seen in APS patients is the focus of this review.
Genes and the environment in APS
Various animal models and family and population studies have been used to highlight HLA associations with the disease and the occurrence of aPL in patients. That is to say that Major Histocompatibility Complex (MHC) genes may influence not only autoantibody production but also disease expression itself [35]. Another important consideration is the occurrence of prothrombotic genetic markers that may modify disease expression in APS patients especially when associated with acquired factors such as aPL [36]. These pathogenic aPL are thought to be produced by exposure to certain viral or bacterial products with sequence similarity to host antigens inducing a break in tolerance [37]. As stated previously, aPL represent a heterogenous group of antibodies with many different antigenic targets and the clinical experience is that not all aPL are pathogenic; making it likely that only a limited number of aPL induced by certain viral or bacterial products are important in disease [37, 38]. The roles that genetic and environmental factors play in APS development are summarized in Table 1.
Animal genetic studies in APS
There are relatively few animal studies that have assessed the genetic basis for the development of APS. The spontaneous production of IgG aCL antibodies, which exhibit co-factor (β2GPI) dependent binding to cardiolipin, has been detected in NZW × BXSB F1 (W/B F1) male mice [39]. W/B F1 mice are SLE-prone mice, which develop several autoantibodies, circulating immune complexes, and nephritis in addition to a high incidence of degenerative coronary vascular disease with myocardial infarction and thrombocytopenia and thus represent a model of lupus associated APS [39–41]. Interestingly, analysis of the genes utilized in the production of pathogenic aCL in these mice showed preferential usage of certain VH and Vκ genes, whereas other non-pathogenic aCL utilize random V gene combinations [42]. This possibly indicates that pathogenic aCL production in these mice is antigen-driven rather than germ line encoded.
In 1998, Ida et al. analyzed APS disease features in BXSB and NZW mice and their progeny [43]. Although male BXSB parental mice showed similar disease features to their male NZW × BXSB F1 progeny, these features were of decreased frequency and intensity and disease was not apparent in female parental NZW or female NZW × BXSB F1 progeny. These findings suggest that genes from the BXSB strain determines, while NZW genes serve to upregulate or modify, APS disease characteristics in their progeny and that modifying alleles such as BXSB Y-linked autoimmune accelerator gene (Y aa) may also play a role [43–45]. In the same study, genome-wide analysis using microsatellite markers was used to map BXSB alleles affecting development of aCL, anti-platelet antibodies, thrombocytopenia, and myocardial infarction in NZW × (NZW × BXSB) F1 backcross male progeny [43]. This analysis showed that the generation of each disease character was controlled by two independently segregating major dominant alleles producing full expression as a complementary gene action. Although there was complete genetic concordance between anti-platelet Abs and thrombocytopenia, other disease characteristics were independently controlled by different combinations of two dominant alleles suggesting that no single genetic factor can explain the pathogenesis of APS [43].
The presence of IgG aCL antibodies has also been demonstrated in other lupus-prone mice including the MRL/MP/lpr/lpr (MRL/lpr) and MRL/+/+ mice [46]. Similar to aCL produced in W/B F1 mice, those produced in MRL/lpr mice show non-random VH and Vκ gene usage and also evidence of somatic mutation indicating a role for antigen-driven affinity maturation [47]. aCL are also produced in normal C57BL/6 J mice with estrogen treatment increasing the incidence and levels of these antibodies, underscoring the role that environmental factors such as hormones may play in modifying genetic susceptibility in APS patients [48]. It is important to note however that aCL produced in these estrogen treated C57BL/6 J mice and those in MRL/lpr mice are not β2GPI dependent but rather show decreased binding to cardiolipin in the presence of human β2GPI [49]. Interestingly, NZW × NZB F1 mice, another classic murine model of SLE, fail to produce aCL Abs despite the production of other autoantibodies such as anti-dsDNA [46].
Human family and population studies: HLA and non-HLA associations
Multiple HLA-DR and DQ associations with the occurrence of aPL antibodies have been described but small patient sample sizes and difficulties regarding obtaining appropriately ethnically matched control populations make interpretation problematic [35, 36]. A familial clustering of individuals with persistently false positive tests for syphilis in whom overt autoimmune disease developed years later was perhaps the first indication of familial APS [50]. Since 1980, several studies have described families with high incidences of primary APS associated with LA, aCL and other autoantibodies [51–53]. The increased incidence of aCL antibodies in first-degree relatives of patients with primary or secondary APS with SLE has also been demonstrated [54, 55]. A 1998 study which assessed seven families with a high incidence of primary APS, 30 of 101 family members meeting diagnostic criteria, suggested either a dominant or co-dominant model for inheritance of the disease by segregation analysis but failed to find linkage to HLA and other candidate genes including β2GPI and Fas [56]. Other family studies however have reported several HLA associations. The paternal haplotype A30; Cw3; B60; DR4; DRw53; DQw3 has been shown to be associated with aCL in an English Canadian family; both in asymptomatic individuals and those with APS secondary to SLE and autoimmune thyroid disease [57]. The occurrence of LA in families with haplotypes containing either DR4 or DR7 has also been demonstrated [58, 59]. In a family study in which all members had SLE and presented with various APS manifestations, a mother and her twins shared a haplotype that included DR4, DRw53 and DQw7 [60].
Non-familial population studies also highlight several HLA associations of APS. A 1991 study of 20 patients with SLE and LA demonstrated an association with HLA-DQw7 (HLA-DQB1*0301) linked to HLA-DR4 and/or -DR5 [61]. In 13 English patients with primary APS, DR4 and DRw53 were found with increased frequency [62]. Other HLA loci associated with primary APS include DRB1*04, DR7, DQB1*0301/4, DQB1*0604/5/6/7/9, DQA1*0102, and DQA1*0301/2 [63–65]. In a large Italian study in SLE patients, aCL were positively associated with HLA-DRB1*04, -DRB1*07, -DQA1*0201,-DQA1*0301,-DQB1*0302,-DRB3*0301 and anti-β2GPI positively associated with DQB1*0302 [66]. The association of aCL with DRB1*09 has been reported in Japanese patients with APS secondary to SLE [67] Anti-β2GPI in Caucasian and Mexican Americans is strongly associated with HLA-DR4 haplotypes, especially those carrying HLA-DQ8 (DQB1*0302) while in African American and white British patients with primary APS, anti-β2GPI is strongly associated with the HLA-DRB1*1302;DQB1*0604/0605 haplotype [63, 68]. The association of C4A or C4B null alleles with the presence of aCL has been reported in black American populations, however, patients in the Hopkins Lupus Cohort who were homozygous for C4A deficiency had a lower frequency of aCL and LA than patients without this deficiency [69–71].
Other genes outside the MHC region also contribute to both autoantibody production and disease expression in APS. A polymorphism in domain 5 of β2GPI, valine instead of leucine at position 247, is found more frequently in patients with APS than matched controls and is associated with anti-β2GPI production in these patients [72–74]. One study found an increased frequency of this polymorphism in patients with arterial thrombosis than those without [75]. There are other prothrombotic genetic factors that can modify disease expression in APS patients. Those genetic factors clearly related to thrombophilia that have been seen in APS patients include factor V Leiden and prothrombin mutations and antithrombin III, protein C and protein S deficiencies [75]. The gain of function factor V Leiden G1691A (FVL) mutation is highly prevalent in Caucasian populations with population frequencies ranging from 1 to 15% [76, 77]. Several reports have demonstrated an increased incidence of thrombosis in APS patients with FVL mutation when compared to those without but the mutation seems to have a more moderate effect on the development of thrombosis in APS than in the general population [78–80]. The G20210A prothrombin mutation (F2 G20210A) is associated with venous thromboembolism in the general population but there have been conflicting reports of the increased risk of thrombosis related to this gene mutation in APS patients. Initial reports indicated no increased risk but some of the subsequent studies have demonstrated the association between the mutation and thrombosis in APS patients, the first case being described in a young female with SLE-associated APS homozygous for the G20210A mutation [81–84]. Protein C, S and antithrombin III deficiencies are uncommon diseases making it difficult for an accurate assessment to be made of the relative contributions of these mutations and aPL in thrombus generation in APS. However there have been reports of increased thrombosis rates in APS patients with protein C and protein S deficiency [85, 86]. Other polymorphisms that potentially impact the risk of thrombosis in APS patients affect platelet glycoproteins GP Ia/IIa and GP IIb/IIIa, platelet Fcγ receptor IIa, tissue factor pathway inhibitor, thermolabile variant of methylenetetrahydrofolate reductase, type-I plasminogen activator inhibitor, tumor necrosis factor α, thrombomodulin, annexin A5, p-selectin, p-selectin glycoprotein ligand-1, toll-like receptor 4, factor XIII and CD40 [87–97].
Origin of aPL: environmental factors
The processes underlying the production of aPL in APS patients remain undetermined. When these antibodies were first described, aPL were defined as antibodies reacting to cardiolipin, however, it is now well accepted that these antibodies recognize various PL and protein antigenic complexes [4–7]. Indeed, as stated previously, the main antigenic target for these antibodies is β2GPI, an abundant serum protein that is a necessary co-factor for aPL binding to phospholipid. In fact, efforts to induce high-titer production of pathogenic aPL in animal models succeeded only after immunization with heterologous β2GPI rather than pure phospholipids [4, 98]. This led researchers to believe that perhaps in vivo binding of foreign PL-binding proteins resembling β2GPI to self-phospholipids in APS patients may lead to the formation of immunogenic complexes against which aPL is produced. A synthesized 15 amino acid peptide, GDKV, which spanned an area of the fifth domain of β2GPI known to be a major PL-binding site of the molecule, was able to induce pathogenic aPL and anti-β2GPI production in immunized mice [99]. A monoclonal Ab with aPL and anti-β2GPI activity generated from these GDKV-immunized mice was shown to be pathogenic using in vivo models for thrombus enhancement and microcirculation [100]. Similar results were subsequently obtained using peptides from microorganisms with functional and sequence similarity to that of the PL-binding site of β2GPI [101]. The peptides TIFI and VITT from cytomegalovirus (CMV), TADL from adenovirus (AdV) and SGDF from Bacillus subtilis all had greater degrees of PL-binding compared to GDKV and induced high-titer aPL and anti-β2GPI production in mice. Subsequent in vivo and in vitro experiments confirmed the pathogenicity of antibodies induced in TIFI-immunized mice [101–103].
Further supporting evidence for molecular mimicry as a possible mechanism for APS development was provided by a study evaluating the APS-related pathogenic potential of microorganisms carrying sequences related to a hexapeptide, TLRVYK, known to be specifically recognized by a pathogenic monoclonal anti-β2GPI Ab [104]. Following immunization with Haemophilus influenzae, Neisseria gonorrhoeae or tetanus toxoid; high titers of antibodies with anti-peptide (TLRVYK) and anti-β2GPI activity were observed in BALB/c mice. These affinity-purified antibodies were then infused into naive mice at day 0 of pregnancy. At day 15, these mice had significant thrombocytopenia, prolonged activated partial thromboplastin times (aPTT) and increased frequency of fetal loss compared to controls.
Infections are thought perhaps to be the most prominent environmental trigger for aPL production and APS development. Syphilis was the first infectious disease recognized to be linked to aPL production and these infectious type aPL were initially thought to be non-pathogenic [105–107]. However, several subsequent reports have shown that many infections not only trigger aPL production but are associated with the development of APS manifestations as well [108]. This is perhaps best exemplified by catastrophic APS, a rare presentation of APS characterized by multiple small vessel occlusions affecting multiple organ systems with a high mortality rate, which is strongly linked to preceding infections and/or trauma [15]. CMV, parvovirus B19, Human immunodeficiency virus (HIV), Hepatitis B and C viruses, Human T cell lymphoma/leukemia virus (HTLV) and Varicella Zoster Virus (VZV) are just a few of the infectious agents that have reported associations with aPL production and APS manifestations [109]. In addition to molecular mimicry, infectious agents can potentially induce autoimmune responses by selectively activating or destroying unique lymphocyte subsets, directing cytokine/chemokine release or exposing cryptic autoantigens during cell necrosis and/or apoptosis [110–112]. Other potential environmental triggers of APS development include vaccination, drug therapy and certain malignancies. However, to date there is no conclusive evidence linking vaccination to the development of APS [113, 114]. The ability of drugs to bind and perhaps alter the processing and presentation of self-antigens such that cryptic antigens are presented makes the development of an autoimmune response possible [115]. Indeed, agents such as chlorpromazine, amoxicillin, phenytoin, chlorothiazide, propranolol, oral contraceptives, quinine, alpha-interferon and infliximab have been associated with the presence of aPL but data regarding the prevalence of drug-induced aPL in APS is still lacking [105, 116]. The presence of aPL has been reported in patients with both solid and hematological malignancies and the significance of this finding lies in the increased risk for thrombosis and the potential for precipitating CAPS in these patients. The mechanisms leading to aPL production remain unclarified but may result from an immune response directed against tumor antigens or perhaps against neoantigens formed due to immunomodulatory drug therapy such as interferon-α (IFNα) [117].
The relative degree to which genetic and environmental factors influence susceptibility to APS development is still uncertain. It is likely that there is a complex interplay of multiple environmental factors in a genetically susceptible patient to produce the varied autoantibodies and myriad clinical manifestations typical of this disease. Improved understanding of the relative contributions of these many factors would certainly aid in prevention and management of these patients.
Thrombogenic mechanisms in APS pathophysiology
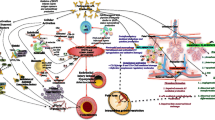
There is overwhelming evidence of the thrombogenic capacity of aPL provided by both in vitro studies and in vivo animal models [118]. The main targets of aPL action, β2GPI and prothrombin (PT), are proteins that interact with many factors involved in hemostasis making the central role that aPL-mediated thrombosis plays in APS unsurprising [4, 5, 119]. Platelet, endothelial cell and monocyte activation occurs in conjunction with disruption of natural anticoagulant and fibrinolytic systems in response to aPL resulting in a procoagulant phenotype in APS patients (Fig. 1) [118]. However, the clinical observation that thrombosis is only occasionally observed despite the persistent presence of aPL suggests that the procoagulant state induced by these antibodies (‘first hit’) only leads to thrombosis in the presence of an inciting factor (‘second hit’) such as inflammatory responses or trauma [120]. The role that inflammatory responses play in thrombus formation in APS is discussed later.

Thrombogenic mechanisms of antiphospholipid antibodies (aPL). Y− inhibitory aPL action Y+ aPL-induced activation, Straight green arrow activation, broken red arrow inhibition. AnnA2 annexin A2, APC activated protein C, ApoER2′ apolipoprotein E receptor 2′, AT-III antithrombin III, β-2GPI beta-2 glycoprotein, C5a activated complement component 5, C5aR C5a receptor, Flt-1 tyrosine kinase receptor, GPIb-V-IX glycoprotein Ib-V-IX, GPIIb/IIIa glycoprotein IIb/IIIa, ICAM-1 intercellular adhesion molecule-1, IL interleukin, LPa lipoprotein a, MAC membrane attack complex, NF-κB nuclear factor-kappa B, p38 MAPK p38 mitogen activated protein kinase, PAI-1 plasminogen activator inhibitor-1, PMN polymorphonuclear leukocyte, TF tissue factor, TLR4 toll-like receptor 4, TNFα tumor necrosis factor alpha, tPA tissue plasminogen activator, TXB2 thromboxane-B2, VCAM-1 vascular cellular adhesion molecule – 1, VEGF vascular endothelial growth factor
Cells of the coagulation system
A role for platelet activation in the pathophysiology of APS was initially suggested by the frequent finding of thrombocytopenia in APS patients and APS animal models [121]. Elevated urinary secretion of 11-dehydro-thromboxane B2 (11-dehydro-TXB2), a major platelet derived thromboxane metabolic breakdown product, in APS patients with elevated LA levels indicates this as well [122]. Binding of aPL Abs to platelets is dependent on both the presence of dimeric β2GPI and the exposure of anionic phospholipids, especially phospatidylserine (PS), on platelets which occurs after stimulation by agonists such as thrombin, collagen, and adenosine diphosphate (ADP) [123, 124]. aPL Abs enhance the expression of GPIIb/IIIa, a major fibrinogen receptor, on platelets and Pierangeli et al. showed that aPL-mediated thrombus formation was significantly reduced in GPIIb/IIIa deficient (β3-null) mice and mice treated with a monoclonal anti-GPIIb/IIIa antibody [125, 126]. The intracellular mechanisms through which aPL activate platelets was also elucidated by this group. They showed that aPL-induced TXB2 production in platelets through the activation of p38 mitogen activated protein kinase (MAPK) and subsequent phosphorylation of cytosolic phospholipase A2 (cPLA2). Other MAPK pathways in platelets, such as ERK-1 (p44 MAPK) and ERK-2 (p42 MAPK), have a potential role in signaling after initial activation through p38 MAPK [126]. Researchers have demonstrated the role that apolipoprotein E receptor 2′ (ApoER2′) and the GPIbα subunit of the GPIb-V-IX receptor on the platelet membrane play in aPL-mediated platelet activation through the p38 MAPK pathway and subsequent thrombus formation in several studies [124, 127]. Interestingly, these 2 receptors are able to form a complex on the platelet membrane and recently Urbanus et al. reported that anti-β2GPI/dimeric β2GPI complex mediated signaling through both these receptors is required for platelet activation [128]. This suggests that perhaps there is a critical role for complex formation of these two receptors in aPL-mediated platelet activation. A recent study has demonstrated a putative role for platelet factor 4 (PF4), a CXC chemokine secreted and bound by platelets, in the stabilization of dimeric β2GPI and subsequent binding to ant-β2GPI Abs and exposed phospholipids and receptors on the platelet surface [129].
Like platelet activation, a role for vascular endothelial cell and monocyte activation in aPL-mediated thrombogenesis has been described. Several in vitro studies have shown that endothelial cells express significantly higher amounts of the adhesion molecules vascular cell adhesion molecule 1 (VCAM-1), intercellular cell adhesion molecule 1 (ICAM-1) and E-selectin [130–132]. Employing in vitro and in vivo experiments utilizing ICAM-1, VCAM-1, E-selectin and P-selectin knockout mice, Pierangeli et al. demonstrated that the ability of human polyclonal and monoclonal aPL to activate endothelium promoting leukocyte adhesion and thrombus formation is mediated by ICAM-1, E-selectin, P-selectin and VCAM-1 [133]. This group and others have also demonstrated the upregulation of tissue factor (TF) expression and microparticle formation with associated increases in interleukin-6 (IL-6) and IL-8 secretion in endothelial cells and monocytes treated with aPL [134–137]. This TF upregulation in monocytes may occur as a result of stimulation of the Flt-1 tyrosine kinase receptor by vascular endothelial growth factor (VEGF); increased plasma levels of VEGF and surface expression of both VEGF and Flt-1 on monocytes being noted in APS patients [138]. The aPL-induced activation and TF upregulation in both endothelial cells and monocytes has been shown to be dependent on p38 MAPK activation and nuclear factor κB (NF-κB) [139, 140]. Many researchers have provided evidence that upregulated TF mRNA and antigen expression and TF pathway activation plays a key role in APS thrombotic manifestations. Indeed, Pierangeli et al. found, in an ongoing clinical trial, that mean serum levels of soluble TF, tumor necrosis factor-α (TNFα) and VEGF were significantly elevated in APS patients compared to controls and treatment with fluvastatin, a statin with efficacy in treating APS, resulted in significant decreases of these pro-inflammatory markers in most APS patients [141].
There is substantial evidence to show that β2GPI binds to endothelial cells, providing suitable epitopes for aPL binding to endothelial cells. However, the full details regarding the identity of these receptors that bind β2GPI and the mechanisms by which signaling cascade activation occurs have not been elucidated [142]. Annexin A2 and toll-like receptor 4 (TLR4) have been identified as candidate receptors on both endothelial cells and monocytes and ApoER2′ on endothelial cells as well [139, 143]. Annexin A2 is able to bind anti-β2GPI/β2GPI complexes on both endothelial cells and monocytes leading to activation and expression of a procoagulant phenotype [143]. Romay-Penabad et al. have also demonstrated the protective effect of annexin A2 deficiency in mice against the development of aPL-induced thrombosis [144]. However, annexin A2 on the cell surface lacks an intracellular tail meaning that a co-receptor would be required for intracellular signal transduction and subsequent cell activation as a result of anti-β2GPI/β2GPI complex binding [143]. Raschi et al. showed that the myeloid differentiation factor 88 (MyD88) signaling cascade, which is important in TLR signaling, is triggered by binding of aPL on human endothelial cells in vitro [139]. A subsequent in vivo study by Pierangeli et al. in lipopolysaccharide (LPS) non-responsive mice (LPS −/−) displaying a single point mutation on the TLR4 gene demonstrated reduced TF expression, endothelial cell activation and thrombosis in response to aPL [97]. These findings indicate that TLR4 may serve as a co-receptor for annexin A2 binding of anti-β2GPI/β2GPI complexes and subsequent endothelial cell activation. There is also evidence for annexin A2 and TLR4 acting as co-receptors for these complexes on human monocytes [145]. A very interesting study recently demonstrated that IgGs from APS patients with differing clinical manifestations of the disease vary in their ability to activate NF-κB and p38 MAPK and increase TF production in monocytes and that this was possibly due to differential TLR4 activation [146]. Of note, TLR2 has been shown to play a role in anti-β2GPI mediated activation of fibroblasts through a MyD88 dependent pathway [147]. Since TLR2 and TLR4 have several shared ligands and TLR2 is also present on endothelial cells, monocytes and platelets, it is possible that TLR2 is involved in aPL-mediated activation of these cells as well [148]. In fact, a recently published study presented evidence for TLR2 being the major TLR involved in aPL-induced endothelial cell activation [149]. Recently, Doring et al. showed that monoclonal aPL and polyclonal IgG from APS patients induced TNFα production in monocytes by activating TLR8, a member of the endogenous group of TLRs [150]. Previous evidence provided by the same group had suggested a role for endogenous stimulation of both TLR7 and TLR8 in the production of pro-inflammatory mediators in APS patients [151]. ApoER2′ is also expressed on endothelial cells and in vitro studies utilizing anti- ApoER2′ antibodies have shown partial inhibition of β2GPI dependent aPL binding to and subsequent activation of endothelial cells [152]. A recent in vivo study by Romay-Penabad et al. has demonstrated in ApoER2′ deficient (−/−) mice, a significant reduction in thrombus formation and TF production induced by polyclonal IgG aPL, a murine anti-β2GPI monoclonal Ab (E7) and a constructed β2GPI dimer compared to wild type controls. A similar effect was noted in wild type mice treated with soluble binding domain I of ApoER2′ (sBD1), an inhibitor of ApoER2′, thus highlighting the importance of this receptor in the pathogenesis of APS [153].
Anticoagulation and fibrinolytic systems
Interference with natural anticoagulant and fibrinolytic systems also plays a role in thrombus formation in APS patients [152, 154]. Activated protein C (APC), in conjunction with its main co-factor protein S, is an important anticoagulant that functions by binding and inactivating the activated forms of coagulation factors V (FVa) and VIII (FVIIIa) [155]. Anti-β2GPI/β2GPI complexes potentially affect the assembly and function of APC thus inducing a procoagulant phenotype [156]. Interestingly, anti-PT antibodies in APS patients may also affect APC function [156]. The clinical relevance of these antibodies, as evidenced by association with thrombosis and LA activity, seems to be seen with anti-PT complexed to PS rather than anti-PT itself [157]. Antithrombin III (AT-III) is another natural anticoagulant that performs its function by inactivating the activated forms of factors XII (FXIIa), XI (XIa), X (FXa), IX (FIXa), VII (FVIIa) and thrombin (FIIa), these actions being potentiated by heparin [158]. The inhibition of AT-III activity in vitro by polyclonal IgG and IgM and purified anti-heparan sulfate antibodies from APS patients has been described [159–161]. Annexin A5 is a calcium dependent cationic protein that binds anionic PL, especially PS, with high affinity and functions as a protective anticoagulant shield by competing for sites on PL membranes where coagulation factors normally assemble into active complexes. This protein is found in abundance in placental tissue and is an important natural mediator of anticoagulation at this site [162]. The disruption of this anticoagulant’s function and its possible importance in aPL-mediated pregnancy morbidity is discussed later in this review.
The fibrinolytic system is activated when the proenzyme plasminogen is converted to plasmin which then degrades fibrin, a major component of thrombi. This process is primarily mediated through the action of tissue plasminogen activator (tPA) or urokinase plasminogen activator, the former being more important in coagulation homeostasis. The activity of tPA itself is primarily regulated by the serine protease inhibitor, plasminogen activator inhibitor type I (PAI-1) [154]. Anti-tPA antibodies correlated with reduced tPA plasma levels and the ability to bind and inactivate tPA have been described in APS patients [163]. Elevated PAI-1 levels and decreased tPA release after venous occlusion in APS patients has also been reported [164]. Atrium et al. also reported elevated levels of lipoprotein a (LPa), which shares structural similarity to plasminogen, in the plasma of APS patients. Their experiments suggested that LPa may compete with plasmin or plasminogen for binding to fibrin and others have demonstrated LPa upregulation of PAI-1 expression by human endothelial cells [165]. Some aPL have been reported to bind plasmin thus inhibiting fibrinolysis; one study reporting a frequency of 28% of these antibodies in APS patients [166].
In addition to the role of annexin A2 as a receptor for anti-β2GPI/β2GPI complex mediated endothelial cell activation, experimental evidence has shown that cell surface annexin A2 stimulates the activation of plasminogen by tPA [167]. Antibodies that act against annexin A2 and correlate with a history of thrombosis have been shown to be significantly more prevalent in APS patients than in healthy individuals, patients with non-autoimmune thrombosis and SLE patients without thrombosis [168]. IgG anti-annexin A2 Abs have been shown to limit plasmin generation in in vitro experiments on human umbilical vein endothelial cells by 34–83% [9]. Interestingly, β2GPI has demonstrated the capacity to block PAI-1 activity and also to directly increase plasmin generation by binding to plasminogen [169–171]. These mechanisms of β2GPI mediated fibrinolysis enhancement have been inhibited by monoclonal anti-β2GPI and polyclonal IgG from APS patients [172].
Thrombosis in aPL-mediated pregnancy morbidity
Due to the prothrombotic nature of APS, impairment of maternal-fetal blood exchange as a result of thrombus formation in the uteroplacental vasculature was thought to be the main pathogenic mechanism underlying pregnancy morbidity in this condition [173]. Histological examination has supported this hypothesis; placental thrombosis and infarction being demonstrated in APS patients with first and second trimester abortions [174, 175]. IgG fractions from LA positive APS patients are able to induce a procoagulant phenotype with significant increases in thromboxane synthesis in placental explants from normal human pregnancies [176]. As stated previously annexin A5 is thought to be an important anticoagulant during pregnancy acting as a shield on potentially thrombogenic anionic membrane surfaces in the placenta [162]. Rand et al. have reported significantly lower levels of annexin A5 covering the intervillous surfaces of placentas in women with aPL when compared to controls [177]. In vitro studies have also demonstrated displacement of annexin V from trophoblast and endothelial cell monolayers by aPL while murine studies have demonstrated the necessity of this protein in maintaining placental integrity [178, 179]. Anti-annexin A5 Abs have been reported in APS patients at frequencies up to 30% and several studies have demonstrated the association of these Abs with recurrent fetal loss in APS patients [180, 181].
Despite experimental models providing evidence for a role of thrombosis in APS-related pregnancy losses, epidemiological studies fail to demonstrate this consistently. In fact, histological evidence of thrombosis in the uteroplacental circulation cannot be demonstrated in the majority of placentas from APS patients [182]. Other theories have thus been put forward to explain APS-related pregnancy morbidity such as defective trophoblast invasion and decidual transformation in early pregnancy and placental injury as a result of local inflammatory events [183]. It is likely that abnormalities of early trophoblast invasion and defective placentation rather than thrombosis may be the primary pathological mechanism involved in first trimester losses in these patients (Fig. 2) [184].

Pathogenic mechanisms leading to obstetric complications in APS. EGF-like GF epidermal growth factor-like growth factor, hCG human chorionic gonadotrophin, MMP matrix metalloproteinases, TF tissue factor, VEGF vascular endothelial growth factor
Non-thrombotic mechanisms in APS pathophysiology
Cellular dysfunction in obstetric APS
There is evidence for a direct effect of aPL on trophoblasts resulting in increased apoptosis and abnormal proliferation, human chorionic gonadotrophin (hCG) release, invasiveness and adhesion molecule expression. There is also evidence for an effect of aPL on maternal spiral artery transformation and the maturation and differentiation of maternal decidual endometrial cells [183]. The direct reactivity of aPL on these cells is supported by the fact that β2GPI is normally expressed on trophoblast membranes under physiological conditions which also explains the placental tropism of aPL. There is a high level of tissue remodeling during placentation so that trophoblasts, which are able to synthesize β2GPI, normally express anionic phospholipids on the outer leaflet of the cell membrane allowing β2GPI binding [185].
Normal placentation involves a dynamic and complex interaction between trophoblast and decidual cells requiring finely regulated cell surface adhesion and signaling molecule expression, activation of matrix metalloproteinases (MMPs), angiogenesis and spiral artery transformation [186]. In vitro studies utilizing murine and human monoclonal aPL and polyclonal IgG Abs from APS patients have demonstrated β2GPI dependent binding of these Abs to trophoblast monolayers [187, 188]. Bound murine monoclonal anti-PS Abs have been shown to react with syncytiotrophoblast and to prevent intertrophoblast fusion, trophoblast invasiveness and hCG secretion [189, 190]. Comparable results were reported using polyclonal IgG fractions from APS patients and IgM monoclonal anti-β2GPI Abs [187]. Similarly, anti-β2GPI mAbs have demonstrated an inhibitory effect on the proliferation of a human choriocarcinoma cell line and on extravillous trophoblast differentiation [189, 191]. In an in vitro model of trophoblast invasion, aPL induced trophoblast expression of particular integrins and cadherins, affecting decidual invasion [192]. Di Simone et al. showed recently that decreased expression of heparan binding epidermal growth factor-like growth factor (EGF-like GF), an important factor in blastocyst implantation, might also play a role in defective placentation [193]. Increased apoptosis has been demonstrated in rat embryos and placental explant cultures incubated with polyclonal IgG from women with SLE/APS and associated pregnancy loss as well as in rat embryos incubated with monoclonal anti-PS Abs [194, 195]. However, human monoclonal IgM anti-β2GPI Abs failed to induce apoptosis in trophoblast cell monolayers despite affecting Bcl-2 and Bax expression [196].
It has been shown that aPL, particularly anti-β2GPI, also react with human stromal decidual cells in addition to trophoblasts thus affecting the maternal side of the placenta. Polyclonal and monoclonal β2GPI dependent aPL can bind stromal decidua cell monolayers and induce a pro-inflammatory phenotype characterized by increased ICAM-1 expression and TNFα secretion [197]. Impaired endometrial differentiation as well as diminished expression of the complement regulatory protein DAF (decay accelerating factor) have been demonstrated in endometrial biopsy samples from APS patients with recurrent pregnancy loss [198]. In a recent study assessing in vitro human endometrial endothelial cell (HEEC) angiogenesis and in vivo angiogenesis in a murine model, aPL were shown to significantly decrease the number and total length of tubule formation, VEGF and MMP production and NF-κB DNA binding activity in HEEC. Newly formed vessels were also reduced in aPL inoculated mice suggesting that inhibition of angiogenesis is a potential mechanism of defective placentation in APS patients [199].
For the most part the candidate cell membrane receptors and the signaling pathways involved in β2GPI dependent aPL effects on trophoblast and stromal decidual cells remain unknown. Both annexin A2 and TLR4 have been reported to be expressed on trophoblast membranes and a recent study by Mulla et al. has demonstrated that aPL triggers a placental inflammatory response via the TLR4/MyD88 pathway compromising trophoblast survival [200–202]. The same group subsequently showed that anti-β2GPI Abs limit trophoblast cell migration by down-regulating trophoblast IL-6 secretion and signal transducer and activator of transcription 3 (STAT-3) activation [203]. Inhibitory antibodies to candidate receptors in decidual cells (i.e., TLR4, annexin A2 and ApoER2′) have induced partial inhibition of anti-β2GPI mediated binding and cellular activation suggesting that perhaps more than one receptor mediates the interaction between β2GPI and cells of the decidua and trophoblast [197].
Non-thrombotic pathogenic mechanisms in other organ systems
Thrombotic pathogenic mechanisms have failed to explain several other clinical manifestations in APS apart from those related to pregnancy morbidity. Renal involvement in APS, although involving thrombosis in glomeruli and larger vessels, is also associated with non-thrombotic kidney lesions that are associated with an increased risk of end-stage renal disease development. Endothelial cell dysfunction induced by aPL is likely to play a major role [204]. Central nervous system (CNS) involvement in APS, although commonly due to stroke or transient ischemic attack (TIA), is not always associated with a clearly identified ischemic event [205]. Both cognitive dysfunction and magnetic resonance imaging (MRI) white matter hypodensities can occur in aPL positive patients but little is known of the pathogenic mechanisms that lead to the development of these manifestations. An association exists among cognitive dysfunction, livedo reticularis and white matter MRI lesions suggesting a microangiopathic mechanism [206]. In addition to small vessel thrombosis, cognitive dysfunction in APS may be due to aPL binding to CNS cells causing permeabilization and depolarization [207]. Diffuse alveolar hemorrhage (DAH) is an unusual disease presentation in APS. Although some case reports of APS patients with DAH describe lung biopsy specimens with microvascular thrombosis, others revealed pulmonary capillaritis as the main pathogenic finding with no evidence of thrombosis [208, 209]. Furthermore, the fact that appropriately anticoagulated APS patients can present with DAH suggests a vasculitic, non-thrombotic pathogenesis [210]. Several in vivo and in vitro studies have highlighted the contribution of aPL to atherosclerotic plaque formation through endothelial cell and monocyte activation. The formation of atherogenic oxidized low-density lipoprotein (LDL)-β2GPI complexes and aPL promoting the binding of these complexes to macrophages plays a central role in this process [211, 212].
Inflammation in APS pathology
Inflammatory mediators in APS-related pregnancy morbidity
Several pathogenic changes in maternal immune responses occur during pregnancy in order to protect the developing fetus from attack by the maternal immune system. Key features of this change include the increased prevalence of a Th2 type cytokine profile and an anti-inflammatory environment as dictated by a delicate dynamic equilibrium between pro and anti-inflammatory mediators at different stages of gestation. Acute inflammatory events are generally believed to be responsible for a negative pregnancy outcome and it is therefore reasonable that APS-related pregnancy morbidity may be associated with a pro-inflammatory state in uteroplacental tissue [213]. Experimental models have demonstrated a role for complement, TF, TNFα and CC chemokines in aPL-mediated fetal loss [214, 215].
Initial murine models used to evaluate the role of complement activation in aPL-induced fetal loss had focused on complement components C3 and C5. Passive transfer of large amounts of human IgG-aPL from APS patients with recurrent miscarriage to pregnant naïve mice induced extensive placental damage resulting in increased resorption and fetal growth retardation at day 15 of pregnancy [214]. Placental inflammation in these mice was characterized by recruitment of neutrophils, upregulated TF and TNFa secretion, decidual focal necrosis and apoptosis, loss of fetal membrane elements and complement deposition. Complement receptor 1 related gene/protein y (Crry)-Ig, which is an inhibitor of classical and alternative pathway complement C3 convertases, was shown to reduce the frequency of fetal resorption, prevent growth restriction in surviving fetuses and limit the development of placental lesions [216]. The importance of C3 in aPL-induced pregnancy complications was also confirmed by the resistance of C3 deficient mice to these antibody effects. Interestingly, C4 or factor B deficient mice show similar resistance pointing to the involvement of all complement pathways in aPL-mediated pregnancy morbidity [217]. This indicates perhaps that the classical and/or lectin pathways generate C3, a central component of all the complement pathways, at sites of injury, which then activates a positive feedback loop through the alternative pathway thus amplifying the response.
Additional murine studies have demonstrated the key role that complement component C5 plays in aPL-induced placental injury; aPL-induced pregnancy complications were reduced in C5 or C5a receptor (C5aR) deficient mice and in mice treated with monoclonal anti-C5 Abs or a highly specific peptide antagonist of C5aR (C5aR-AP) [217]. These findings highlight the critical importance of C5a-C5aR interaction in inducing aPL antibody mediated placental injury. C6 deficient mice on the other hand are not protected from aPL-mediated fetal loss indicating that the membrane attack complex (MAC, C5b-9) is unlikely to play an essential role in this process [218]. Interestingly, deciduas from C5 −/− mice treated with aPL had normal morphology with absent neutrophil infiltration and in similar experiments mice depleted of neutrophils did not have pregnancy loss, fetal growth restriction or inflammatory infiltrates [217]. In addition, murine studies utilizing mice treated with monoclonal anti-TF Abs or TF deficient mice were similarly protected from aPL-induced obstetric complications [218]. Redecha et al. showed that a neutrophil oxidative burst associated with TF production was an essential factor in IgG aPL-mediated fetal loss in a murine model and that this pro-inflammatory response could be induced by C5a activation of neutrophils [218]. Studies have also suggested a possible role for C5a activation of monocytes resulting in soluble VEGF receptor-1 (sVEGFR-1) and subsequently diminished VEGF levels in conditions associated with fetal loss [219]. All these findings together suggest a mechanism by which aPL bind trophoblast and induce complement activation, generating C5a and subsequent C5a-C5aR interactions including those on neutrophils and monocytes. C5a induces TF expression and superoxide production in neutrophils causing oxidative damage and possibly sVEGFR-1 secretion in monocytes causing diminished VEGF levels and inadequate placental development and perfusion. This results in placental injury, fetal growth restriction, or resorption. Pro-inflammatory chemokines have also been shown to be important factors in the development of aPL-induced pregnancy complications. Mice deficient in D6, an abundant chemokine receptor in placental tissue that targets inflammatory CC chemokines for destruction, are more susceptible to fetal loss when passively infused with human IgG aPL from women compared to wild type mice [215]. It is important to note that the efficacy of heparin in preventing obstetric complications in women with APS has been shown not to be a function of its ability to inhibit thrombosis, as was once believed, but rather its ability to limit aPL-induced complement activation and subsequent placental inflammation [220].
Conclusive immunohistological evidence of complement activation in abortive material and placentas from women with APS is still lacking. While complement deposition in abortive specimens from APS patients has been reported in some case series, others were unable to demonstrate this pathological finding [221, 222]. Although increased inflammatory infiltrates have been noted in placental biopsies taken from women with APS compared to controls, these inflammatory cells consist predominantly of macrophages, not the polymorphonuclear leukocytes typical of murine models of aPL-induced placental injury [223]. Further analysis of the applicability of murine models to aPL-induced obstetric complications in APS patients is thus required to translate these findings into the development of targeted therapies for these patients. In that line, a prospective multicentre observational study entitled PROMISSE (PRedictors of PRegnancy Outcome: bioMarkers in antiphospholipid antibody Syndrome and SLE - NCT00198068) has been initiated to examine the role of complement as a potential surrogate marker that predicts poor pregnancy outcomes in APS patients. This study is scheduled for completion in 2013 [224].
Role of complement in endothelial cell activation and thrombosis
In addition to its role in aPL-mediated pregnancy morbidity, complement also plays an important role in endothelial cell activation and thrombosis. In murine models of thrombus formation due to aPL action, mice deficient in C3 and C5 were found to be resistant to thrombosis and endothelial cell activation. Monoclonal Abs against C5 reduced thrombus formation and endothelial cell activation in CD1 mice injected intraperitoneally with IgG aPL [225] C5aR deficient and C6 deficient mice and those treated with a C5aR antagonist were similarly protected from aPL-induced endothelial cell activation and thrombosis thus highlighting the importance of the membrane attack complex (MAC) in addition to C5a-C5aR interactions in this pathogenic process [226–228]. Complement activation, through the binding of C5 and MAC to receptors on endothelial cells, can upregulate TF expression and so contribute to thrombus formation [134]. It is likely that complement activation and subsequent generation of potent vasoactive mediators occurs as a necessary intermediary step between endothelial cell and platelet activation by aPL and thrombosis. In fact, a recent cross-sectional retrospective study found that hypocomplementemia was common in patients with primary APS and that it was likely the result of complement activation rather than deficiency. However, no correlation was found between reduced complement levels and thrombotic or obstetric manifestations in these patients [229].
Conclusion
The pathogenic mechanisms by which aPL induce disease expression in APS are manifold and the corollary of this is the exceptional variability in the clinical manifestations observed in these patients. The specific genetic and environmental interactions that occur in individual patients are likely to be particularly important in determining the makeup of the heterogenous aPL milieu found in each, potentially impacting which aPL-induced effects are preferentially activated. Unfortunately, the molecular mechanisms that lead to aPL development and aPL-induced disease expression remain incompletely understood.
The recent elucidation of some of the cell surface interactions of aPL and subsequent intracellular events has led to the identification of novel targets for the treatment and prevention of APS disease manifestations, especially thrombosis [230]. This is quite important as typical anticoagulation therapy with heparin and/or warfarin is sometimes ineffective and has a myriad of side effects, not the least of which is the increased propensity for hemorrhage. The development of therapies that target specific aPL-receptor interactions and distinctive intracellular signaling pathways would ideally result in fewer adverse side effects without sacrificing therapeutic efficacy.
The future success of this ‘immunomodulatory’ approach to the treatment of APS relies on the improved understanding of the molecular events that occur in the disease, which can then inform basic and clinical research for drug development and implementation.
References
Harris EN (1987) Syndrome of the black swan. Br J Rheumatol 26:324–326
Wilson WA, Ghavari AE, Koike T et al (1999) International concensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum 42:1309–1311
Miyakis S, Lockshin MD, Atsumi I et al (2006) International concensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 4:295–306
McNeil HP, Simpson RJ, Cherterman CN et al (1990) Antiphospholipid antibodies are directed against a complex antigen that includes lipid binding inhibitor of coagulation: β2 glycoprotein I (apolipoprotein H). Proc Natl Acad Sci USA 87:4120–4124
Galli M, Comfurius P, Maassen C et al (1990) Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet 335(8705):1544–1547
Amengual O, Atsumi T, Koike T (2004) Antiprothombin antibodies and the diagnosis of antiphospholipid syndrome. Clin Immunol 112(2):144–149
Permpikul P, Rao LV, Rapaport SI (1994) Functional and binding studies of the roles of prothrombin and beta 2-glycoprotein I in the expression of lupus anticoagulant activity. Blood 83(10):2878–2892
Chen PP, Giles I (2010) Antibodies to serine proteases in the antiphospholipid syndrome. Curr Rheumatol Rep 12(1):45–52
Cesarman-Maus G, Ríos-Luna NP, Deora AB et al (2006) Autoantibodies against the fibrinolytic receptor, annexin 2, in antiphospholipid syndrome. Blood 107(11):4375–4382
Rand JH, Wu XX, Quinn AS, Taatjes DJ (2008) Resistance to annexin A5 anticoagulant activity: a thrombogenic mechanism for the antiphospholipid syndrome. Lupus 17(10):922–930
Boey ML et al (1983) Thrombosis in SLE: striking association with the presence of circulating lupus anticoagulant. Br Med J 287:101–102
Ginsburg KS, Liang MH, Newcomer L et al (1992) Anticardiolipin antibodies and the risk for ischemic stroke and venous thrombosis. Ann Intern Med 117:997–1002
McClain MT, Arbuckle MR, Heinlen LD et al (2004) The prevalence, onset and clinical significance of antiphospholipid antibodies prior to diagnosis of systemic lupus erythematosus. Arthritis Rheum 50:1226–1232
Petri M (2010) Update on anti-phospholipid antibodies in SLE: the Hopkins’ Lupus Cohort. Lupus 19:419–423
Asherson RA (2005) Multiorgan failure and antiphospholipid antibodies: the catastrophic antiphospholipid (Asherson’s) syndrome. Immunobiology 210:727–733
Tincani A, Balestieri G, Danieli E et al (2003) Pregnancy complications of the antiphospholipid syndrome. Autoimmunity 36:27–32
Levine JS, Branch DW, Rauch J (2002) The antiphospholipid syndrome N Engl J Med 346:752–763
Sailer T, Zaglami C, Kurz C et al (2006) Anti-beta(2)-glycoprotein-I antibodies are associated with pregnancy loss in women with the lupus anticoagulant. Thromb Haemost 5:796–801
Erkan D, Lockshin MD (2010) Non-criteria manifestations of antiphospholipid syndrome. Lupus 19:424–427
Cervera R, Piette JC, Font J et al (2002) Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1, 000 patients. Arthritis Rheum 46:1019–1027
Galli M, Finazzi G, Barbui T (1996) Thrombocytopenia in the antiphospholipid syndrome. Br J Hematol 93:1–5
Harris EN, Asherson RA, Hughes GR (1988) Antiphospholipid antibodies–autoantibodies with a difference. Annu Rev Med 39:261–271
Gharavi AE, Pierangeli SS (1998) Origin of antiphospholipid antibodies: induction of aPL by viral peptides. Lupus 7(Suppl 2):S52–S54
Sherer Y, Blank M, Shoenfeld Y (2007) Antiphospholipid syndrome (APS): where does it come from? Best Pract Res Clin Rheumatol 21(6):1071–1078
Meroni PL, Raschi E, Camera M et al (2000) Endothelial activation by aPL: a potential pathogenetic mechanism for the clinical manifestations of the syndrome. J Autoimmun 15(2):237–240
Urbanus RT, Derksen RH, de Groot PG (2008) Platelets and the antiphospholipid syndrome. Lupus 17(10):888–894
Nojima J, Suehisa E, Kuratsune H et al (1999) Platelet activation induced by combined effects of anticardiolipin and lupus anticoagulant IgG antibodies in patients with systemic lupus erythematosus—possible association with thrombotic and thrombocytopenic complications. Thromb Haemost 81(3):436–441
Krone KA, Allen KL, McCrae KR (2010) Impaired fibrinolysis in the antiphospholipid syndrome. Curr Rheumatol Rep 12(1):53–57
Horstman LL, Jy W, Bidot CJ et al (2009) Antiphospholipid antibodies: paradigm in transition. J Neuroinflammation 6:3
D’Ippolito S, Di Simone N, Di Nicuolo F et al (2007) Antiphospholipid antibodies: effects on trophoblast and endothelial cells. Am J Reprod Immunol 58(2):150–158
Di Simone N, Luigi MP, Marco D et al (2007) Pregnancies complicated with antiphospholipid syndrome: the pathogenic mechanism of antiphospholipid antibodies: a review of the literature. Ann N Y Acad Sci 1108:505–514
Katsiari CG, Giavri I, Mitsikostas DD et al (2010) Acute transverse myelitis and antiphospholipid antibodies in lupus. No evidence for anticoagulation. Eur J Neurol. doi:10.1111/j.1468-1331.2010.03208.x. (in press)
Ames PR, Antinolfi I, Ciampa A et al (2008) Primary antiphospholipid syndrome: a low-grade auto-inflammatory disease? Rheumatology (Oxford) 47(12):1832–1837
Abrahams VM (2009) Mechanisms of antiphospholipid antibody-associated pregnancy complications. Thromb Res 124(5):521–525
Sebastiani GD, Galeazzi M (2009) Genetic aspects of the antiphospholipid syndrome: HLA associations. Handbook of systemic autoimmune diseases. Antiphospholipid Syndrome Syst Autoimmune Dis 10:81–89
Castro-Marrero J, Balada E, Vilardell-Tarrés M et al (2009) Genetic risk factors of thrombosis in the antiphospholipid syndrome. Br J Haematol 147(3):289–296
Gharavi AE, Pierangeli SS, Harris EN (2001) Origin of antiphospholipid antibodies. Rheum Dis Clin North Am 27(3):551–563
Gharavi AE, Pierangeli SS, Harris EN (2003) Viral origin of antiphospholipid antibodies: endothelial cell activation and thrombus enhancement by CMV peptide-induced APL antibodies. Immunobiology 207(1):37–42
Hashimoto Y, Kawamura M, Ichikawa K et al (1992) Anticardiolipin antibodies in NZW × BXSB F1 mice. A model of antiphospholipid syndrome. J Immunol 149(3):1063–1068
Hang LM, Izui S, Dixon FJ (1981) (NZW × BXSB)F1 hybrid. A model of acute lupus and coronary vascular disease with myocardial infarction. J Exp Med 154(1):216–221
Oyaizu N, Yasumizu R., Miyama-Inaba M, Nomura S, Yoshida H, Miyama S, Shibata Y, Mitsuoka S, Yasunaga K, Mori S, Good RA, Ikehara S (1988) (NZW × BXSB) F1 mouse, a new animal model of idlopathic thrombocytopenic purpura. J Exp Med 167(6):2017–2022
Kita Y, Sumida T, Iwamoto I, Yoshida S, Koike T (1994) V gene analysis of anti-cardiolipin antibodies from (NZW × BXSB) F1 mice. Immunology 82(3):494–501
Ida A, Hirose S, Hamano Y et al (1998) Multigenic control of lupus-associated antiphospholipid syndrome in a model of (NZW × BXSB) F1 mice. Eur J Immunol 28(9):2694–2703
Izui S, Masuda K, Yoshida H (1984) Acute SLE in F1 hybrids between SB/Le and NZW mice: Prominently enhanced formation of gp70 immune complexes by a Y chromosome-associated factor from SB/Le mice. J Immunol 132:701–710
Izui S, Higaki M, Morrow D, Merino R (1988) The Y chromosome from autoimmune BXSB/MpJ mice induces a lupus-like syndrome in (NZW × C57BL/6)F1 male mice, but not in C57BL/6 male mice. Eur J Immunol 18(6):911–915
Gharavi AE, Mellors RC, Elkon KB (1989) IgG anti-cardiolipin antibodies in murine lupus. Clin Exp Immunol 78(2):233–238
Kita Y, Sumida T, Ichikawa K, Maeda T, Yonaha F, Iwamoto I, Yoshida S, Koike T (1994) V gene analysis of anticardiolipin antibodies from MRL-lpr/lpr mice
Ahmed SA, Verthelyi D (1993) Antibodies to cardiolipin in normal C57BL/6 J mice: induction by estrogen but not dihydrotestosterone. J Autoimmun 6(3):265–279
Verthelyi D, Ansar Ahmed S (1997) Characterization of estrogen-induced autoantibodies to cardiolipin in non-autoimmune mice. J Autoimmun 10(2):115–125
Harvey AM, Shulman LE (1966) Connective tissue disease and the chronic biologic false-positive test for syphilis (BFP reaction). Med Clin North Am 50(5):1271–1279
Exner T, Barber S, Kronenberg H, Rickard KA (1980) Familial association of the lupus anticoagulant. Br J Haematol 45(1):89–96
Jolidon RM, Knecht H, Humair L, de Torrente A (1991) Different clinical presentations of a lupus anticoagulant in the same family. Klin Wochenschr 69(8):340–344
Matthey F, Walshe K, Mackie IJ, Machin SJ (1989) Familial occurrence of the antiphospholipid syndrome. J Clin Pathol 42(5):495–497
Mackworth-Young C, Chan J, Harris N et al (1987) High incidence of anticardiolipin antibodies in relatives of patients with systemic lupus erythematosus. J Rheumatol 14(4):723–726
Goldberg SN, Conti-Kelly AM, Greco TP (1995) A family study of anticardiolipin antibodies and associated clinical conditions. Am J Med 99(5):473–479
Goel N, Ortel TL, Bali D et al (1999) Familial antiphospholipid antibody syndrome: criteria for disease and evidence for autosomal dominant inheritance. Arthritis Rheum 42(2):318–327
Dagenais P, Urowitz MB, Gladman DD, Norman CS (1992) A family study of the antiphospholipid syndrome associated with other autoimmune diseases. J Rheumatol 19(9):1393–1396
Rouget JP, Goudemand J, Montreuil G et al (1982) Lupus anticoagulant: a familial observation. Lancet 2(8289):105
Mackie IJ, Colaco CB, Machin SJ (1987) Familial lupus anticoagulants. Br J Haematol 67(3):359–363
May KP, West SG, Moulds J, Kotzin BL (1993) Different manifestations of the antiphospholipid antibody syndrome in a family with systemic lupus erythematosus. Arthritis Rheum 36(4):528–533
Arnett FC, Olsen ML, Anderson KL, Reveille JD (1991) Molecular analysis of major histocompatibility complex alleles associated with the lupus anticoagulant. J Clin Invest 87(5):1490–1495
Asherson RA, Doherty DG, Vergani D et al (1992) Major histocompatibility complex associations with primary antiphospholipid syndrome. Arthritis Rheum 35(1):124–125
Caliz R, Atsumi T, Kondeatis E et al (2001) HLA class II gene polymorphisms in antiphospholipid syndrome: haplotype analysis in 83 Caucasoid patients. Rheumatology (Oxford) 40(1):31–36
Bertolaccini ML, Atsumi T, Caliz AR et al (2000) Association of antiphosphatidylserine/prothrombin autoantibodies with HLA class II genes. Arthritis Rheum 43(3):683–688
Vargas-Alarcon G, Granados J, Bekker C et al (1995) Association of HLA-DR5 (possibly DRB1*1201) with the primary antiphospholipid syndrome in Mexican patients. Arthritis Rheum 38(9):1340–1341
Galeazzi M, Sebastiani GD, Tincani A et al (2000) HLA class II alleles associations of anticardiolipin and anti-beta2GPI antibodies in a large series of European patients with systemic lupus erythematosus. Lupus 9(1):47–55
Hashimoto H, Yamanaka K, Tokano Y et al (1998) HLA-DRB1 alleles and beta 2 glycoprotein I-dependent anticardiolipin antibodies in Japanese patients with systemic lupus erythematosus. Clin Exp Rheumatol 16(4):423–427
Arnett FC, Thiagarajan P, Ahn C, Reveille JD (1999) Associations of anti-beta2-glycoprotein I autoantibodies with HLA class II alleles in three ethnic groups. Arthritis Rheum 42(2):268–274
Wilson WA, Perez MC, Michalski JP, Armatis PE (1988) Cardiolipin antibodies and null alleles of C4 in black Americans with systemic lupus erythematosus. J Rheumatol 15(12):1768–1772
Wilson WA, Scopelitis E, Michalski JP et al (1995) Familial anticardiolipin antibodies and C4 deficiency genotypes that coexist with MHC DQB1 risk factors. J Rheumatol 22(2):227–235
Petri M, Watson R, Winkelstein JA, McLean RH (1993) Clinical expression of systemic lupus erythematosus in patients with C4A deficiency. Medicine (Baltimore) 72(4):236–244
Hirose N, Williams R, Alberts AR et al (1999) A role for the polymorphism at position 247 of the beta2-glycoprotein I gene in the generation of anti-beta2-glycoprotein I antibodies in the antiphospholipid syndrome. Arthritis Rheum 42(8):1655–1661
Atsumi T, Tsutsumi A, Amengual O et al (1999) Correlation between beta2-glycoprotein I valine/leucine247 polymorphism and anti-beta2-glycoprotein I antibodies in patients with primary antiphospholipid syndrome. Rheumatology (Oxford) 38(8):721–723
Prieto GA, Cabral AR, Zapata-Zuñiga M et al (2003) Valine/valine genotype at position 247 of the beta2-glycoprotein I gene in Mexican patients with primary antiphospholipid syndrome: association with anti-beta2-glycoprotein I antibodies. Arthritis Rheum 48(2):471–474
Reverter JC, Tassies MD (2009) Genetic aspects of the antiphospholipid syndrome: associations with clinical manifestations. Handbook of Systemic Autoimmune Diseases. Antiphospholipid Syndrome in Systemic Autoimmune Disease 10:91–103
Rees DC, Cox M, Clegg JB (1995) World distribution of factor V Leiden. Lancet 346(8983):1133–1134
Franco RF, Elion J, Tavella MH et al (1999) The prevalence of factor V Arg306– > Thr (factor V Cambridge) and factor V Arg306– > Gly mutations in different human populations. Thromb Haemost 81(2):312–313
Schütt M, Klüter H, Hagedorn-Greiwe M et al (1998) Familial coexistence of primary antiphospholipid syndrome and factor V Leiden. Lupus 7(3):176–182
Brenner B, Vulfsons SL, Lanir N, Nahir M (1996) Coexistence of familial antiphospholipid syndrome and factor V Leiden: impact on thrombotic diathesis. Br J Haematol 94(1):166–167
Chopra N, Koren S, Greer WL et al (2002) Factor V Leiden, prothrombin gene mutation, and thrombosis risk in patients with antiphospholipid antibodies. J Rheumatol 29(8):1683–1688
Bentolila S, Ripoll L, Drouet L et al (1997) Lack of association between thrombosis in primary antiphospholipid syndrome and the recently described thrombophilic 3′-untranslated prothrombin gene polymorphism. Thromb Haemost 78(5):1415
Bertolaccini ML, Atsumi T, Hunt BJ et al (1998) Prothrombin mutation is not associated with thrombosis in patients with antiphospholipid syndrome. Thromb Haemost 80(1):202–203
Ruiz-Argüelles GJ, Garcés-Eisele J, Ruiz-Delgado GJ, Alarcón-Segovia D (1999) The G20210A polymorphism in the 3′-untranslated region of the prothrombin gene in Mexican mestizo patients with primary antiphospholipid syndrome. Clin Appl Thromb Hemost 5(3):158–160
Sivera P, Bosio S, Bertero MT et al (2000) G20210A homozygosity in antiphospholipid syndrome secondary to systemic lupus erythematosus. Haematologica 85(1):109–110
de Visser MC, Rosendaal FR, Bertina RM (1999) A reduced sensitivity for activated protein C in the absence of factor V Leiden increases the risk of venous thrombosis. Blood 93(4):1271–1276
Erkan D, Zhang HW, Shriky RC, Merrill JT (2002) Dual antibody reactivity to beta2-glycoprotein I and protein S: increased association with thrombotic events in the antiphospholipid syndrome. Lupus 11(4):215–220
Ames PR, Margaglione M, Tommasino C et al (2001) Impact of plasma homocysteine and prothrombin G20210 A on primary antiphospholipid syndrome. Blood Coagul Fibrinolysis 12(8):699–704
Lincz LF, Adams MJ, Scorgie FE et al (2007) Polymorphisms of the tissue factor pathway inhibitor gene are associated with venous thromboembolism in the antiphospholipid syndrome and carriers of factor V Leiden. Blood Coagul Fibrinolysis 18(6):559–564
Jiménez S, Tàssies D, Espinosa G et al (2008) Double heterozygosity polymorphisms for platelet glycoproteins Ia/IIa and IIb/IIIa increases arterial thrombosis and arteriosclerosis in patients with the antiphospholipid syndrome or with systemic lupus erythematosus. Ann Rheum Dis 67(6):835–840
Yasuda S, Tsutsumi A, Atsumi T et al (2002) Gene polymorphisms of tissue plasminogen activator and plasminogen activator inhibitor-1 in patients with antiphospholipid antibodies. J Rheumatol 29(6):1192–1197
Diz-Kucukkaya R, Hancer VS, Inanc M et al (2004) Factor XIII Val34Leu polymorphism does not contribute to the prevention of thrombotic complications in patients with antiphospholipid syndrome. Lupus 13(1):32–35
de Laat B, Derksen RH, Mackie IJ et al (2006) Annexin A5 polymorphism (-1C– > T) and the presence of anti-annexin A5 antibodies in the antiphospholipid syndrome. Ann Rheum Dis 65(11):1468–1472
Diz-Kucukkaya R, Inanc M, Afshar-Kharghan V et al (2007) P-selectin glycoprotein ligand-1 VNTR polymorphisms and risk of thrombosis in the antiphospholipid syndrome. Ann Rheum Dis 66(10):1378–1380
de la Red G, Tàssies D, Espinosa G et al (2009) Factor XIII-A subunit Val34Leu polymorphism is associated with the risk of thrombosis in patients with antiphospholipid antibodies and high fibrinogen levels. Thromb Haemost 101(2):312–316
Bugert P, Pabinger I, Stamer K et al (2007) The risk for thromboembolic disease in lupus anticoagulant patients due to pathways involving P-selectin and CD154. Thromb Haemost 97(4):573–580
Bertolaccini ML, Atsumi T, Lanchbury JS et al (2001) Plasma tumor necrosis factor alpha levels and the -238*A promoter polymorphism in patients with antiphospholipid syndrome. Thromb Haemost 85(2):198–203
Pierangeli SS, Vega-Ostertag ME, Raschi E et al (2007) Toll-like receptor and antiphospholipid mediated thrombosis: in vivo studies. Ann Rheum Dis 66(10):1327–1333
Gharavi AE, Sammaritano LR, Wen J, Elkon KB (1992) Induction of antiphospholipid autoantibodies by immunization with beta 2 glycoprotein I (apolipoprotein H). J Clin Invest 90(3):1105–1109
Gharavi AE, Pierangeli SS, Colden-Stanfield M et al (1999) GDKV-induced antiphospholipid antibodies enhance thrombosis and activate endothelial cells in vivo and in vitro. J Immunol 163(5):2922–2927
Gharavi AE, Pierangeli SS, Gharavi EE et al (1998) Thrombogenic properties of antiphospholipid antibodies do not depend on their binding to beta2 glycoprotein 1 (beta2GP1) alone. Lupus 7(5):341–346
Gharavi EE, Chaimovich H, Cucurull E et al (1999) Induction of antiphospholipid antibodies by immunization with synthetic viral and bacterial peptides. Lupus 8(6):449–455
Gharavi AE, Pierangeli SS, Espinola RG et al (2002) Antiphospholipid antibodies induced in mice by immunization with a cytomegalovirus-derived peptide cause thrombosis and activation of endothelial cells in vivo. Arthritis Rheum 46(2):545–552
Gharavi AE, Vega-Ostertag M, Espinola RG et al (2004) Intrauterine fetal death in mice caused by cytomegalovirus-derived peptide induced aPL antibodies. Lupus 13(1):17–23
Blank M, Krause I, Fridkin M et al (2002) Bacterial induction of autoantibodies to beta2-glycoprotein-I accounts for the infectious etiology of antiphospholipid syndrome. J Clin Invest 109(6):797–804
Levy Y, Almog O, Gorshtein A, Shoenfeld Y (2006) The environment and antiphospholipid syndrome. Lupus 15(11):784–790
Wasserman A (1907) Über Entwicklung und den Gegenwartingen Stand der Serodiagnostic gegenüber Syphilis, Berl. Klin Wochenschr 44:1599–1634
Harris EN, Gharavi AE, Boey ML et al (1983) Anticardiolipin antibodies: detection by radioimmunoassay and association with thrombosis in systemic lupus erythematosus. Lancet 2(8361):1211–1214
Uthman IW, Gharavi AE (2002) Viral infections and antiphospholipid antibodies. Semin Arthritis Rheum 31(4):256–263
Sène D, Piette JC, Cacoub P (2009) Antiphospholipid antibodies, antiphospholipid syndrome and viral infections. Rev Med Int 30(2):135–141
van de Berg PJ, Heutinck KM, Raabe R et al (2010) Human cytomegalovirus induces systemic immune activation characterized by a type 1 cytokine signature. J Infect Dis 202(5):690–699
Prandota J (2003) Possible pathomechanism of autoimmune hepatitis. Am J Ther 10(1):51–57
Nakagawa K, Harrison LC (1996) The potential roles of endogenous retroviruses in autoimmunity. Immunol Rev 152:193–236
Molina V, Shoenfeld Y (2005) Infection, vaccines and other environmental triggers of autoimmunity. Autoimmunity 38(3):235–245
Martinuc Porobic J, Avcin T, Bozic B et al (2005) Anti-phospholipid antibodies following vaccination with recombinant hepatitis B vaccine. Clin Exp Immunol 142(2):377–380
Uetrecht J (2005) Current trends in drug-induced autoimmunity. Autoimmun Rev 4(5):309–314
Lillicrap DP, Pinto M, Benford K et al (1990) Heterogeneity of laboratory test results for antiphospholipid antibodies in patients treated with chlorpromazine and other phenothiazines. Am J Clin Pathol 93(6):771–775
Tincani A, Taraborelli M, Cattaneo R (2010) Antiphospholipid antibodies and malignancies. Autoimmun Rev 9(4):200–202
Palomo I, Segovia F, Ortega C, Pierangeli S (2009) Antiphospholipid syndrome: a comprehensive review of a complex and multisystemic disease. Clin Exp Rheumatol 27(4):668–677
Miyakis S, Giannakopoulos B, Krilis SA (2004) Beta 2 glycoprotein I—function in health and disease. Thromb Res 114(5–6):335–346
Shoenfeld Y, Blank M, Cervera R et al (2006) Infectious origin of the antiphospholipid syndrome. Ann Rheum Dis 65(1):2–6
Harris EN, Asherson RA, Gharavi AE et al (1985) Thrombocytopenia in SLE and related autoimmune disorders: association with anticardiolipin antibody. Br J Haematol 59(2):227–230
Lellouche F, Martinuzzo M, Said P et al (1991) Imbalance of thromboxane/prostacyclin biosynthesis in patients with lupus anticoagulant. Blood 78(11):2894–2899
Khamashta MA, Harris EN, Gharavi AE et al (1988) Immune mediated mechanism for thrombosis: antiphospholipid antibody binding to platelet membranes. Ann Rheum Dis 47(10):849–854
Lutters BC, Derksen RH, Tekelenburg WL et al (2003) Dimers of beta 2-glycoprotein I increase platelet deposition to collagen via interaction with phospholipids and the apolipoprotein E receptor 2′. J Biol Chem 278(36):33831–33838
Espinola RG, Pierangeli SS, Gharavi AE, Harris EN (2002) Hydroxychloroquine reverses platelet activation induced by human IgG antiphospholipid antibodies. Thromb Haemost 87(3):518–522
Pierangeli SS, Vega-Ostertag M, Harris EN (2004) Intracellular signaling triggered by antiphospholipid antibodies in platelets and endothelial cells: a pathway to targeted therapies. Thromb Res 114(5–6):467–476
Shi T, Giannakopoulos B, Yan X et al (2006) Anti-beta2-glycoprotein I antibodies in complex with beta2-glycoprotein I can activate platelets in a dysregulated manner via glycoprotein Ib-IX-V. Arthritis Rheum 54(8):2558–2567
Urbanus RT, Pennings MT, Derksen RH, de Groot PG (2008) Platelet activation by dimeric beta2-glycoprotein I requires signaling via both glycoprotein Ibalpha and apolipoprotein E receptor 2’. J Thromb Haemost 6(8):1405–1412
Sikara MP, Routsias JG, Samiotaki M et al (2010) {beta}2 Glycoprotein I ({beta}2GPI) binds platelet factor 4 (PF4): implications for the pathogenesis of antiphospholipid syndrome. Blood 115(3):713–723
Pierangeli SS, Colden-Stanfield M, Liu X et al (1999) Antiphospholipid antibodies from antiphospholipid syndrome patients activate endothelial cells in vitro and in vivo. Circulation 99(15):1997–2002
Meroni PL, Raschi E, Camera M et al (2000) Endothelial activation by aPL: a potential pathogenetic mechanism for the clinical manifestations of the syndrome. J Autoimmun 15(2):237–240
Simantov R, Lo SK, Gharavi A et al (1996) Antiphospholipid antibodies activate vascular endothelial cells. Lupus 5(5):440–441
Pierangeli SS, Espinola RG, Liu X, Harris EN (2001) Thrombogenic effects of antiphospholipid antibodies are mediated by intercellular cell adhesion molecule-1, vascular cell adhesion molecule-1, and P-selectin. Circ Res 88(2):245–250
Amengual O, Atsumi T, Khamashta MA, Hughes GR (1998) The role of the tissue factor pathway in the hypercoagulable state in patients with the antiphospholipid syndrome. Thromb Haemost 79(2):276–281
Kornberg A, Blank M, Kaufman S, Shoenfeld Y (1994) Induction of tissue factor-like activity in monocytes by anti-cardiolipin antibodies. J Immunol 153(3):1328–1332
Reverter JC, Tàssies D, Font J et al (1998) Effects of human monoclonal anticardiolipin antibodies on platelet function and on tissue factor expression on monocytes. Arthritis Rheum 41(8):1420–1427
Alijotas-Reig J, Palacio-Garcia C, Vilardell-Tarres M (2009) Circulating microparticles, lupus anticoagulant and recurrent miscarriages. Eur J Obstet Gynecol Reprod Biol 145(1):22–26
Cuadrado MJ, Buendía P, Velasco F et al (2006) Vascular endothelial growth factor expression in monocytes from patients with primary antiphospholipid syndrome. J Thromb Haemost 4(11):2461–2469
Raschi E, Testoni C, Bosisio D et al (2003) Role of the MyD88 transduction signaling pathway in endothelial activation by antiphospholipid antibodies. Blood 101(9):3495–3500
Vega-Ostertag ME, Ferrara DE, Romay-Penabad Z et al (2007) Role of p38 mitogen-activated protein kinase in antiphospholipid antibody-mediated thrombosis and endothelial cell activation. J Thromb Haemost 5(9):1828–1834
Jajoria P, Murthy V, Papalardo E, Romay-Penabad Z, Gleason C, Pierangeli SS (2009) Statins for the treatment of antiphospholipid syndrome? Ann N Y Acad Sci 1173:736–745
Fischetti F, Durigutto P, Pellis V et al (2005) Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood 106(7):2340–2346
Zhang J, McCrae KR (2005) Annexin A2 mediates endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood 105(5):1964–1969
Romay-Penabad Z, Montiel-Manzano MG, Shilagard T, Papalardo E et al (2009) Annexin A2 is involved in antiphospholipid antibody-mediated pathogenic effects in vitro and in vivo. Blood 114(14):3074–3083
Sorice M, Longo A, Capozzi A et al (2007) Anti-beta2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis Rheum 56(8):2687–2697
Lambrianides A, Carroll CJ, Pierangeli SS et al (2010) Effects of polyclonal IgG derived from patients with different clinical types of the antiphospholipid syndrome on monocyte signaling pathways. J Immunol 184(12):6622–6628
Satta N, Dunoyer-Geindre S, Reber G et al (2007) The role of TLR2 in the inflammatory activation of mouse fibroblasts by human antiphospholipid antibodies. Blood 109(4):1507–1514
Verstak B, Nagpal K, Bottomley SP et al (2009) MyD88 adapter-like (Mal)/TIRAP interaction with TRAF6 is critical for TLR2- and TLR4-mediated NF-kappaB proinflammatory responses. J Biol Chem 284(36):24192–24203
Alard JE, Gaillard F, Daridon C et al (2010) TLR2 is one of the endothelial receptors for beta 2-glycoprotein I. J Immunol 185(3):1550–1557
Döring Y, Hurst J, Lorenz M et al (2010) Human antiphospholipid antibodies induce TNFalpha in monocytes via Toll-like receptor 8. Immunobiology 215(3):230–241
Hurst J, Prinz N, Lorenz M et al (2009) TLR7 and TLR8 ligands and antiphospholipid antibodies show synergistic effects on the induction of IL-1beta and caspase-1 in monocytes and dendritic cells. Immunobiology 214(8):683–691
Raschi E, Broggini V, Grossi C, Borghi MO, Pierangeli SS, Meroni PL (2009) Mechanisms of action of antiphospholipid antibodies. Handbook of systemic autoimmune diseases. Antiphospholipid Syndr Syst Autoimmun Dis 10:55–67
Romay-Penabad Z, Aguilar-Valenzuela R, Urbanus RT, Derksen H et al (2010) Apolipoprotein E receptor 2′ is involved in the thrombotic complications in a murine model of the antiphospholipid syndrome. Blood. (in press)
Krone KA, Allen KL, McCrae KR (2010) Impaired fibrinolysis in the antiphospholipid syndrome. Curr Rheumatol Rep 12(1):53–57
Griffin JH, Fernandez JA, Gale AJ, Mosnier LO (2007) Activated protein C. J Thromb Haemost 5(Suppl 1):73–80
Urbanus RT, de Laat B (2010) Antiphospholipid antibodies and the protein C pathway. Lupus 19(4):394–399
Atsumi T, Ieko M, Bertolaccini ML et al (2000) Association of autoantibodies against the phosphatidylserine-prothrombin complex with manifestations of the antiphospholipid syndrome and with the presence of lupus anticoagulant. Arthritis Rheum 43(9):1982–1993
High KA (1988) Antithrombin III, protein C, and protein S. Naturally occurring anticoagulant proteins. Arch Pathol Lab Med 112(1):28–36
Kolev K, Léránt I, Skopál J et al (2005) Impaired inactivation by antithrombin and hirudin and preserved fibrinogen-clotting activity of thrombin in complex with anti-thrombin antibody from a patient with antiphospholipid syndrome. Thromb Haemost 94(1):82–87
Chamley LW, McKay EJ, Pattison NS (1993) Inhibition of heparin/antithrombin III cofactor activity by anticardiolipin antibodies: a mechanism for thrombosis. Thromb Res 71(2):103–111
Shibata S, Harpel PC, Gharavi A, Rand J, Fillit H (1994) Autoantibodies to heparin from patients with antiphospholipid antibody syndrome inhibit formation of antithrombin III-thrombin complexes. Blood 83(9):2532–2540
Krikun G, Lockwood CJ, Wu XX et al (1994) The expression of the placental anticoagulant protein, annexin V, by villous trophoblasts: immunolocalization and in vitro regulation. Placenta 15(6):601–612
Cugno M, Cabibbe M, Galli M et al (2004) Antibodies to tissue-type plasminogen activator (tPA) in patients with antiphospholipid syndrome: evidence of interaction between the antibodies and the catalytic domain of tPA in 2 patients. Blood 103(6):2121–2126
Ames PR, Tommasino C, Iannaccone L et al (1996) Coagulation activation and fibrinolytic imbalance in subjects with idiopathic antiphospholipid antibodies—a crucial role for acquired free protein S deficiency. Thromb Haemost 76(2):190–194
Atsumi T, Khamashta MA, Andujar C et al (1998) Elevated plasma lipoprotein(a) level and its association with impaired fibrinolysis in patients with antiphospholipid syndrome. J Rheumatol 25(1):69–73
Yang CD, Hwang KK, Yan W, Gallagher K et al (2004) Identification of anti-plasmin antibodies in the antiphospholipid syndrome that inhibit degradation of fibrin. J Immunol 172(9):5765–5773
Cesarman GM, Guevara CA, Hajjar KA (1994) An endothelial cell receptor for plasminogen/tissue plasminogen activator (t-PA). II. Annexin II-mediated enhancement of t-PA-dependent plasminogen activation. J Biol Chem 269(33):21198–21203
Ao W, Zheng H, Chen XW et al (2010) Anti-annexin II antibody is associated with thrombosis and/or pregnancy morbidity in antiphospholipid syndrome and systemic lupus erythematosus with thrombosis. Rheumatol Int (in press)
Takeuchi R, Atsumi T, Ieko M et al (2002) Suppressed intrinsic fibrinolytic activity by monoclonal anti-beta-2 glycoprotein I autoantibodies: possible mechanism for thrombosis in patients with antiphospholipid syndrome. Br J Haematol 119(3):781–788
Ieko M, Ichikawa K, Atsumi T et al (2000) Effects of beta2-glycoprotein I and monoclonal anticardiolipin antibodies on extrinsic fibrinolysis. Semin Thromb Hemost 26(1):85–90
López-Lira F, Rosales-León L, Martínez VM, Ruiz Ordaz BH (2006) The role of beta2-glycoprotein I (beta2GPI) in the activation of plasminogen. Biochim Biophys Acta 1764(4):815–823
Bu C, Gao L, Xie W et al (2009) beta2-glycoprotein i is a cofactor for tissue plasminogen activator-mediated plasminogen activation. Arthritis Rheum 60(2):559–568
De Wolf F, Carreras LO, Moerman P et al (1982) Decidual vasculopathy and extensive placental infarction in a patient with repeated thromboembolic accidents, recurrent fetal loss, and a lupus anticoagulant. Am J Obstet Gynecol 142(7):829–834
Hanly JG, Gladman DD, Rose TH et al (1988) Lupus pregnancy. A prospective study of placental changes. Arthritis Rheum 31(3):358–366
Nayar R, Lage JM (1996) Placental changes in a first trimester missed abortion in maternal systemic lupus erythematosus with antiphospholipid syndrome; a case report and review of the literature. Hum Pathol 27(2):201–206
Peaceman AM, Rehnberg KA (1993) The effect of immunoglobulin G fractions from patients with lupus anticoagulant on placental prostacyclin and thromboxane production. Am J Obstet Gynecol 169(6):1403–1406
Rand JH, Wu XX, Guller S et al (1994) Reduction of annexin-V (placental anticoagulant protein-I) on placental villi of women with antiphospholipid antibodies and recurrent spontaneous abortion. Am J Obstet Gynecol 171(6):1566–1572
Rand JH, Wu XX, Guller S et al (1997) Antiphospholipid immunoglobulin G antibodies reduce annexin-V levels on syncytiotrophoblast apical membranes and in culture media of placental villi. Am J Obstet Gynecol 177(4):918–923
Wang X, Campos B, Kaetzel MA, Dedman JR (1999) Annexin V is critical in the maintenance of murine placental integrity. Am J Obstet Gynecol 180(4):1008–1016
Nojima J, Kuratsune H, Suehisa E et al (2001) Association between the prevalence of antibodies to beta(2)-glycoprotein I, prothrombin, protein C, protein S, and annexin V in patients with systemic lupus erythematosus and thrombotic and thrombocytopenic complications. Clin Chem 47(6):1008–1015
Donohoe S, Kingdom JC, Mackie IJ et al (2000) Ontogeny of beta 2 glycoprotein I and annexin V in villous placenta of normal and antiphospholipid syndrome pregnancies. Thromb Haemost 84(1):32–38
Out HJ, Kooijman CD, Bruinse HW, Derksen RH (1991) Histopathological findings in placentae from patients with intra-uterine fetal death and anti-phospholipid antibodies. Eur J Obstet Gynecol Reprod Biol 41(3):179–186
Di Simone N, Luigi MP, Marco D et al (2007) Pregnancies complicated with antiphospholipid syndrome: the pathogenic mechanism of antiphospholipid antibodies: a review of the literature. Ann N Y Acad Sci 1108:505–514
Sebire NJ, Fox H, Backos M et al (2002) Defective endovascular trophoblast invasion in primary antiphospholipid antibody syndrome-associated early pregnancy failure. Hum Reprod 17(4):1067–1071
McIntyre JA (1992) Immune recognition at the maternal-fetal interface: overview. Am J Reprod Immunol 28(3–4):127–131
Castellucci M, De Matteis R, Meisser A et al (2000) Leptin modulates extracellular matrix molecules and metalloproteinases: possible implications for trophoblast invasion. Mol Hum Reprod 6(10):951–958
Di Simone N, Meroni PL, de Papa N et al (2000) Antiphospholipid antibodies affect trophoblast gonadotropin secretion and invasiveness by binding directly and through adhered beta2-glycoprotein I. Arthritis Rheum 43(1):140–150
Katsuragawa H, Kanzaki H, Inoue T et al (1997) Monoclonal antibody against phosphatidylserine inhibits in vitro human trophoblastic hormone production and invasion. Biol Reprod 56(1):50–58
Adler RR, Ng AK, Rote NS (1995) Monoclonal antiphosphatidylserine antibody inhibits intercellular fusion of the choriocarcinoma line, JAR. Biol Reprod 53(4):905–910
Rote NS, Vogt E, DeVere G et al (1998) The role of placental trophoblast in the pathophysiology of the antiphospholipid antibody syndrome. Am J Reprod Immunol 39(2):125–136
Chamley LW, Duncalf AM, Mitchell MD, Johnson PM (1998) Action of anticardiolipin and antibodies to beta2-glycoprotein-I on trophoblast proliferation as a mechanism for fetal death. Lancet 352(9133):1037–1038
Di Simone N, Castellani R, Caliandro D, Caruso A (2002) Antiphospholipid antibodies regulate the expression of trophoblast cell adhesion molecules. Fertil Steril 77(4):805–811
Di Simone N, Marana R, Castellani R et al (2010) Decreased expression of heparin-binding epidermal growth factor-like growth factor as a newly identified pathogenic mechanism of antiphospholipid-mediated defective placentation. Arthritis Rheum 62(5):1504–1512
Ornoy A, Yacobi S, Matalon ST et al (2003) The effects of antiphospholipid antibodies obtained from women with SLE/APS and associated pregnancy loss on rat embryos and placental explants in culture. Lupus 12(7):573–578
Matalon ST, Shoenfeld Y, Blank M et al (2004) Antiphosphatidylserine antibodies affect rat yolk sacs in culture: a mechanism for fetal loss in antiphospholipid syndrome. Am J Reprod Immunol 51(2):144–151
Di Simone N, Castellani R, Raschi E et al (2006) Anti-beta-2 glycoprotein I antibodies affect Bcl-2 and Bax trophoblast expression without evidence of apoptosis. Ann N Y Acad Sci 1069:364–376
Borghi MO, Raschi E, Scurati S et al (2007) Effects of a toll-like receptor antagonist and anti-annexin A2 antibodies on binding and activation of decidual cells by anti-β2glycoprotein I antibodies. Clin Exp Rheumatol 2:35
Francis J, Rai R, Sebire NJ et al (2006) Impaired expression of endometrial differentiation markers and complement regulatory proteins in patients with recurrent pregnancy loss associated with antiphospholipid syndrome. Mol Hum Reprod 12(7):435–442
Di Simone N, Di Nicuolo F, D’Ippolito S et al (2010) Antiphospholipid antibodies affect human endometrial angiogenesis. Biol Reprod 83(2):212–219
Riley JK, Nelson DM (2009) Toll-like Receptors in Pregnancy Disorders and Placental Dysfunction. Clin Rev Allergy Immunol (in press)
Bogic LV, Ohira RH, Yamamoto SY et al (1999) Tissue plasminogen activator and its receptor in the human amnion, chorion, and decidua at preterm and term. Biol Reprod 60(4):1006–1012
Mulla MJ, Brosens JJ, Chamley LW et al (2009) Antiphospholipid antibodies induce a pro-inflammatory response in first trimester trophoblast via the TLR4/MyD88 pathway. Am J Reprod Immunol 62(2):96–111
Mulla MJ, Myrtolli K, Brosens JJ et al (2010) Antiphospholipid antibodies limit trophoblast migration by reducing IL-6 production and STAT3 activity. Am J Reprod Immunol 63(5):339–348
Amigo MC (2006) Kidney disease in antiphospholipid syndrome. Rheum Dis Clin North Am 32(3):509–522
Mayer M, Cerovec M, Rados M, Cikes N (2010) Antiphospholipid syndrome and central nervous system. Clin Neurol Neurosurg 112(7):602–608
Tektonidou MG, Varsou N, Kotoulas G et al (2006) Cognitive deficits in patients with antiphospholipid syndrome: association with clinical, laboratory, and brain magnetic resonance imaging findings. Arch Intern Med 166(20):2278–2284
Chapman J, Cohen-Armon M, Shoenfeld Y, Korczyn AD (1999) Antiphospholipid antibodies permeabilize and depolarize brain synaptoneurosomes. Lupus 8(2):127–133
Gertner E (1999) Diffuse alveolar hemorrhage in the antiphospholipid syndrome: spectrum of disease and treatment. J Rheumatol 26(4):805–807
Deane KD, West SG (2005) Antiphospholipid antibodies as a cause of pulmonary capillaritis and diffuse alveolar hemorrhage: a case series and literature review. Semin Arthritis Rheum 35(3):154–165
Asherson RA, Cervera R, Wells AU (2005) Diffuse alveolar hemorrhage: a nonthrombotic antiphospholipid lung syndrome? Semin Arthritis Rheum 35(3):138–142
Matsuura E, Kobayashi K, Hurley BL, Lopez LR (2006) Atherogenic oxidized low-density lipoprotein/beta2-glycoprotein I (oxLDL/beta2GPI) complexes in patients with systemic lupus erythematosus and antiphospholipid syndrome. Lupus 15(7):478–483
Kobayashi K, Lopez LR, Matsuura E (2007) Atherogenic antiphospholipid antibodies in antiphospholipid syndrome. Ann N Y Acad Sci 1108:489–496
Chaouat G (2007) The Th1/Th2 paradigm: still important in pregnancy? Semin Immunopathol 29(2):95–113
Holers VM, Girardi G, Mo L et al (2002) Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J Exp Med 195(2):211–220
Martinez de la Torre Y, Buracchi C, Borroni EM et al (2007) Protection against inflammation- and autoantibody-caused fetal loss by the chemokine decoy receptor D6. Proc Natl Acad Sci USA 104(7):2319–2324
Quigg RJ, Kozono Y, Berthiaume D et al (1998) Blockade of antibody-induced glomerulonephritis with Crry-Ig, a soluble murine complement inhibitor. J Immunol 160(9):4553–4560
Girardi G, Berman J, Redecha P et al (2003) Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest 112(11):1644–1654
Redecha P, Tilley R, Tencati M et al (2007) Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood 110(7):2423–2431
Girardi G, Yarilin D, Thurman JM et al (2006) Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J Exp Med 203(9):2165–2175
Girardi G, Redecha P, Salmon JE (2004) Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat Med 10(11):1222–1226
Shamonki JM, Salmon JE, Hyjek E, Baergen RN (2007) Excessive complement activation is associated with placental injury in patients with antiphospholipid antibodies. Am J Obstet Gynecol196(2):167.e1–5
Cavazzana I, Manuela N, Irene C et al (2007) Complement activation in anti-phospholipid syndrome: a clue for an inflammatory process? J Autoimmun 28(2–3):160–164
Stone S, Pijnenborg R, Vercruysse L et al (2006) The placental bed in pregnancies complicated by primary antiphospholipid syndrome. Placenta 27(4–5):457–467
Salmon JE, Girardi G, Theodore E (2007) Woodward Award: antiphospholipid syndrome revisited: a disorder initiated by inflammation. Trans Am Clin Climatol Assoc 118:99–114
Pierangeli SS, Girardi G, Vega-Ostertag M et al (2005) Requirement of activation of complement C3 and C5 for antiphospholipid antibody-mediated thrombophilia. Arthritis Rheum 52(7):2120–2124
Romay-Penabad Z, Liu XX, Montiel-Manzano G, Papalardo De Martínez E, Pierangeli SS (2007) C5a receptor-deficient mice are protected from thrombophilia and endothelial cell activation induced by some antiphospholipid antibodies. Ann N Y Acad Sci 1108:554–566
Carrera-Marin AL, Romay-Penabad Z, Qu HC et al (2009) A C5a receptor antagonist ameliorates in vivo effects of antiphospholipid antibodies. Arthritis Rheum 60:s767 (abstract)
Carrera-Marin AL, Aguilar-Valenzuela R, Romay-Penabad Z et al (2010) Involvement of C6 and the MAC on in vivo antiphospholipid-mediated pathogenic effects. Lupus 19:507 (abstract)
Oku K, Atsumi T, Bohgaki M et al (2009) Complement activation in patients with primary antiphospholipid syndrome. Ann Rheum Dis 68(6):1030–1035
Pierangeli SS, Vega-Ostertag ME, González EB (2007) New targeted therapies for treatment of thrombosis in antiphospholipid syndrome. Expert Rev Mol Med 9(30):1–15
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Willis, R., Pierangeli, S.S. Pathophysiology of the antiphospholipid antibody syndrome. Autoimmun Highlights 2, 35–52 (2011). https://doi.org/10.1007/s13317-011-0017-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13317-011-0017-9