
Abstract
Traumatic brain injury (TBI) is a leading cause of mortality, sensorimotor morbidity, and neurocognitive disability. Neuroinflammation is one of the key drivers causing secondary brain injury after TBI. Therefore, attenuation of the inflammatory response is a potential therapeutic goal. This review summarizes the most important neuroinflammatory pathophysiology resulting from TBI and the clinical trials performed to attenuate neuroinflammation. Studies show that non-selective attenuation of the inflammatory response, in the early phase after TBI, might be detrimental and that there is a gap in the literature regarding pharmacological trials targeting specific pathways. The complement system and its crosstalk with the coagulation system play an important role in the pathophysiology of secondary brain injury after TBI. Therefore, regaining control over the complement cascades by inhibiting overshooting activation might constitute useful therapy. Activation of the complement cascade is an early component of neuroinflammation, making it a potential target to mitigate neuroinflammation in TBI. Therefore, we have described pathophysiological aspects of complement inhibition and summarized animal studies targeting the complement system in TBI. We also present the first clinical trial aimed at inhibition of complement activation in the early days after brain injury to reduce the risk of morbidity and mortality following severe TBI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Traumatic brain injury (TBI) presents a great challenge to public health worldwide. TBI is responsible for over a third of all traumatic deaths, and each year, 80–90.000 new cases of long-term disability due to TBI occur in the USA [1]. The dynamic pathophysiology that evolves over time after trauma to the central nervous system (CNS), consisting of primary injury by the direct traumatic impact, followed by secondary brain tissue injury driven substantially by host responses, makes it a highly complex problem to tackle compared to trauma in other organs [1]. Primary damage develops due to direct and contrecoup mechanical forces on the brain, including damage to neurons, axons, and glial cells and shearing of blood vessels causing hemorrhage [2]. The damage causes a breakdown of the blood–brain barrier (BBB), and changes in blood supply result in mitochondrial and subsequent energy production impairment, release of neurotransmitters and free radicals, immune cell activation and infiltration, apoptosis, and cytokine release [2, 3]. This is the initiation of the secondary brain-injury period, which occurs after a latency interval of minutes to several hours, and is probably the most important to focus on. Neuroinflammation develops over hours to days after the trauma and results in edema formation and subsequent increased intracranial pressure (ICP). Increased ICP causes additional impairment of cerebral blood flow and oxygen delivery and may contribute to brain herniation, requiring additional neuro-interventions that complicate the hospital course and final recovery [4].
TBI-induced neuroinflammation has been hypothesized to contribute very substantially to the pathological progression of brain injury, in addition to the primary injury itself [5, 6]. It is a complex interaction between the cellular components of the CNS (neurons, astrocytes, microglia), cytokines, and chemokines, in concert with influx of peripheral immune cells. Neuroinflammation is beneficial to promote clearance of debris and regeneration, but it can also cause collateral damage when dysregulated and excessive, leading to secondary brain injury. The fact that the intracranial space is inherently non-compliant, being enclosed by the rigid skull, with the evident advantage to primarily protect the brain, is a clear and unique disadvantage compared with trauma to other vital organs, when swelling occurs. Every organ develops edema in response to significant trauma, but when brain tissue develops edema facilitated by neuroinflammation, the rigid and protective skull bone is a barrier to allow for this swelling. Progressive brain edema and subsequent tissue swelling, that is refractory to ICP-lowering medical ICU treatments, will ultimately elicit brain ischemia due to impeded cerebral blood flow caused by high ICP, unless decompressive craniectomy (DC) is performed in the secondary injury phase after TBI, as has only recently been studied in a well-performed clinical trial [7].
After the (sub)acute phase of injury, a prolonged state of chronic inflammation may linger for years after TBI and predispose patients to develop other neurodegenerative disorders, such as dementia [8, 9]. Probably chronic traumatic encephalopathy is caused by a similar pathophysiology which has been shown to occur after recurrent mild trauma, like sport-related injuries [10].
Early attenuation of neuroinflammation is therefore considered an important target for TBI treatment, especially in the early in-hospital phase. Despite the vast amount of research performed to improve our understanding of the pathophysiology in TBI, the field has repeatedly experienced collective failures to translate research from animals to successful therapeutic application in humans [11, 12]. This review will summarize the most important drivers of neuroinflammation in TBI and previous trials aiming to attenuate these drivers. The main focus of the review will be the role of the complement system in post-traumatic neuroinflammation and future directions in research on complement inhibition.
Neuroinflammation in the Clinical Setting
The primary effects of moderate and severe TBI include diffuse injuries such as diffuse axonal injury (DAI) and focal brain damage, such as epidural and subdural hematomas (ASDH) and intracerebral hematomas/contusions (tICH). In the first hours after head trauma, expansion of hematomas is the main threat, whereas during the following days, the pathophysiological consequences of neuroinflammation may subsequently increase ICP [13]. International guidelines recommend monitoring of ICP in all patients with severe TBI at high risk of secondary injuries and abnormalities on computed tomography (CT) [14]. Currently, invasive monitoring with an intraparenchymally placed sensor is the most reliable and most applied method to monitor ICP. If ICP can be maintained below a threshold of between 20 and 25 mm Hg with general supportive intensive care treatments, including appropriate pain control, ventilator support, careful fluid, and temperature management, this portends a better prognosis [15]. Management of ICP has evolved towards a “staircase” approach with an escalating treatment intensity, including cerebrospinal fluid (CSF) drainage, deeper sedation, hyperosmolar therapy to dehydrate the brain and prevent a rise in ICP, evacuation of hematomas by craniotomy and, in case of raised ICP refractory to medical managements, including barbiturates, and in the end: DC [13, 14]. High mortality has been related to the occurrence of increased ICP, and although a lower mortality has been reported when treating ICP with a DC, these patients will have higher rates of vegetative state and severe neurological disability [7]. Importantly, this current clinical practice has concentrated on trying to mitigate ICP to minimize the extent of secondary injury once this process has already started, rather than focused on managing neuroinflammatory pathways leading to a rise in ICP and thus to prevent secondary injury.
Molecular Mechanism of Neuroinflammation After TBI
Cerebral ischemia and direct traumatic apoptosis after TBI lead to disruption of cerebral energy metabolism due to depletion of cellular adenosine triphosphate (ATP) stores. In this state, cells produce energy via the less efficient pathway of anaerobic glycolysis. Cell metabolic impairment is followed by membrane depolarization and release of excitatory glutamate [16]. Excess accumulation of extracellular glutamate causes neuronal excitotoxicity, which is a major contributor to post-traumatic neurodegeneration. It results in excessive calcium influx within the neuronal cytoplasm due to increased activation of the N-methyl-D-aspartate (NMDA) receptors and voltage-dependent ion channels [17]. The increased intracellular Ca2+ levels lead to mitochondrial damage, lipid peroxidation, production of free radicals, and activation of caspases and other proteases involved in membrane and nucleosomal DNA changes. This results in synapse elimination and neuronal apoptosis [17, 18].
Cell injury in the brain results in the release of intracellular components such as ATP, other damage-associated molecular patterns (DAMPs), and complement components which activate pattern recognition receptors (PRRs) on glial cells. As such, injury of brain cells initiates and perpetuates a post-traumatic neuroinflammatory response [19, 20].
Microglia are brain cells acting as support cells for neurons and other metabolic and immunological processes and play a critical role in neuroinflammation as the first line of defense. Microglial cell activation is prominent at the perilesional area and in ipsilateral and contralateral regions of the contusion area [21]. In the post-traumatic phase, microglial cells undergo morphological transformations and changes in their gene expression repertoire to produce a wide spectrum of pro- or anti-inflammatory cytokines (Fig. 1). A proinflammatory M1 phenotype characterized by the production of IL-1β and TNF-α, nitric oxide and reactive oxygen species (ROS), and an anti-inflammatory M2 phenotype characterized by the production of IL-10, IL-13 and transforming growth factor (TGF)-β have been previously described for the activated microglia [22]. However, nowadays is it well recognized that the M1 and M2 states are the extremes of a continuum of activation states and intermediate phenotypes are present [23]. In TBI, microglial polarization is being skewed towards a proinflammatory state varying with increasing time after the trauma [24]. Similar to microglia, a proinflammatory and an anti-inflammatory state have been described for activated astrocytes [25]. These proinflammatory astrocytes secrete many cytokines and other factors that may greatly enhance the inflammatory response [25]. Astrocytes may respond to trauma with proliferation followed by assembly of a dense barrier, known as the glial scar, aiming to protect healthy tissue from nearby areas of neuroinflammation [26].

Pathophysiology of neuroinflammation after TBI
Important factors in the promotion of neuroinflammation are proinflammatory cytokines. Well known is IL-1β, involved in oligodendrocyte damage and early microglia activation. Levels of IL-1β correlate with Glasgow Coma Scale (GCS) scores, ICP, and outcome [27, 28]. Production of IL-1β by glia requires an activated NLRP3 inflammasome, which catalyzes the cleavage of pro-interleukins into their active forms [29]. Deactivation of inflammasomes results in the alleviation of brain edema, reduction of lesion volume, and improvement of long-term motor and cognitive function in experimental TBI in animals [30, 31]. TNFα is another key cytokine in post-traumatic cerebral neuroinflammation. Human carriers of two TNF alleles, resulting in higher TNFα production in response to TBI, had a higher probability of poor outcome after TBI [32, 33].
Besides their role in neuroinflammation, proinflammatory cytokines may also challenge the integrity of the BBB vasculature. This could lead to vasogenic brain edema and penetration of serum proteins into brain interstitium, such as complement components and fibrin which can further activate glial cells [34]. The BBB consists of endothelial cells, astrocytic endfeet, and pericytes, and its integrity results from the selectivity of the tight junctions between the endothelial cells to restrict the passage of solutes [35]. Disruption of the BBB integrity is primarily caused by damage of these tight junction proteins, especially occludin and claudin-5, and it is further challenged by post-traumatic systemic inflammation which promotes leukocyte chemotaxis and transendothelial migration [36, 37].
Leukocyte recruitment into the CNS is mediated by upregulation of endothelial and leukocyte adhesion molecules and activated complement fragments, as discussed later on [38]. Neutrophils are the first-line transmigrated immune cells and can be found as early as 2 h after injury and peak within 24–48 h, before rapidly declining over the following days [39]. Cerebral accumulation of neutrophils has been associated with increased secondary brain damage and adverse outcome [40]. The early neutrophil recruitment is followed by infiltration of lymphocytes and monocyte-derived macrophages. Of note, the neutrophil-to-lymphocyte ratio has been reported as an objective, low-cost, and early predictor of inflammation and clinical outcome in TBI patients [41].
The Complement System in TBI—Pathophysiology and Animal Models
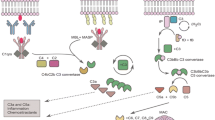
The complement system functions to eliminate foreign pathogens and substances and remove debris and apoptotic cells [42]. Complement activation can be mediated in three distinct pathways: the classical, lectin, and alternative pathways (Fig. 2), all resulting in the formation of the membrane attack complex (MAC). The host is protected against overactivation of the complement cascade by the complement regulatory proteins. These include the C1-inhibitor (C1-INH), which inactivates the C1r, C1s, and MASPs, the decay-accelerating factor (DAF or CD55) which accelerates the breakdown of C3 and C5 convertases, factor H (fH) leading to breakdown of the C3 convertase, membrane cofactor protein (MCP or CD46) aimed to cleave C3b, and CD59 protein which prevents MAC formation.

Pathways of the complement system
Complement has a key contribution to pathophysiological events that sequester the initial impact of injury and may radiate damage from the contusion core to the penumbra after TBI. For instance, glial synthesis of complement C3 and clusterin, a regulator of complement, was found in the vicinity of experimental brain contusion [43]. Further, C3d and C9 were localized on neurons in the vicinity of experimental brain contusion, suggesting that these neurons are targets of complement proteins [43].
High-quality preclinical evidence suggests that inhibition of various complement factors can improve neurological performance and reduce inflammation (Table 1). For example, our data on the closed head injury model of TBI showed that pharmacological inhibition of MAC formation improved neurological performance in mice by reducing inflammasome activation and preventing microglial activation and axonal damage [44]. In line with these data, mice with a genetic deletion in the CD59a gene, being deficient for a major regulator of MAC formation and therefore resulting in excessive MAC formation, showed increased neuronal cell death and brain tissue destruction following head trauma [45, 46].
Facilitation of phagocytosis is another important contribution of complement activation to secondary damage in TBI. Cerebral biosynthesis of most complement proteins is induced in response to trauma and adds to the pool of complement proteins which penetrate the compromised BBB [47]. Intracerebral complement activation leads to the generation of complement opsonins which facilitate the clearance of debris at the site of injury by microglia and macrophages bearing the CR3 receptor [48]. In addition, elevated levels of C1q within the cerebral parenchyma promote the transformation of microglia to the proinflammatory phenotype [49]. Lastly, C1q and C3 play a key role in microglia-mediated synapse elimination which is a prominent neurodegenerative mechanism after head trauma [50].
Activated microglial cells induce a proinflammatory phenotype to astrocytes via secretion of C1q, IL-1α, and TNFα, contributing to the rapid death or functional disability of neurons and oligodendrocytes [25]. Proinflammatory astrocytes, in turn, secrete many complement components, such as C3, that enhance the neuroinflammatory response [25, 51].
In addition to the complement components which are synthesized by neurons and glial cells within the CNS [52, 53], there is an influx of complement factors from the blood due to breakdown of the BBB after TBI [54, 55]. The anaphylatoxins C3a and C5a act as chemoattractants for immune cells expressing the relevant receptors such as the granulocytes which are present within TBI lesions [56]. In addition, in vitro studies showed that C5a activates the expression of beta2-integrin on neutrophils promoting their adhesion to the inflamed endothelium of the BBB and stimulates the secretion of the proinflammatory mediators TNFα and IL1 by human mononuclear cells [57,58,59]. Notably, animal studies showed that genetic deletion of the genes encoding for the C3 or the C5 component or pharmacological inhibition of C3 or C5 resulted in a reduction of neutrophil infiltration, injury size, microglial activation. and brain edema leading to significantly improved neurological outcomes [60,61,62,63,64].
The Interaction of the Complement System with the Coagulation System
In TBI, hemostasis is often derailed, either leading to a hypo-coagulopathic state on one end of the spectrum, causing cerebral bleeding disorders leading to progression of contusions into growing t-ICHs and ASDHs, and to a hyper-coagulopathic state that contributes to ischemic lesions due to (micro)vascular thrombosis in lesioned areas [65]. The intimate interaction and co-evolution of the coagulation system together with the complement system is widely appreciated within the basic science research field. The complement system has been found to increase tissue factor activity, thereby activating the extrinsic coagulation pathway, and form activated thrombin from prothrombin. Moreover, complement factors increase platelet activity and aggregation and prothrombinase activity, including von Willebrand factor and P-selectin. Classical and lectin pathway activation has been reported to be associated with increased odds of venous thromboembolism in the clinical setting [66, 67]. Moreover, in sepsis patients, disseminated intravascular coagulation (DIC) was correlated to the degree of complement activation [68]. It is also known that MAC attenuates endothelium-dependent relaxation leading to a hypertensive state [69]. This evidence, reviewed in [70], suggests that complement overactivation shortly after TBI could potentially lead to increased coagulation activity, i.e., a prothrombotic state often seen days to weeks following TBI. Nevertheless, the correlation between trajectories of complement activity and markers of hemostasis and platelet function, specifically in TBI, warrants more research.
Complement Activation in TBI—Human Studies
In TBI patients, high levels of C4, C3, and MAC have been found in serum [54, 71,72,73,74,75,76], and upregulation of factor B, C3, and MAC was detected in the CSF of severe TBI patients [55, 77]. Moreover, increased immunoreactivity was found in resected contused tissue for C1q, C3b, C3d, and MAC within/on neurons located in the penumbra area [78, 79]. Intracerebral deposition of MBL, ficolin-2 and 3, and MASP-2 and 3 was found after TBI within the vasculature and in the injured perivascular tissue [79, 80]. High levels of complement proteins were strongly associated with lower GCS scores and independently predict mortality or unfavorable clinical outcomes in TBI [73, 75]. A proteomics study using human frontotemporal cortex samples showed a consistent overexpression of C4a, C4b, C3, C7, and C9 [81]. More recently, microvesicles and exosomes were analyzed in the CSF of TBI patients and mass spectrometry-based proteomic identification of proteins indicated presence of complement C1q [82]. Furthermore, plasma astrocyte-derived exosomes (ADEs) protein levels of C4b, factor D, Bb, MBL, C3b, and MAC were significantly higher and those of the regulatory proteins CR1 and CD59 lower in the first week of TBI compared to controls [83].
The complement system is further triggered by secondary insults [84]. Expression of complement proteins C3, C8a, and C9 is still increased in the plasma of TBI patients at 1, 3, and 6 months after injury compared to controls, suggesting persistent complement activation during the subacute and chronic phase [85]. Prolonged complement activation has been linked to early-onset cognitive decline, behavior disorders, and predisposition to dementia syndromes like Alzheimer’s disease [86].
Clinical Trials to Control Neuroinflammation: What Has Been Tried So Far?
Studies on the dysregulated inflammatory response are important to serve as a roadmap for future clinical trials aiming at “targeted” pharmacological neuroprotection and improved neurological recovery after TBI. Although all of the described interventions within trials up to now (Table 2) have shown to be effective in preclinical and small single center phase II trials, successful translation to phase III clinical trials showing efficacy of these treatments has not yet been accomplished. Multiple explanations have been proposed to explain these failures. First, the heterogeneity of the TBI population and the large treatment variation in the management of TBI indicate that large sample sizes are warranted to achieve any statistical significant difference between groups [87, 88]. Second, there is a growing recognition of the problem of age and sex bias on the outcomes in neurotrauma research, as for example, fewer women than men are recruited in clinical trials (Table 2) [89, 90]. Third, most trials focused on “delayed” outcome metrics, such as the Glasgow Outcome Scale Extended (GOSE) at 6 months, as primary endpoint, whereas animal models focus on the direct impact of a therapy on microglial activation, edema formation, or neuronal death.
Last and most importantly, most trials did not focus on a targeted dysregulated part of neuroinflammation after the first TBI impact. The steroid trials, as described in Table 2, show that broad inhibition of the immune response can be deleterious after TBI, and therefore a more targeted approach focused on a specific neuroinflammatory pathway and during a limited period of time may be more successful in improving outcomes [91, 92]. Current and future clinical trials aiming to reduce secondary brain injury should focus on targeting well-defined specific pathways with a closely related endpoint to the therapeutic mechanism of action to test efficacy. This should be based on both thorough and sufficient preclinical testing in multiple injury models (including different age ranges and sex), together with a detailed insight into the most important drivers of TBI pathophysiology for each individual patient.
Complement Inhibition in the Clinical Setting—Future Directions
Currently, no clinical trials are present in literature aiming to inhibition complement activation in brain injury. Only a few drugs, C1-esterase inhibitors (C1-INH), Cinryze, Berinert and Ruconest, and C5-inhibitors, eculizumab and ravulizumab, are approved complement inhibitory drugs, but many others are in clinical development. The indications for the current approved drugs are hereditary angioedema (C1-INH) and paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome, and neuromyelitis optica (C5). A trial with C5-antibodies in patients with aneurysmal subarachnoid hemorrhage (SAH) is now recruiting participants [93]. As it has been reported that inhibition more upstream in the complement cascade is necessary to prevent the amplification of a feedforward mechanism of neuroinflammation that persists throughout the chronic phase [94], C1-INH might be more effective to attenuate complement overactivation. C1-INH is a potent multi-target serpin, which effectively inhibits activation of the classical, lectin, and alternative pathways [95,96,97]. Administration of C1-INH in animal models showed reduced contusion volume and brain water content and improvement of cognitive and motor function [98, 99]. In addition to its role in complement inhibition, C1-INH is a known inhibitor of FXIIa, FXIa, FXII, thrombin, kallikrein, HMWK prekallikrein complexes, and plasmin which inhibit fibrinolysis, contact activation, and coagulation [100, 101]. Efficacy and an excellent safety profile of high doses of C1-INH have been reported in off-label trials in sepsis and ischemia–reperfusion injury patients [102, 103]. Therefore, we are currently recruiting TBI patients in the Complement Inhibition: Attacking Overshooting inflammation @fter Traumatic Brain Injury (CIAO@TBI) trial to assess the safety and efficacy of C1-INH in this patient population [104]. In this trial, patients will be randomized to either receive one dose of 1600 IU C1-INH or a placebo injection. The primary outcome is the therapy intensity level scale that measures all ICP-directed interventions. This study will provide insight in the promising role of complement inhibition in brain injury. In the meantime, more research is warranted towards defining the inflammatory phenotypes of our patients based on injury characteristics (e.g., age, sex, and injury severity), imaging, and biomarkers to eventually being able to target inflammation with personalized immunomodulatory treatments,
Conclusion
Neuroinflammation is one of the “nonsurgical” key drivers causing secondary brain injury after TBI. Attenuation of the inflammatory response is a potential therapeutic target. This review covers the most important neuroinflammatory drivers resulting from TBI and summarizes the clinical work performed to date directed to attenuate neuroinflammation. The complement system play an important role in the pathophysiology of TBI, and therefore therapies targeting this pathway might contribute to future targeted therapy, currently evaluated in a clinical trial.
Availability of Data and Materials
Not applicable.
Abbreviations
- C1-INH:
-
C1-inhibitor
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- GCS:
-
Glasgow Coma Scale
- GOSE:
-
Glasgow Outcome Scale Extended
- ICP:
-
Intracranial pressure
- TBI:
-
Traumatic brain injury
- TXA:
-
Tranexamic acid
References
Hardcastle N, Benzon HA, Vavilala MS. Update on the 2012 guidelines for the management of pediatric traumatic brain injury - information for the anesthesiologist. Paediatr Anaesth. 2014;24(7):703–10.
Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7(8):728–41.
Hanrahan F, Campbell M. Neuroinflammation. In: Laskowitz D, Grant G, editors. Translational research in traumatic brain injury. Boca Raton (FL): CRC Press/Taylor and Francis Group; 2016. Chapter 6. Available from: https://www.ncbi.nlm.nih.gov/books/NBK326720/.
Krakau K, et al. Nutritional treatment of patients with severe traumatic brain injury during the first six months after injury. Nutrition. 2007;23(4):308–17.
Wofford KL, Loane DJ, Cullen DK. Acute drivers of neuroinflammation in traumatic brain injury. Neural Regen Res. 2019;14(9):1481–9.
Patterson ZR, Holahan MR. Understanding the neuroinflammatory response following concussion to develop treatment strategies. Front Cell Neurosci. 2012;6:58.
Hutchinson PJ, et al. Trial of decompressive craniectomy for traumatic intracranial hypertension. N Engl J Med. 2016;375(12):1119–30.
Fann JR, et al. Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry. 2018;5(5):424–31.
Pischiutta F, et al. Single severe traumatic brain injury produces progressive pathology with ongoing contralateral white matter damage one year after injury. Exp Neurol. 2018;300:167–78.
Pierre K, et al. Chronic traumatic encephalopathy: update on current clinical diagnosis and management. Biomedicines. 2021;9(4).
Bragge P, et al. A state-of-the-science overview of randomized controlled trials evaluating acute management of moderate-to-severe traumatic brain injury. J Neurotrauma. 2016;33(16):1461–78.
Maas AI, Roozenbeek B, Manley GT. Clinical trials in traumatic brain injury: past experience and current developments. Neurotherapeutics. 2010;7(1):115–26.
Stocchetti N, Maas AI. Traumatic intracranial hypertension. N Engl J Med. 2014;370(22):2121–30.
Carney N, et al. Guidelines for the management of severe traumatic brain injury, Fourth Edition. Neurosurgery. 2017;80(1):6–15.
Alam HB, et al. Western Trauma Association critical decisions in trauma: management of intracranial hypertension in patients with severe traumatic brain injuries. J Trauma Acute Care Surg. 2020;88(2):345–51.
Guerriero RM, Giza CC, Rotenberg A. Glutamate and GABA imbalance following traumatic brain injury. Curr Neurol Neurosci Rep. 2015;15(5):27.
Margolick J, et al. A systematic review of the risks and benefits of venous thromboembolism prophylaxis in traumatic brain injury. Can J Neurol Sci. 2018;45(4):432–44.
Suliman HB, Piantadosi CA. Mitochondrial quality control as a therapeutic target. Pharmacol Rev. 2016;68(1):20–48.
Laird MD, et al. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia. 2014;62(1):26–38.
Wang KY, et al. Plasma high-mobility group box 1 levels and prediction of outcome in patients with traumatic brain injury. Clin Chim Acta. 2012;413(21–22):1737–41.
Zhao J, et al. Glial response in early stages of traumatic brain injury. Neurosci Lett. 2019;708:134335.
Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol. 2016;173(4):692–702.
Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13.
Izzy S, et al. Time-dependent changes in microglia transcriptional networks following traumatic brain injury. Front Cell Neurosci. 2019;13:307.
Liddelow SA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–7.
Anderson MA, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016;532(7598):195–200.
Flygt J, et al. Neutralization of interleukin-1beta following diffuse traumatic brain injury in the mouse attenuates the loss of mature oligodendrocytes. J Neurotrauma. 2018;35(23):2837–49.
Shiozaki T, et al. Cerebrospinal fluid concentrations of anti-inflammatory mediators in early-phase severe traumatic brain injury. Shock. 2005;23(5):406–10.
Lin C, et al. Omega-3 fatty acids regulate NLRP3 inflammasome activation and prevent behavior deficits after traumatic brain injury. Exp Neurol. 2017;290:115–22.
Xu X, et al. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol Dis. 2018;117:15–27.
Ismael S, Nasoohi S, Ishrat T. MCC950, the selective inhibitor of nucleotide oligomerization domain-like receptor protein-3 inflammasome, protects mice against traumatic brain injury. J Neurotrauma. 2018;35(11):1294–303.
Waters RJ, et al. Cytokine gene polymorphisms and outcome after traumatic brain injury. J Neurotrauma. 2013;30(20):1710–6.
Rowe RK, et al. Novel TNF receptor-1 inhibitors identified as potential therapeutic candidates for traumatic brain injury. J Neuroinflammation. 2018;15(1):154.
Tanno H, et al. Breakdown of the blood-brain barrier after fluid percussive brain injury in the rat. Part 1: Distribution and time course of protein extravasation. J Neurotrauma. 1992;9(1):21–32.
Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015;7(1):a020412.
Nag S, Venugopalan R, Stewart DJ. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. 2007;114(5):459–69.
Cunningham TL, et al. Correlations between blood-brain barrier disruption and neuroinflammation in an experimental model of penetrating ballistic-like brain injury. J Neurotrauma. 2014;31(5):505–14.
Scholz M, et al. Neutrophils and the blood-brain barrier dysfunction after trauma. Med Res Rev. 2007;27(3):401–16.
Clark RS, et al. Neutrophil accumulation after traumatic brain injury in rats: comparison of weight drop and controlled cortical impact models. J Neurotrauma. 1994;11(5):499–506.
Vaibhav K, et al. Neutrophil extracellular traps exacerbate neurological deficits after traumatic brain injury. Sci Adv. 2020;6(22):eaax8847.
Sabouri E, et al. Neutrophil-to-lymphocyte ratio and traumatic brain injury: a review study. World Neurosurg. 2020;140:142–7.
Sarma JV, Ward PA. The complement system. Cell Tissue Res. 2011;343(1):227–35.
Bellander BM, et al. Activation of the complement cascade and increase of clusterin in the brain following a cortical contusion in the adult rat. J Neurosurg. 1996;85(3):468–75.
Fluiter K, et al. Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. J Immunol. 2014;192(5):2339–48.
Stahel PF, et al. Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. J Neuroinflammation. 2009;6:2.
Ruseva MM, et al. An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice. Proc Natl Acad Sci U S A. 2015;112(46):14319–24.
Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Mol Immunol. 2011;48(14):1592–603.
Ricklin D, et al. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–97.
Färber K, et al. C1q, the recognition subcomponent of the classical pathway of complement, drives microglial activation. J Neurosci Res. 2009;87(3):644–52.
Fumagalli S, et al. The ischemic environment drives microglia and macrophage function. Front Neurol. 2015;6:81.
Clark DPQ, et al. Inflammation in traumatic brain injury: roles for toxic A1 astrocytes and microglial-astrocytic crosstalk. Neurochem Res. 2019;44(6):1410–24.
Levi-Strauss M, Mallat M. Primary cultures of murine astrocytes produce C3 and factor B, two components of the alternative pathway of complement activation. J Immunol. 1987;139(7):2361–6.
Gasque P, Fontaine M, Morgan BP. Complement expression in human brain. Biosynthesis of terminal pathway components and regulators in human glial cells and cell lines. J Immunol. 1995;154(9):4726–33.
Nekludov M, et al. Coagulation abnormalities associated with severe isolated traumatic brain injury: cerebral arterio-venous differences in coagulation and inflammatory markers. J Neurotrauma. 2007;24(1):174–80.
Stahel PF, et al. Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood-brain barrier dysfunction in patients with traumatic brain injury. J Neurotrauma. 2001;18(8):773–81.
Hajishengallis G, Lambris JD. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol. 2010;31(4):154–63.
Jagels MA, Daffern PJ, Hugli TE. C3a and C5a enhance granulocyte adhesion to endothelial and epithelial cell monolayers: epithelial and endothelial priming is required for C3a-induced eosinophil adhesion. Immunopharmacology. 2000;46(3):209–22.
Okusawa S, et al. C5a stimulates secretion of tumor necrosis factor from human mononuclear cells in vitro. Comparison with secretion of interleukin 1 beta and interleukin 1 alpha. J Exp Med. 1988;168(1):443–8.
Okusawa S, et al. C5a induction of human interleukin 1. Synergistic effect with endotoxin or interferon-gamma. J Immunol. 1987;139(8):2635–40.
Yang S, et al. The role of complement C3 in intracerebral hemorrhage-induced brain injury. J Cereb Blood Flow Metab. 2006;26(12):1490–5.
Sewell DL, et al. Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J Neuroimmunol. 2004;155(1–2):55–63.
Leinhase I, et al. Reduced neuronal cell death after experimental brain injury in mice lacking a functional alternative pathway of complement activation. BMC Neurosci. 2006;7:55.
You Z, et al. Reduced tissue damage and improved recovery of motor function after traumatic brain injury in mice deficient in complement component C4. J Cereb Blood Flow Metab. 2007;27(12):1954–64.
Garrett MC, et al. Synergistic neuroprotective effects of C3a and C5a receptor blockade following intracerebral hemorrhage. Brain Res. 2009;1298:171–7.
Maegele M, et al. Coagulopathy and haemorrhagic progression in traumatic brain injury: advances in mechanisms, diagnosis, and management. Lancet Neurol. 2017;16(8):630–47.
Hoiland II, et al. Associations between complement pathways activity, mannose-binding lectin, and odds of unprovoked venous thromboembolism. Thromb Res. 2018;169:50–6.
Liang RA, et al. Plasma levels of mannose-binding lectin and future risk of venous thromboembolism. J Thromb Haemost. 2019;17(10):1661–9.
Abe T, et al. Complement activation in human sepsis is related to sepsis-induced disseminated intravascular coagulation. Shock. 2020.
Stahl GL, Reenstra WR, Frendl G. Complement-mediated loss of endothelium-dependent relaxation of porcine coronary arteries. Role of the terminal membrane attack complex. Circ Res. 1995;76(4):575–83.
Fletcher-Sandersjöö A, Maegele M, Bellander BM. Does complement-mediated hemostatic disturbance occur in traumatic brain injury? A literature review and observational study protocol. Int J Mol Sci. 2020;21(5).
Becker P, et al. Complement activation following head and brain trauma. Anaesthesist. 1987;36(6):301–5.
Wang JW, et al. Humoral and cellular immunity changed after traumatic brain injury in human patients. Ann Clin Lab Sci. 2017;47(1):10–6.
Yu W, et al. High levels of serum mannose-binding lectins are associated with the severity and clinical outcomes of severe traumatic brain injury. Clin Chim Acta. 2015;451(Pt B):111–6.
Pan JW, et al. Low serum ficolin-3 levels are associated with severity and poor outcome in traumatic brain injury. J Neuroinflammation. 2015;12:226.
Osthoff M, et al. Association of lectin pathway protein levels and genetic variants early after injury with outcomes after severe traumatic brain injury: a prospective cohort study. J Neurotrauma. 2017;34(17):2560–6.
Parry J, et al. Soluble terminal complement activation fragment sC5b-9: a new serum biomarker for traumatic brain injury? Eur J Trauma Emerg Surg. 2020.
Kossmann T, et al. Elevated levels of the complement components C3 and factor B in ventricular cerebrospinal fluid of patients with traumatic brain injury. J Neuroimmunol. 1997;73(1–2):63–9.
Bellander BM, et al. Complement activation in the human brain after traumatic head injury. J Neurotrauma. 2001;18(12):1295–311.
De Blasio D, et al. Human brain trauma severity is associated with lectin complement pathway activation. J Cereb Blood Flow Metab. 2019;39(5):794–807.
Longhi L, et al. Mannose-binding lectin is expressed after clinical and experimental traumatic brain injury and its deletion is protective. Crit Care Med. 2014;42(8):1910–8.
Xu B, et al. Protein profile changes in the frontotemporal lobes in human severe traumatic brain injury. Brain Res. 2016;1642:344–52.
Manek R, et al. Protein Biomarkers and neuroproteomics characterization of microvesicles/exosomes from human cerebrospinal fluid following traumatic brain injury. Mol Neurobiol. 2018;55(7):6112–28.
Goetzl EJ, et al. Traumatic brain injury increases plasma astrocyte-derived exosome levels of neurotoxic complement proteins. Faseb j. 2020;34(2):3359–66.
Bellander BM, et al. Secondary insults following traumatic brain injury enhance complement activation in the human brain and release of the tissue damage marker S100B. Acta Neurochir (Wien). 2011;153(1):90–100.
Bao W, et al. Complement cascade on severe traumatic brain injury patients at the chronic unconscious stage: implication for pathogenesis. Expert Rev Mol Diagn. 2018;18(8):761–6.
Alawieh A, et al. Complement drives synaptic degeneration and progressive cognitive decline in the chronic phase after traumatic brain injury. J Neurosci. 2021;41(8):1830–43.
Maas AIR, et al. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017;16(12):987–1048.
Huijben JA, et al. Variation in general supportive and preventive intensive care management of traumatic brain injury: a survey in 66 neurotrauma centers participating in the Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury (CENTER-TBI) study. Crit Care. 2018;22(1):90.
Gupte R, et al. Sex differences in traumatic brain injury: what we know and what we should know. J Neurotrauma. 2019;36(22):3063–91.
Delage C, et al. Traumatic brain injury: an age-dependent view of post-traumatic neuroinflammation and its treatment. Pharmaceutics. 2021;13(10).
Edwards P, et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet. 2005;365(9475):1957–9.
Cox CS Jr, Juranek J, Bedi S. Clinical trials in traumatic brain injury: cellular therapy and outcome measures. Transfusion. 2019;59(S1):858–68.
Koopman I, Rinkel GJE, Vergouwen MDI. CompLement C5 antibodies for decreasing brain injury after aneurysmal subarachnoid haemorrhage (CLASH): study protocol for a randomised controlled phase II clinical trial. Trials. 2020;21(1):969.
Alawieh A, et al. Identifying the role of complement in triggering neuroinflammation after traumatic brain injury. J Neurosci. 2018;38(10):2519–32.
Matsushita M, et al. Proteolytic activities of two types of mannose-binding lectin-associated serine protease. J Immunol. 2000;165(5):2637–42.
Jiang H, et al. Complement 1 inhibitor is a regulator of the alternative complement pathway. J Exp Med. 2001;194(11):1609–16.
Nielsen EW, et al. Effect of supraphysiologic levels of C1-inhibitor on the classical, lectin and alternative pathways of complement. Mol Immunol. 2007;44(8):1819–26.
Longhi L, et al. C1-inhibitor attenuates neurobehavioral deficits and reduces contusion volume after controlled cortical impact brain injury in mice. Crit Care Med. 2009;37(2):659–65.
Weiss E, et al. Effect of complement C1-esterase inhibitor on brain edema and inflammation after mild traumatic brain injury in an animal model. Clin Exp Emerg Med. 2020;7(2):87–94.
Zeerleder S. C1-inhibitor: more than a serine protease inhibitor. Semin Thromb Hemost. 2011;37(4):362–74.
Brown EW, Ravindran S, Patston PA. The reaction between plasmin and C1-inhibitor results in plasmin inhibition by the serpin mechanism. Blood Coagul Fibrinolysis. 2002;13(8):711–4.
Caliezi C, et al. C1-inhibitor in patients with severe sepsis and septic shock: beneficial effect on renal dysfunction. Crit Care Med. 2002;30(8):1722–8.
de Zwaan C, et al. Continuous 48-h C1-inhibitor treatment, following reperfusion therapy, in patients with acute myocardial infarction. Eur Heart J. 2002;23(21):1670–7.
van Erp IAM, et al. Safety and efficacy of C1-inhibitor in traumatic brain injury (CIAO@TBI): study protocol for a randomized, placebo-controlled, multi-center trial. Trials. 2021;22(1):874.
De Blasio D, et al. Pharmacological inhibition of mannose-binding lectin ameliorates neurobehavioral dysfunction following experimental traumatic brain injury. J Cereb Blood Flow Metab. 2017;37(3):938–50.
Hicks RR, et al. Vaccinia virus complement control protein enhances functional recovery after traumatic brain injury. J Neurotrauma. 2002;19(6):705–14.
Kaczorowski SL, et al. Effect of soluble complement receptor-1 on neutrophil accumulation after traumatic brain injury in rats. J Cereb Blood Flow Metab. 1995;15(5):860–4.
Kotwal GJ, Lahiri DK, Hicks R. Potential intervention by Vaccinia virus complement control protein of the signals contributing to the progression of central nervous system injury to Alzheimer’s disease. Ann N Y Acad Sci. 2002;973:317–22.
Krukowski K, et al. Traumatic brain injury in aged mice induces chronic microglia activation, synapse loss, and complement-dependent memory deficits. Int J Mol Sci. 2018;19(12).
Leinhase I, et al. Inhibition of the alternative complement activation pathway in traumatic brain injury by a monoclonal anti-factor B antibody: a randomized placebo-controlled study in mice. J Neuroinflammation. 2007;4:13.
Leinhase I, et al. Pharmacological complement inhibition at the C3 convertase level promotes neuronal survival, neuroprotective intracerebral gene expression, and neurological outcome after traumatic brain injury. Exp Neurol. 2006;199(2):454–64.
Li Y, et al. Protective effects of decay-accelerating factor on blast-induced neurotrauma in rats. Acta Neuropathol Commun. 2013;1:52.
Mercurio D, et al. Targeted deletions of complement lectin pathway genes improve outcome in traumatic brain injury, with MASP-2 playing a major role. Acta Neuropathol Commun. 2020;8(1):174.
Neher MD, et al. Deficiency of complement receptors CR2/CR1 in Cr2−/− mice reduces the extent of secondary brain damage after closed head injury. J Neuroinflammation. 2014;11:95.
Pillay NS, Kellaway LA, Kotwal GJ. Vaccinia virus complement control protein significantly improves sensorimotor function recovery after severe head trauma. Brain Res. 2007;1153:158–65.
Pillay NS, Kellaway LA, Kotwal GJ. Administration of Vaccinia virus complement control protein shows significant cognitive improvement in a mild injury model. Ann N Y Acad Sci. 2005;1056:450–61.
Rancan M, et al. Central nervous system-targeted complement inhibition mediates neuroprotection after closed head injury in transgenic mice. J Cereb Blood Flow Metab. 2003;23(9):1070–4.
Rich MC, et al. Site-targeted complement inhibition by a complement receptor 2-conjugated inhibitor (mTT30) ameliorates post-injury neuropathology in mouse brains. Neurosci Lett. 2016;617:188–94.
Yager PH, et al. Mannose binding lectin gene deficiency increases susceptibility to traumatic brain injury in mice. J Cereb Blood Flow Metab. 2008;28(5):1030–9.
VanLandingham JW, et al. Neurosteroids reduce inflammation after TBI through CD55 induction. Neurosci Lett. 2007;425(2):94–8.
Drew PD, Chavis JA. Female sex steroids: effects upon microglial cell activation. J Neuroimmunol. 2000;111(1–2):77–85.
He J, et al. Progesterone and allopregnanolone reduce inflammatory cytokines after traumatic brain injury. Exp Neurol. 2004;189(2):404–12.
Brotfain E, et al. Neuroprotection by estrogen and progesterone in traumatic brain injury and spinal cord injury. Curr Neuropharmacol. 2016;14(6):641–53.
Cutler SM, et al. Progesterone improves acute recovery after traumatic brain injury in the aged rat. J Neurotrauma. 2007;24(9):1475–86.
Skolnick BE, et al. A clinical trial of progesterone for severe traumatic brain injury. N Engl J Med. 2014;371(26):2467–76.
Wright DW, et al. Very early administration of progesterone for acute traumatic brain injury. N Engl J Med. 2014;371(26):2457–66.
Kunz M, et al. Bradykinin in blood and cerebrospinal fluid after acute cerebral lesions: correlations with cerebral edema and intracranial pressure. J Neurotrauma. 2013;30(19):1638–44.
Chao H, et al. Activation of bradykinin B2 receptor induced the inflammatory responses of cytosolic phospholipase A2 after the early traumatic brain injury. Biochim Biophys Acta Mol Basis Dis. 2018;1864(9 Pt B):2957–71.
Trabold R, et al. The role of bradykinin B(1) and B(2) receptors for secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab. 2010;30(1):130–9.
Zweckberger K, Plesnila N. Anatibant, a selective non-peptide bradykinin B2 receptor antagonist, reduces intracranial hypertension and histopathological damage after experimental traumatic brain injury. Neurosci Lett. 2009;454(2):115–7.
Hellal F, et al. Detrimental role of bradykinin B2 receptor in a murine model of diffuse brain injury. J Neurotrauma. 2003;20(9):841–51.
Shakur H, et al. The BRAIN TRIAL: a randomised, placebo controlled trial of a Bradykinin B2 receptor antagonist (Anatibant) in patients with traumatic brain injury. Trials. 2009;10:109.
Fee D, et al. Activated/effector CD4+ T cells exacerbate acute damage in the central nervous system following traumatic injury. J Neuroimmunol. 2003;136(1–2):54–66.
Mazzeo AT, et al. Safety and tolerability of cyclosporin a in severe traumatic brain injury patients: results from a prospective randomized trial. J Neurotrauma. 2009;26(12):2195–206.
Shohami E, Novikov M, Bass R. Long-term effect of HU-211, a novel non-competitive NMDA antagonist, on motor and memory functions after closed head injury in the rat. Brain Res. 1995;674(1):55–62.
Shohami E, et al. Cytokine production in the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-alpha inhibitor and an effective neuroprotectant. J Neuroimmunol. 1997;72(2):169–77.
Gallily R, et al. Protection against septic shock and suppression of tumor necrosis factor alpha and nitric oxide production by dexanabinol (HU-211), a nonpsychotropic cannabinoid. J Pharmacol Exp Ther. 1997;283(2):918–24.
Maas AI, et al. Efficacy and safety of dexanabinol in severe traumatic brain injury: results of a phase III randomised, placebo-controlled, clinical trial. Lancet Neurol. 2006;5(1):38–45.
Knoller N, et al. Dexanabinol (HU-211) in the treatment of severe closed head injury: a randomized, placebo-controlled, phase II clinical trial. Crit Care Med. 2002;30(3):548–54.
Lieutaud T, et al. Characterization of the pharmacokinetics of human recombinant erythropoietin in blood and brain when administered immediately after lateral fluid percussion brain injury and its pharmacodynamic effects on IL-1beta and MIP-2 in rats. J Neurotrauma. 2008;25(10):1179–85.
Chen G, et al. Inhibitory effect on cerebral inflammatory agents that accompany traumatic brain injury in a rat model: a potential neuroprotective mechanism of recombinant human erythropoietin (rhEPO). Neurosci Lett. 2007;425(3):177–82.
Blixt J, Gunnarson E, Wanecek M. Erythropoietin attenuates the brain edema response after experimental traumatic brain injury. J Neurotrauma. 2018;35(4):671–80.
Juul SE, et al. Microarray analysis of high-dose recombinant erythropoietin treatment of unilateral brain injury in neonatal mouse hippocampus. Pediatr Res. 2009;65(5):485–92.
Zhou ZW, et al. Erythropoietin regulates immune/inflammatory reaction and improves neurological function outcomes in traumatic brain injury. Brain Behav. 2017;7(11):e00827.
Hellewell SC, et al. Erythropoietin improves motor and cognitive deficit, axonal pathology, and neuroinflammation in a combined model of diffuse traumatic brain injury and hypoxia, in association with upregulation of the erythropoietin receptor. J Neuroinflammation. 2013;10:156.
Menon DK, Maas AI. EPO in traumatic brain injury: two strikes…but not out? Lancet. 2015;386(10012):2452–4.
Nichol A, et al. Erythropoietin in traumatic brain injury (EPO-TBI): a double-blind randomised controlled trial. Lancet. 2015;386(10012):2499–506.
Robertson CS, et al. Effect of erythropoietin and transfusion threshold on neurological recovery after traumatic brain injury: a randomized clinical trial. JAMA. 2014;312(1):36–47.
Clausen F, et al. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci. 2009;30(3):385–96.
Tehranian R, et al. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J Neurotrauma. 2002;19(8):939–51.
Clausen F, et al. Neutralization of interleukin-1beta reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur J Neurosci. 2011;34(1):110–23.
Thome JG, et al. Contributions of interleukin-1 receptor signaling in traumatic brain injury. Front Behav Neurosci. 2019;13:287.
Helmy A, et al. Recombinant human interleukin-1 receptor antagonist promotes M1 microglia biased cytokines and chemokines following human traumatic brain injury. J Cereb Blood Flow Metab. 2016;36(8):1434–48.
Helmy A, et al. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J Cereb Blood Flow Metab. 2014;34(5):845–51.
Tomura S, et al. Effects of therapeutic hypothermia on inflammasome signaling after traumatic brain injury. J Cereb Blood Flow Metab. 2012;32(10):1939–47.
Zhang F, et al. Moderate hypothermia inhibits microglial activation after traumatic brain injury by modulating autophagy/apoptosis and the MyD88-dependent TLR4 signaling pathway. J Neuroinflammation. 2018;15(1):273.
Cooper DJ, et al. Effect of early sustained prophylactic hypothermia on neurologic outcomes among patients with severe traumatic brain injury: the POLAR randomized clinical trial. JAMA. 2018;320(21):2211–20.
Clifton GL, et al. Early induction of hypothermia for evacuated intracranial hematomas: a post hoc analysis of two clinical trials. J Neurosurg. 2012;117(4):714–20.
Clifton GL, et al. Very early hypothermia induction in patients with severe brain injury (the National Acute Brain Injury Study: Hypothermia II): a randomised trial. Lancet Neurol. 2011;10(2):131–9.
Tao L, et al. Neuroprotective effects of metformin on traumatic brain injury in rats associated with NF-kappaB and MAPK signaling pathway. Brain Res Bull. 2018;140:154–61.
Hill JL, et al. Traumatic brain injury decreases AMP-activated protein kinase activity and pharmacological enhancement of its activity improves cognitive outcome. J Neurochem. 2016;139(1):106–19.
Chen X, et al. Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-kappaB pathway following experimental traumatic brain injury. J Neuroinflammation. 2017;14(1):143.
Zhu HT, et al. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway in experimental traumatic brain injury. J Neuroinflammation. 2014;11:59.
Taheri A, et al. A randomized controlled trial on the efficacy, safety, and pharmacokinetics of metformin in severe traumatic brain injury. J Neurol. 2019;266(8):1988–97.
Li B, et al. Simvastatin attenuates microglial cells and astrocyte activation and decreases interleukin-1beta level after traumatic brain injury. Neurosurgery. 2009;65(1):179–86.
Xu X, et al. Anti-inflammatory and immunomodulatory mechanisms of atorvastatin in a murine model of traumatic brain injury. J Neuroinflammation. 2017;14(1):167.
Farzanegan GR, et al. Effects of atorvastatin on brain contusion volume and functional outcome of patients with moderate and severe traumatic brain injury; a randomized double-blind placebo-controlled clinical trial. J Clin Neurosci. 2017;44:143–7.
Sanchez-Aguilar M, et al. Effect of rosuvastatin on cytokines after traumatic head injury. J Neurosurg. 2013;118(3):669–75.
Yin J, et al. Hypertonic saline alleviates brain edema after traumatic brain injury via downregulation of aquaporin 4 in rats. Med Sci Monit. 2018;24:1863–70.
Huang LQ, et al. Hypertonic saline alleviates cerebral edema by inhibiting microglia-derived TNF-alpha and IL-1beta-induced Na-K-Cl cotransporter up-regulation. J Neuroinflammation. 2014;11:102.
Bulger EM, et al. Out-of-hospital hypertonic resuscitation following severe traumatic brain injury: a randomized controlled trial. JAMA. 2010;304(13):1455–64.
Ng SY, et al. Attenuation of microglial activation with minocycline is not associated with changes in neurogenesis after focal traumatic brain injury in adult mice. J Neurotrauma. 2012;29(7):1410–25.
Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166(12):7527–33.
Lee SM, et al. Minocycline inhibits apoptotic cell death via attenuation of TNF-alpha expression following iNOS/NO induction by lipopolysaccharide in neuron/glia co-cultures. J Neurochem. 2004;91(3):568–78.
Meythaler J, et al. Safety and feasibility of minocycline in treatment of acute traumatic brain injury. Brain Inj. 2019;33(5):679–89.
Acknowledgements
The authors would like to thank Fleur Fisher for designing the figures. The authors would like to thank Jan Schoones for his help with conducting the literature search.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
This work was supported by Hersenstichting Nederland (Dutch Brain Foundation) for the Complement Inhibition: Attacking Overshooting inflammation @fter Traumatic Brain Injury (CIAO@TBI) trial.
Author information
Authors and Affiliations
Contributions
IVE, IM, and KF wrote the manuscript. All authors contributed to and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
FB is founder of Complement Pharma BV, and KF is paid consultant for Complement Pharma BV. All other authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
van Erp, I.A.M., Michailidou, I., van Essen, T.A. et al. Tackling Neuroinflammation After Traumatic Brain Injury: Complement Inhibition as a Therapy for Secondary Injury. Neurotherapeutics 20, 284–303 (2023). https://doi.org/10.1007/s13311-022-01306-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-022-01306-8